

Effect of biogeochemical redox processes on the fate and transport of As and U at an abandoned uranium mine site: an X-ray absorption spectroscopy study
Lyndsay D. Troyer A , James J. Stone B and Thomas Borch A C DA Department of Chemistry, Colorado State University, 1872 Campus Delivery, Fort Collins, CO 80523, USA.
B Department of Civil and Environmental Engineering South Dakota School of Mines and Technology, 501 East Saint Joseph Street, Rapid City, SD 57701, USA.
C Present address: Department of Soil and Crop Sciences, Colorado State University, 1170 Campus Delivery, Fort Collins, CO 80523, USA.
D Corresponding author. Email: borch@colostate.edu
Environmental Chemistry 11(1) 18-27 https://doi.org/10.1071/EN13129
Submitted: 15 July 2013 Accepted: 30 September 2013 Published: 28 January 2014
Environmental context. Uranium and arsenic, two elements of human health concern, are commonly found at sites of uranium mining, but little is known about processes influencing their environmental behaviour. Here we focus on understanding the chemical and physical processes controlling uranium and arsenic transport at an abandoned uranium mine. We find that the use of sedimentation ponds limits the mobility of uranium; however, pond conditions at our site resulted in arsenic mobilisation. Our findings will help optimise restoration strategies for mine tailings.
Abstract. Although As can occur in U ore at concentrations up to 10 wt-%, the fate and transport of both U and As at U mine tailings have not been previously investigated at a watershed scale. The major objective of this study was to determine primary chemical and physical processes contributing to transport of both U and As to a down gradient watershed at an abandoned U mine site in South Dakota. Uranium is primarily transported by erosion at the site, based on decreasing concentrations in sediment with distance from the tailings. Sequential extractions and U X-ray absorption near-edge fine structure (XANES) fitting indicate that U is immobilised in a near-source sedimentation pond both by prevention of sediment transport and by reduction of UVI to UIV. In contrast to U, subsequent release of As to the watershed takes place from the pond partially due to reductive dissolution of Fe oxy(hydr)oxides. However, As is immobilised by adsorption to clays and Fe oxy(hydr)oxides in oxic zones and by formation of As–sulfide mineral phases in anoxic zones down gradient, indicated by sequential extractions and As XANES fitting. This study indicates that As should be considered during restoration of uranium mine sites in order to prevent transport.
Introduction
With the Cold War came a boom in U mining that left behind a legacy of environmental problems.[1] At that time, no government requirements for mine tailing reclamation were in place. Unreclaimed tailings left U ore exposed to weathering, resulting in the spread of U and other metals to surface water, groundwater and sediments surrounding mine sites.
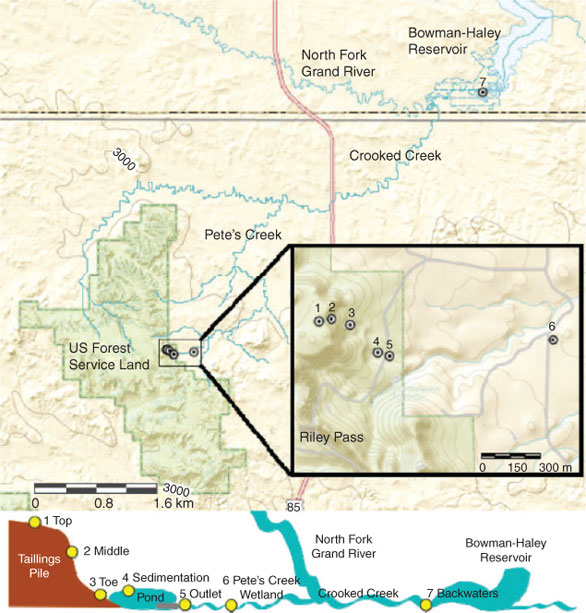
The North Cave Hills in Harding Country, South Dakota is one of those unreclaimed U mine tailings sites. Open-pit mining began at this site in 1955 after U-containing lignite seams were discovered and continued through to 1967.[2] Because of the lack of reclamation requirements, loose piles of overburden and ashed material were left behind, allowing for the release of heavy metals, metalloids and radionuclides into the surrounding watershed. Previous work at the site identified the major metals in sediments in the surrounding watershed to include Cu, Pb, Se, Th, As, U and V.[3] Of these elements, U and As have the greatest potential to threaten the local water supply because of their redox sensitivity, solubility and their concentration levels at the North Cave Hills. Uranium and As were previously found in near-source and watershed sediments above their respective background concentrations of 4 and 27 mg kg–1.[2] Near-source surface water U and As concentrations as measured by Larson et al. were above the Environmental Protection Agency’s (EPA) maximum contaminant levels (MCL), which are 30 and 10 ppb for U and As.[4] Down gradient of the tailings is a wetland followed by Crooked Creek that flows to the Bowman-Haley Reservoir, which is open to the public for recreation (Fig. 1). Larson et al.[4] have previously shown that heavy metal accumulation in the reservoir is the result of local U mining activity.[5]
|
U and As have differences in mobility under similar redox conditions. Under oxic conditions and environmentally relevant pHs, U is present as aqueous UVI species, including UO2(OH)20 and UO2(OH)3–.[6] When carbonate and calcium are also present, UVI can form uranyl-carbonato or uranyl-calcium-carbonato complexes including (UO2)2CO3(OH)3–, UO2(CO3)22–, UO2(CO3)34–, Ca2UO2(CO3)30 and CaUO2(CO3)32–.[1,7] These complexes have high solubility, limiting UVI adsorption to Fe oxy(hydr)oxides and sediments when calcium and carbonate are present.[6,8,9] Under anoxic conditions, U can be present as molecular UIV, nano-uraninite, or the most stable and least soluble of the reduced forms, crystalline uraninite (UO2).[10–13] Although U is less mobile in its reduced forms than in its oxidised forms, As is generally more mobile in its reduced form as AsIII.[14–16] Arsenic can adsorb to iron oxides under environmental pH as both AsIII (H2AsO30) and AsV (H2AsO4–).[17] Although AsIII has a greater sorption affinity to iron oxides than AsV at pH 7, AsV binds more strongly and is not as easily desorbed and released into surface waters.[17,18] In addition to having greater mobility, AsIII is more toxic to humans than AsV.[19] AsIII–sulfide mineral phases with low solubility, such as realgar (As4S4) and orpiment (As2S3), can also form under anoxic conditions, but because Fe–sulfide mineral formation is favoured, AsIII is most commonly found in its dissolved form.[20,21]
Iron and sulfate biogeochemical cycling can control the mobility of U and As. Studies of abiotic reduction of UVI by FeII in soils have shown that UVI can be reduced by adsorbed FeII and by structural FeIII, to a limited extent.[22,23] Uranium can also be reduced by precipitated or biogenically produced Fe–sulfide and by aqueous sulfide under anoxic conditions.[15,24–27] Enzymatic reduction of UVI has been observed by iron and sulfate reducing bacteria.[28,29] Although both AsIII and AsV can be adsorbed to iron minerals, As can be released, not only by desorption, but by microbial reduction of iron mineral phases, resulting in mineral dissolution.[30] Arsenic has been shown to be released during microbial reduction of sulfate to sulfide, resulting in thioarsenate formation.[31] In addition, AsV can be reduced by dissimilatory metal reducing bacteria or by bacteria as a detoxification mechanism to form less toxic methylated AsIII.[32–34] Thermodynamically, AsV reduction is favoured over FeIII and sulfate reduction.[35] Uranium and As may also control the solubility of each other in the environment. Recently, U and As have also been found to form uranyl arsenate surface complexes with aluminium oxide under a range of pHs and uranyl arsenate aqueous complexes and precipitates under acidic conditions in the laboratory.[36,37] Little is known about the formation of uranyl arsenates in the environment, but their effect on mobility of U and As should be considered in environments where both elements are present.
Previous studies at the North Cave Hills have tried to determine controls on U and As transport and fate. Kipp et al. found that both aerobic and anaerobic processes in sediments play a role in U and As transport by comparing their concentrations to that of Th – a metal that is not sensitive to environmental redox changes.[3] Although U and As concentrations fluctuated with distance from the tailings pile, Th, which was used as a naturally occurring conservative tracer, steadily decreased suggesting that, in addition to erosion, redox-promoted transport may be significant.[3] The study also determined that U and As were primarily associated with the Fe oxide fraction of collected soils, further emphasising the need for a better understanding of Fe controls on U and As fate at this site. Larson et al. looked at the seasonal variation of U and As speciation in North Cave Hills sediment pore waters, concluding that seasonal changes in redox conditions will continue to promote release of U and As as a result of reductive Fe oxide dissolution.[4] Aqueous surface water and pore water Fe, U and As measurements in Larson et al. complement sediment concentrations determined in our study.[4]
Although As can occur in U ore at concentrations up to 10 wt-%,[37] the fate and transport of both U and As at U mine tailings has not been previously investigated at any other field sites. However, U geochemistry has been studied extensively in Rifle, Colorado and other mill tailing sites.[1,14,38,39] At these sites, U immobilisation can occur with reduction to UIV or when UVI forms stable precipitates with, e.g. dissolved phosphate.[1,12,40,41] Arsenic mobility at a former U mill site has also been investigated independently of U mobility. For example, Stucker et al. discovered that As was released to Rifle, CO, groundwater during bioremediation of U as a result of, in part, the formation of thioarsenic species.[31] Donahue and Hendry studied As transport at a U mill tailing in Canada and found that As is primarily present as calcium arsenates, which could dissolve into site pore water resulting in concentrations of up to 126 ppm As.[37,42] Arsenic was also found to be adsorbed to ferrihydrite resulting in long-term stability under site conditions, preventing aqueous As transport.[37,43,44] The majority of these studies have focussed on the geochemistry and stability of U or As in tailing piles rather than physical and chemical controls on down-gradient transport of both elements in the same study.
Because of the dissimilarity of these co-contaminant metal redox pairs, and because of negative health effects associated with both U and As, it is essential to understand the processes that promote the retention and redox transformation of U and As within mining-affected watersheds such as the North Cave Hills, South Dakota.[45,46] Thus, the goal of this work is to determine how sorption and redox processes within various sites (e.g. tailings pile, sedimentation pond, wetlands, reservoir) control the down-gradient transport of U and As at an unreclaimed uranium mine tailing site.
Materials and methods
Study site and sample collection
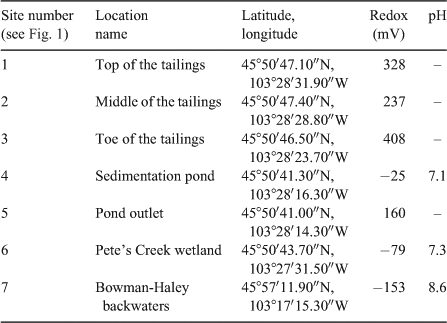
Samples were collected from nine previously established locations including the North Cave Hills mine tailings and the surrounding down gradient watershed.[2–4] Locations were selected based upon variations in redox conditions and U and As concentrations in sediments and porewater from past studies (Fig. 1, Table 1).[2] All samples were collected in late June of 2009, at the beginning of the rainy season (Fig. S1 of the Supplementary material). Three samples were collected from the tailings pile, including the top of the pile (1), the middle of the pile’s eastern slope (2) and the toe of the pile (3) before entering a sedimentation pond. The collection site at the base of the pile was selected because of evidence of recent erosion and rainwater transport. The fourth sampling site is at the inlet to the sedimentation pond located ~0.7-km downstream of the tailings pile (4). The pond is intended to capture sediment from the tailings pile to prevent physical transport (erosion) into Pete’s Creek. The fifth sampling site is at the sedimentation pond’s outlet pipe draining into Pete’s Creek (5). The next sampling location is within the sediment deposition zone surrounding Pete’s Creek referred to as Pete’s Creek Wetland (6), ~2 km from the tailings pile. Pete’s Creek then connects to Crooked Creek, which leads into the Bowman-Haley Reservoir, 45-km down gradient of the tailings pile. The final sampling location is at the inlet to the reservoir, referred to as Bowman-Haley backwaters (7).
|
At the time of sample collection, the oxidation–reduction potential (ORP) was measured using a handheld meter and ORP probe (Hanna Instruments 9025, Hanna Instruments, Woonsocket, RI, USA) by inserting the probe into the moist soil. The pH of surface waters was measured at sampling locations where soils were submerged. Two cores were collected at each sampling location, one preserved anoxically for determination of metal speciation and one collected for determination of total metal content. Soil cores were collected using a stainless steel soil corer lined with a polyethylene tube ~30 cm long. Both soil cores were preserved by immediately capping the tubes and sealing the caps with wax. The soil cores were then stored on ice for up to 2 days and transported to an anaerobic chamber in the laboratory. Because of the loose sediment material at the Bowman-Haley Reservoir, an anoxic soil core was not preserved. Anoxic cores were divided into 5-cm sections by depth and allowed to air dry in the anaerobic chamber. Once dry, soil samples were ground by mortar and pestle and passed through a 2-mm sieve. For long-term storage, samples were transferred into 125-mL airtight crimp-sealed bottles. For some analyses, samples were homogenised by taking equal masses of soil from each of the 5-cm sections of each core. Samples were combined using a mortar and pestle. For the majority of analyses, the top 5 cm of the soil core were used.
Laboratory methods
Soil texture analysis for homogenised dry samples was performed by Colorado State University’s Soil Testing Laboratory. The clay-sized fraction of the homogenised dried soils was separated and prepared for analysis by X-ray diffraction (XRD) with a Scintag X2 theta-theta powder diffractometer (Scintag, Inc., Cupertino, CA, USA) with a Peltier detector and Cu X-ray tube. The X-ray source was operated at 40 kV and 40 mA. Soluble salts and carbonates were removed from soils by washing with 1 M HCl.[47] Soils were washed and centrifuged and the solution was decanted. This process was repeated several times until the soil was no longer suspended in solution. Soil was then transferred into a beaker and 30 % H2O2 was added to remove organic material.[48] The solution was covered with a watch glass and heated to 70 °C until bubbling had subsided. Soils were then passed through a 63-μm sieve (mesh size #230) to remove the sand fraction. The clay fraction was separated from the silt fraction by centrifuging repeatedly with sodium hexametaphosphate at 2 min at 55g at 20 °C until the solution remained clear.[49] Upon isolating the clay-sized fraction, each sample was treated in three different ways, Mg-saturated air-dried,[50] Mg-saturated glycerol solvated[51] and K-saturated air-dried[51] and then filtered onto Isopore membrane filters (5.0 μm, TMTP, EMD Millipore Corporation, Billerica, MA, USA) and transferred to microscope slides,[52] allowing for the identification of smectite, vermiculite, chlorite, mica, gibbsite and kaolinite.
Total sediment metal concentrations were determined by total digestion of a 1.0-g sample from the top 5 cm of soil cores by the method described by Soltanpour et al.[53] The resulting digestions were analysed by inductively coupled plasma mass spectrometry (ICP-MS) (Perkin Elmer Elan DRC II, Perkin Elmer, Waltham, MA) for U and As concentrations and inductively coupled plasma optical emission spectroscopy (ICP-OES) (Thermo Scientific Iris Intrepid II, Thermo Scientific, Waltham, MA) for Fe. Sequential extractions were performed separately for analysis of U and As. Extraction procedures for As were based upon Huang and Kretzschmar.[54] Steps 1, 3, 5, 6 and 7 were respectively performed to extract soluble and exchangeable, manganese oxide-associated, poorly crystalline Fe and Al (hydr)oxide-associated, sulfide-associated and crystalline Fe and Al (hydr)oxide-associated As. To measure concentrations in the residual fraction, 5 mL of concentrated HNO3 was added to the remaining sediment and heated at 85 °C for 3 h, rather than using X-ray fluorescence.[42,55] Sequential U and As extractions were also performed based upon a modified method from Tessier et al.[56] as described in Salome et al.[42] Extracts were also analysed for both U and As by ICP-MS. Additional elemental measurements of the soil samples were performed using laser ablation inductively coupled plasma mass spectrometry (LA-ICP/MS) at the Environmental Molecular Sciences Laboratory (EMSL), part of the Pacific Northwest National Laboratory (PNNL) in Richland, Washington. The primary purpose of the additional measurements was to independently determine the mineral surface concentration of U and As on unaltered samples. Soil samples were formed into pellets suitable for laser ablation using a conventional shop press. The laser ablation system employed was the Analyte G2, manufactured by Photon Machines Inc. (Bozeman, MT, USA). This uses a pulsed UV excimer laser operating at 193 nm with pulse length less than 4 ns and a nominal irradiance of up to 4 GW cm–2. For the results reported here the laser ablation system was operated at 20 Hz with a laser ablation area of 20 μm. The laser was moved in a raster pattern over an ~2 mm2 area of the sample and the resulting ablated particulates were entrained in a flow of He gas at 0.2–0.5 L min–1 and carried to a Thermo Electron Element 2/XR (Thermo Scientific) for measurement of elemental concentrations. The Element was set to rapidly scan over several elements in addition to U including Fe, Mn, As, Sr and Ba. These elements had been independently measured by ICP-OES and were used as internal standards in combination with an external standard calibration curve generated by LA-ICP/MS of NIST610, NIST612 and NIST614 standard reference materials. Standard deviations for the reported values were ~20–25 % and mostly a result of inhomogeneity in the samples.
X-Ray absorption spectroscopy (XAS)
All XAS samples were taken from the homogenised top 5 cm of the dried soil cores. Samples were analysed at the Stanford Synchrotron Radiation Lightsource (SSRL) in Menlo Park, CA. All anoxic soil preparations occurred within an anaerobic chamber containing 95 % N2 and 5 % H2. Samples were placed into a Teflon holder sealed by a Kapton polyimide film to prevent oxidation while minimising X-ray absorption as previously described.[57] Extended X-ray absorption fine structure (EXAFS) spectroscopy was used to characterise Fe mineralogy. X-Ray absorption near-edge fine structure (XANES) spectroscopy was performed to determine the valence state of U and As. XAS was performed at beamline 11–2 (26-pole wiggler), 10–2 (30-pole wiggler) and 7–3 (20-pole wiggler) at SSRL. The ring operates at 3 GeV with a current of 450 mA. During data collection, As samples were maintained at a temperature of 5 K to prevent beam-induced redox reactions using an Oxford Instruments CF1208 continuous flow liquid helium cryostat (Oxford Instruments, Abingdon, UK).[58] Energy selection was accomplished with a Si (220) monochromator. All Fe EXAFS spectra were collected in fluorescence mode with a wide-angle collection ionisation chamber (Lytle detector). U and As XANES spectra were collected in fluorescence mode with a 30-channel Ge detector. EXAFS spectra were collected from –200 to +1000 eV around the K-edge of Fe (7111 eV). For U and As, XANES spectra were collected from –150 to 450 eV around the LIII-edge of U (17 176 eV) and the K-edge of As (11 876 eV). Between two and three spectra were averaged for each sample. U and As spectra were fit by linear combination fitting of normalised XANES spectra in Athena between –20 and +25 eV from 17 178 eV for U and –20 and +35 eV from 11 873 eV for As.[59] Uranium reference compounds included uranyl acetate and uraninite.[60] Arsenic reference compounds included AsIII adsorbed to goethite, AsV adsorbed to goethite[58] and orpiment (As2S3). Standards of arsenic adsorbed to goethite were prepared according to Amstaetter et al. by adding 1.2 mg L–1 of sodium arsenite and sodium arsenate to a 5.4 g L–1 suspension of goethite (Bayferrox 920 Z, LANXESS Deutschland GmbH, Leverkusen, Germany).[58] Orpiment was obtained from the RRUFF database at University of Arizona Mineral Museum and was originally from Mercur, Utah. Linear combination fitting of k3-weighted Fe EXAFS spectra was performed from 3 to 12 k (Å–1) using Athena.[59] All references were chosen based on their likelihood of being a soil component. The Fe EXAFS spectrum of smectite (SAZ-1) was obtained from Dr Peggy O’Day at University of California, Merced, CA.[61] Compounds were only included in the fit if the contribution was a fraction greater than 0.05. Fits were approximately within ±5 % of the mole percentages.[57]
Results and discussion
Geochemical characterisation of field site
Soil redox measurements were taken at each of the sampling locations (Table 1). The redox potential varies from site to site, with the tailings pile and the pond outlet being oxic to suboxic. All of the sites that were under surface water were anoxic: the sedimentation pond, Pete’s Creek wetland and Bowman-Haley backwaters. pH measurements were only taken at locations where surface water was present above the sampled soil core (Table 1). pH readings were 7.1 and 7.3 for near-source sampling locations and 8.6 at the Bowman-Haley backwaters sites. Previous work at the North Cave Hills has found surface waters around the area to be between pH 7 and 10 and to have high alkalinity with CaCO3 concentrations up to 300 mg L–1.[2]
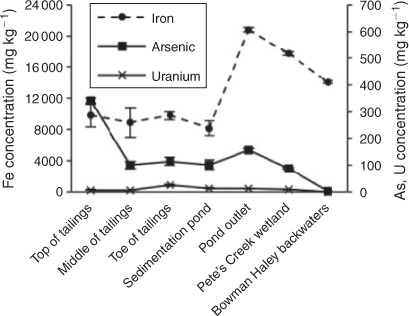
Iron is redox sensitive, abundant in soils and known to influence U and As mobility in soil and sediments.[14] Total Fe concentrations in the top 5 cm of soil from each sampling site are shown in Fig. 2. The concentration of solid phase Fe was greater at sites downstream of the sedimentation pond. The top, middle and bottom of the tailings pile and the sedimentation pond ranged from 8180 to 9780 mg kg–1 total Fe, whereas the samples collected from the pond outlet, Pete’s Creek Wetland and Bowman-Haley backwaters contained 14 100 to 17 700 mg kg–1 total Fe. The highest concentration of Fe was measured at the sedimentation pond’s outlet at 20 800 mg kg–1. The high concentration at the pond outlet indicates that dissolved FeII and suspended iron oxides are accumulating in the pond water and are then carried downstream through the pond outlet. Although relatively low aqueous FeII concentrations (2 mg L–1) were measured in the pond by Larson et al.,[4] oxidative precipitation of dissolved FeII may, in part, be responsible for accumulation of FeIII oxides at this sampling location, Pete’s Creek wetland and Bowman-Haley backwaters sites.[4] Of the total extracted Fe, only 55 % was in crystalline form at the pond outlet compared to 75 % in the pond (Fig. S2 of the Supplementary material). The greater fraction of amorphous and exchangeable Fe at the outlet compared to that at the pond suggests that precipitation of amorphous iron oxides such as ferrihydrite occurs at the outlet (Fig. S2).
|
The distribution of soil iron minerals was determined by linear combination fitting (LCF) of EXAFS data in k-space (Figs S3, S4, Table S1 of the Supplementary material). Based on LCF-EXAFS, the iron mineral composition at the site is primarily made up of iron-bearing silicates including smectite and augite. At all three of the tailings pile sampling locations, the dominant Fe-bearing mineral was smectite (35–54 %), whereas at the down gradient locations the dominant Fe-bearing mineral was augite (31–86 %). In addition to smectite and augite, all of the spectra were fit including ferrosmectite and goethite. Goethite, an iron oxyhydroxide, could form by precipitation at the pond outlet (30 %) following dissolution of amorphous iron hydroxides like ferrihydrite, which can occur in the reducing environment of the sedimentation pond.[62] Clay X-ray diffraction resulted in similar diffraction patterns for all of the sampling sites, confirming the presence of smectite and goethite in site sediments. Analysis of the clay XRD patterns identified mica, kaolinite and gibbsite at all of the sampling locations (Fig. S5 of the Supplementary material).
Geochemical controls on U transport
Uranium surface sediment concentrations vary, based on our limited number of sampling sites, within the tailings pile, but are consistently low at all other sampling locations. The highest concentration of total U among the sampling sites, as determined by total digestion, was found at the toe of the tailings (25 mg kg–1). High concentrations may be found there because of the deposition of sediment resulting from overburden weathering. All sites downstream of the toe of the tailings have total U concentrations between 0 and 15 mg kg–1 in surface sediments as determined by total digestion (Fig. 2). In addition to being determined by digestion, total sediment concentrations for uranium were determined by laser-ablation ICP-MS (Fig. S6a). Sediment U concentrations follow the same trend by site whether determined by total digestion or laser ablation with the exception of two sites, the top of the tailings (7 and 64 mg kg–1 respective to the method of measurement) and the pond outlet (12 and 33 mg kg–1), which were both determined to be greater by laser ablation. LA-ICP/MS determines concentration by ablation of a spot 50 μm in diameter of the sediment sample, so concentrations may be greater if an area containing mineralised U was analysed.[63] With the inhomogeneity of the sediment at the top of the tailings pile, this result is not unexpected. Background level U sediment concentrations were previously determined to be 4 mg kg–1.[2] The Bowman-Haley backwaters site was the only site with U concentrations below background level as measured by total digestion.
Low sediment concentrations down gradient of the tailings pile may be the result of the sedimentation pond, either by serving its purpose to prevent down-gradient sediment transport or by promoting U reduction of dissolved UVI species to uraninite or to biomass associated molecular UIV (Fig. 2).[13,15] The redox conditions of the pond promote microbial reduction of uranyl to uraninite or alternately molecular UIV that is likely to settle to the bottom of the pond and not be further transported.[64][13] The chemical structure of UIV could not be confirmed by the collected XANES spectra. Sediment sulfur concentrations (measured by Larson et al.[4]) and sediment Fe concentrations in the pond could allow for the precipitation of amorphous FeS, following microbial reduction of sulfate and FeIII oxy(hydr)oxides, which can abiotically reduce uranyl to uraninite or non-uraninite UIV.[4,13,15,27,65] Because of the low environmental sediment U concentrations, U XANES spectra were only obtained for the top of the tailings, toe of the tailings, pond and pond outlet (Figs S7, S8, Table S2 of the Supplementary material). Of the collected spectra, the U XANES spectrum from the pond outlet was fit with the greatest percentage of UIV (45.1 %). Similar to the reduced Fe observed at the outlet, the presence of UIV at the outlet supports that uranium reduction is occurring in the pond and uraninite or biomass-associated molecular UIV is settling out at the outlet. The greater concentration of sediment U determined by LA-ICP/MS than by total digestion at the pond outlet also supports the presence of heterogeneously distributed mineralised UIV or mineral-associated UVI. Although UIV forms can be oxidised to soluble UVI with exposure to oxygen, UIV is likely to remain stable in the pond because oxygen exposure would not occur without significant mixing of the pond water.
The mineral-associated U in soils as determined by sequential extractions (Fig. 3a) shows little difference among sites, indicating that U may be transported down gradient of the sedimentation pond mainly as sediment-associated U rather than as dissolved UVI. Sediment-associated U could have been transported by dust or especially before installation or rehabilitation of the sedimentation pond. Sequential extractions show that U is primarily associated with the carbonate and phosphate mineral (acid extractable) fraction of sediment at all of the sampling locations except for the toe of the tailings (Fig. 3a). Based on the carbonate concentration and pH of North Cave Hills surface waters, U is most likely associated with carbonate rather than phosphate, including carbonate minerals such as calcite and aragonite.[56] Uranium in the carbonate mineral fraction is likely incorporated into carbonate minerals in source materials and would not be released down gradient of the pond because calcite and aragonite have low solubility under the neutral to basic pH conditions of the sampling locations.[66,67] Aqueous U concentrations in surface waters (in Larson et al.[4]) support the lack of solubilised U at down gradient sites, with a U concentration 2 times background level (32.7 μg L–1) measured at Pete’s Creek wetland and a below background level concentration (5.14 μg L–1) measured at the Bowman-Haley backwaters during summer sampling.[4] Aqueous UVI is expected to adsorb preferentially to amorphous iron oxides in sediments rather than to carbonate minerals.[66] At the top of the tailings, the pond and the pond outlet, the amount of U associated with the iron mineral fraction increases from the top and middle of the tailings pile, which could indicate that aqueous UVI is adsorbed to iron oxyhydroxides at those sites.[9,68] The sodium acetate fraction (iron mineral-associated fraction) of the Tessier et al. method has also been shown to extract UIV, so increased U extracted in an iron mineral fraction may also support the presence of UIV in the pond and at its outlet.[56,69]

|
Geochemical controls on arsenic transport
Total sediment As at the site does not simply decrease with distance from the tailings pile, indicating that As transport is controlled by chemical processes in addition to physical processes (Fig. 2). Sediment As concentrations measured by total digestion are similar for near-source sites including the tailings pile, the pond and its outlet, ranging between 101 and 155 mg kg–1, with the exception of the top of the tailings pile (Fig. 2). The sediment As concentration at the top of the tailings pile was 346 mg kg–1. Surface sediment concentrations are likely high at this sampling site because of the presence of As-containing minerals that were brought to the surface during mining activity. The second highest As sediment concentration of the sampling locations as measured by total digestion is at the pond outlet (155 mg kg–1). Similar to the U sediment concentrations, As sediment concentrations were measured by both total digestion and laser ablation. Sediment As concentrations measured by the two methods follow the same trend by sampling location with the exception of the sedimentation pond (Fig. S6b of the Supplementary material). The As concentration in the pond was greater when measured by laser ablation (324 mg kg–1) than by total digestion (102 mg kg–1). This is likely attributable to heterogeneous distribution of As in the pond. Of the sites sampled in Larson et al.,[4] surface water As concentrations were highest in the pond (1260 μg L–1).[4]
Reductive dissolution of Fe oxy(hydr)oxides promotes the release of adsorbed As and likely occurs under the reducing conditions of the sedimentation pond, resulting in high aqueous As concentrations in the pond and high sediment As concentrations at the outlet.[18,35,70] The high fraction of amorphous Fe measured at the pond outlet (Fig. S2 of the Supplementary material) supports that reductive dissolution occurs in the pond. Because AsIII is more readily desorbed from Fe oxides than AsV, release of As may also occur upon microbial reduction of AsV to AsIII under the reducing conditions of the pond.[18,33,35] Larson et al.[4] found the As in pore water at the sedimentation pond to be 55–93 % AsIII during summer sampling, supporting that microbial reduction and release from sediment is taking place.[4] Arsenic mobilised in the pond is transported to down gradient locations where it is immobilised in oxic zones as AsIII is oxidised to AsV and adsorption to soil minerals occurs.[33] Downstream of the tailings pile by 2 km, the Pete’s Creek wetland site has an As concentration of 87 mg kg–1, suggesting that transport is limited past the pond. Surface water As concentrations are also lower at the Pete’s Creek wetland site than at the pond (577 μg L–1).[4] At the Bowman-Haley backwaters site, the sediment As concentration is 2 mg kg–1, which is below the mean background level of 26 mg kg–1 established by Stone et al.[2] In addition, no detectable As was measured in surface water samples at the backwaters site.[4]
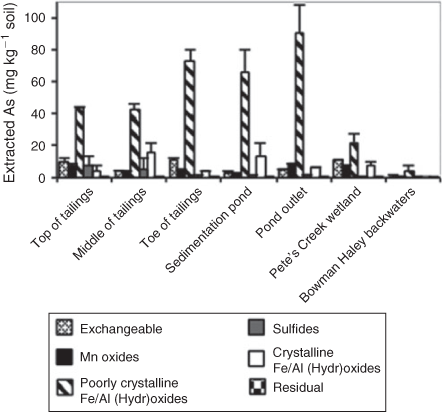
The results of the Huang and Kretzschmar sequential extraction method indicate that As is primarily found associated with the poorly crystalline Fe and Al oxides fraction (ligand-promoted dissolution fraction) for all of the sampling sites with 45 to 80 % of total As present in that fraction (Fig. 4).[54] This fraction may also include amorphous clays minerals, but XRD of the material remaining after each extraction step would need to be performed in order to determine with certainty if clay minerals were dissolved.[54] The other method of extraction based on Tessier et al. shows that As is primarily associated with the residual fraction (38–80 %), which includes phyllosilicates (clays) and inosilicates (Fig. 3b).[56] This result indicates that As may be present at the sampling sites associated with clay minerals. Although clay minerals have a lower affinity for AsIII and AsV than Fe oxy(hydr)oxides, they are the dominant Fe-bearing minerals present in North Cave Hills' soils according to linear combination fitting of Fe EXAFS spectra (Figs S3, S4, Table S1).[71] Adsorption of As to clays at the pond outlet may prevent down gradient As transport because complete desorption of As from clay minerals is not shown to occur under the pH conditions of the North Cave Hills.[71]
|
An As–sulfide mineral phase (orpiment) was found to be present at the top of the tailings pile as determined by As XANES linear-combination fitting, which implies that As minerals were brought to the surface during mining (Fig. 5, Table S3 of the Supplementary material). These As-bearing minerals release As to down gradient areas during weathering or oxidative dissolution at the site’s near-neutral pH conditions.[72,73] In addition to the top of the tailings, fits of As XANES spectra from Pete’s Creek wetland and Bowman-Haley backwaters, both reduced sampling sites, were improved when orpiment (As2S3) was included as a reference compound (Fig. 5, Table S3). Larson et al.[4] determined that formation of orpiment is thermodynamically favourable at the reduced areas surrounding the North Cave Hills based on dissolved AsIII and SO42– concentrations at the Bowman-Haley outlet.[4] Newman et al. found that precipitation of AsIII–sulfides occurs following AsV and SO42– reduction, both of which are likely to take place under the redox conditions of the sites.[74] AsIII–sulfides can also form after adsorbed AsIII is incorporated into the structure of precipitated metal sulfides.[75] Pete’s Creek wetland and Bowman-Haley backwaters have the highest percentage of AsIII of the sampling sites (60–70 %), so incorporation may take place (Fig. 5, Table S3). There is no direct evidence for the presence of metal sulfides at the sites, but the sediment sulfur concentrations in Larson et al.[4] were highest at the Bowman-Haley backwaters site.[4] Although the As XANES spectra were best fit with an orpiment reference, the specific AsIII–sulfide mineral could not be identified conclusively without EXAFS information. However, EXAFS spectra could not be collected for these samples due to low environmental concentrations. Examples of As XANES data and fits can be seen in Fig. S9 of the Supplementary material.
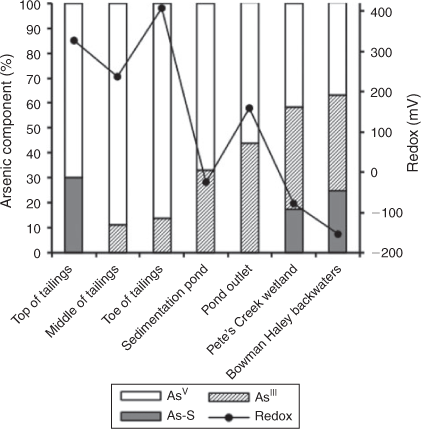
|
Environmental implications
The results of this study illustrate that a combination of wet chemical analysis, XRD and X-ray absorption spectroscopy can be successfully applied to help elucidate major mechanisms controlling the fate and transport of U and As in complex natural systems such as abandoned mining sites. Here we show that sedimentation ponds used to prevent physical transport of U and As from mine tailings are likely to also result in reduction of UVI and AsV. Reduction of UVI to UIV will further limit down gradient transport of aqueous U. In contrast, AsV reduction in the pond is clearly shown to promote aqueous transport of AsIII as a result of either reductive dissolution of Fe oxy(hydr)oxides, an important sorbent for As, or direct reduction of sorbed AsV resulting in formation of aqueous AsIII. Although As is mobilised within the sedimentation pond and accumulating at the pond outlet, sediment and surface water concentrations at the furthest (45 km) down gradient sampling site, the Bowman-Haley backwaters, are below background levels.[4] Our findings suggest that As was immobilised as a result of AsV adsorption to clays or Fe oxy(hydr)oxides in oxic zones or mineralisation of AsIII to AsIII–sulfides in anoxic zones such as within Pete’s Creek wetland. In contrast, the U concentration did not vary much between the sedimentation pond and the down gradient sites. Although U was measured in sediments at 2 times background levels at the Bowman-Haley backwaters site by LA-ICP/MS, U does not pose a risk to human health at that level by any type of acute or chronic exposure.[76] In order to better elucidate the molecular structure and interactions between U and As, and with soil minerals, high quality EXAFS spectra are needed. However, this study shows that it can be very challenging to obtain bulk EXAFS spectra because of low (<100 ppm) environmental concentrations indicating that future studies should include micro-focussed XAS techniques in order to obtain an improved understanding to further advance best managing practices or reclamation strategies for mine tailings.[77]
Supporting material
Figures showing climate data, results of sequential iron extractions, Fe mineral distribution, linear-combination fits of Fe EXAFS, U XANES and As XANES, X-ray diffraction, and total U and As concentrations in soils are available in the Supplementary material, which is available from the journal online (see http://www.publish.csiro.au/?act=view_file&file_id=EN13129_AC.pdf). Also included are tables summarising results of linear-combination fitting of Fe EXAFS, U XANES and As XANES.
Acknowledgements
The authors thank Lance N. Larson for field sampling assistance. They also thank Rong Wei for laboratory assistance with sequential extractions and Damaris L. Roosendaal for assistance with clay X-ray diffraction samples. They thank M. Lizabeth Alexander for sample measurement and analysis by LA-ICP/MS. The LA-ICP/MS work was carried out at the Pacific Northwest National Laboratory’s Environmental Molecular Sciences Laboratory operated for the Department of Energy by Battelle Memorial Institute. The authors also thank John R. Bargar, Juan S. Lezama-Pacheco and Peggy A. O’Day for providing standards for X-ray absorption spectroscopy and Robert T. Downs and the RRUFF library for providing us with an orpiment mineral sample. This work was generously supported by the US Environmental Protection Agency – Region 8 and the US Department of Agriculture/Forest Service – Northern Region. This work was also funded, in part, by a National Science Foundation CAREER Award to Thomas Borch (EAR0847683). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University.
References
[1] A. Abdelouas, Uranium mill tailings: geochemistry, mineralogy, and environmental impact. Elements 2006, 2, 335.| Uranium mill tailings: geochemistry, mineralogy, and environmental impact.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsFant70%3D&md5=5137ca5591d66a85ad172cd4afcdf2f8CAS |
[2] J. J. Stone, L. D. Stetler, A. Schwalm, Final Report: North Cave Hills Abandoned Uranium Mines Impact Investigation. South Dakota School of Mines and Technology 2007, 1.
[3] G. G. Kipp, J. J. Stone, L. D. Stetler, Arsenic and uranium transport in sediments near abandoned uranium mines in Harding County, South Dakota. Appl. Geochem. 2009, 24, 2246.
| Arsenic and uranium transport in sediments near abandoned uranium mines in Harding County, South Dakota.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlyrt7bN&md5=7a283bd78882222151baea6194fe1648CAS |
[4] L. N. Larson, G. G. Kipp, H. V. Mott, J. J. Stone, Sediment pore-water interactions associated with arsenic and uranium transport from the North Cave Hills mining region, South Dakota, USA. Appl. Geochem. 2012, 27, 879.
| Sediment pore-water interactions associated with arsenic and uranium transport from the North Cave Hills mining region, South Dakota, USA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVyjs7w%3D&md5=6ac542acb591203ba03ce038bb017aceCAS |
[5] L. N. Larson, J. J. Stone, Sediment-bound arsenic and uranium within the Bowman-Haley Reservoir, North Dakota. Water Air Soil Pollut. 2011, 219, 27.
| Sediment-bound arsenic and uranium within the Bowman-Haley Reservoir, North Dakota.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntVSks7k%3D&md5=4e34a812d6636388332dfc175519da5dCAS |
[6] T. D. Waite, J. A. Davis, T. Payne, G. A. Waychunas, N. Xu, Uranium(VI) adsorption to ferrihydrite: application of a surface complexation model. Geochim. Cosmochim. Acta 1994, 58, 5465.
| Uranium(VI) adsorption to ferrihydrite: application of a surface complexation model.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXivVCmu78%3D&md5=003b252df95216a8cb87820f4f8cea52CAS |
[7] G. Bernhard, G. Geipel, T. Reich, V. Brendler, S. Amayri, H. Nitsche, Uranyl(VI) carbonate complex formation: Validation of the Ca2UO2(CO3)3 (aq) species. Radiochim. Acta 2001, 89, 511.
| Uranyl(VI) carbonate complex formation: Validation of the Ca2UO2(CO3)3 (aq) species.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXmvFGgsL0%3D&md5=3c6edbcc2f567df97564ecd4cf3fd31bCAS |
[8] B. D. Stewart, M. A. Mayes, S. Fendorf, Impact of uranyl–calcium–carbonato complexes on uranium(VI) adsorption to synthetic and natural sediments. Environ. Sci. Technol. 2010, 44, 928.
| Impact of uranyl–calcium–carbonato complexes on uranium(VI) adsorption to synthetic and natural sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjt1yiug%3D%3D&md5=3aaa01f8be2ded04bc55a24c692162b0CAS | 20058915PubMed |
[9] W. Dong, W. P. Ball, C. Liu, Z. Wang, A. T. Stone, J. Bai, J. M. Zachara, Influence of calcite and dissolved calcium on uranium(VI) sorption to a Hanford subsurface sediment. Environ. Sci. Technol. 2005, 39, 7949.
| Influence of calcite and dissolved calcium on uranium(VI) sorption to a Hanford subsurface sediment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVWrtr3J&md5=1adcd3bd7dc70387d1366291c4c942c9CAS | 16295860PubMed |
[10] J. R. Bargar, R. Bernier-Latmani, D. E. Giammar, B. M. Tebo, Biogenic uraninite nanoparticles and their importance for uranium remediation. Elements 2008, 4, 407.
| Biogenic uraninite nanoparticles and their importance for uranium remediation.Crossref | GoogleScholarGoogle Scholar |
[11] R. Bernier-Latmani, H. Veeramani, E. D. Vecchia, P. Junier, J. S. Lezama-Pacheco, E. I. Suvorova, J. O. Sharp, N. S. Wigginton, J. R. Bargar, Non-uraninite products of microbial UVI reduction. Environ. Sci. Technol. 2010, 44, 9456.
| Non-uraninite products of microbial UVI reduction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVWmsrrP&md5=65d290383fbd5aa689c86352a25956daCAS | 21069950PubMed |
[12] V. Sivaswamy, M. I. Boyanov, B. M. Peyton, S. Viamajala, R. Gerlach, W. A. Apel, R. K. Sani, A. Dohnalkova, K. M. Kemner, T. Borch, Multiple mechanisms of uranium immobilization by Cellulomonas sp. strain ES6. Biotechnol. Bioeng. 2011, 108, 264.
| Multiple mechanisms of uranium immobilization by Cellulomonas sp. strain ES6.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFOjsLnK&md5=14c1ebefa6fea2e94e1fe0ab8d73c1c7CAS | 20872821PubMed |
[13] J. O. Sharp, J. S. Lezama-Pacheco, E. J. Schofield, P. Junier, K.-U. Ulrich, S. Chinni, H. Veeramani, C. Margot-Roquier, S. M. Webb, B. M. Tebo, D. E. Giammar, J. R. Bargar, R. Bernier-Latmani, Uranium speciation and stability after reductive immobilization in aquifer sediments. Geochim. Cosmochim. Acta 2011, 75, 6497.
| Uranium speciation and stability after reductive immobilization in aquifer sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Oqu73F&md5=80170b4fa31b0d41e45f8ed6ebfec9e2CAS |
[14] T. Borch, R. Kretzschmar, A. Kappler, P. Van Cappellen, M. Ginder-Vogel, A. Voegelin, K. M. Campbell, Biogeochemical Redox processes and their impact on contaminant dynamics. Environ. Sci. Technol. 2010, 44, 15.
| Biogeochemical Redox processes and their impact on contaminant dynamics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGhtLnF&md5=1413c3144837751a409a1e64a54400a1CAS | 20000681PubMed |
[15] J. R. Bargar, K. H. Williams, K. M. Campbell, P. E. Long, J. E. Stubbs, E. I. Suvorova, J. S. Lezama-Pacheco, D. S. Alessi, M. Stylo, S. M. Webb, Uranium redox transition pathways in acetate-amended sediments. Proc. Natl. Acad. Sci. USA 2013, 110, 4506.
| Uranium redox transition pathways in acetate-amended sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXms12nsbc%3D&md5=a4597edc20c535d3b3f1c61516e1d8a8CAS |
[16] S. E. Fendorf, H. A. Michael, A. Van Geen, Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science 2010, 328, 1123.
| Spatial and temporal variations of groundwater arsenic in South and Southeast Asia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsVGjtrc%3D&md5=94cd2d7a804ab9f0dd97e9bf90e15635CAS |
[17] S. Dixit, J. G. Hering, Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environ. Sci. Technol. 2003, 37, 4182.
| Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXmtFOltr8%3D&md5=e95cefdfa6e7bd583150adbcd471263fCAS | 14524451PubMed |
[18] K. J. Tufano, C. Reyes, C. W. Saltikov, S. E. Fendorf, Reductive processes controlling arsenic retention: revealing the relative importance of iron and arsenic reduction. Environ. Sci. Technol. 2008, 42, 8283.
| Reductive processes controlling arsenic retention: revealing the relative importance of iron and arsenic reduction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1CgsbfP&md5=3eb971791cc4dd832fa632dd68654eafCAS | 19068807PubMed |
[19] J. Petrick, Monomethylarsonous acid (MMAIII) Is more toxic than arsenite in chang human hepatocytes. Toxicol. Appl. Pharmacol. 2000, 163, 203.
| Monomethylarsonous acid (MMAIII) Is more toxic than arsenite in chang human hepatocytes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhsVSnurw%3D&md5=977f4d170ea38799e9da5017e46ec2f8CAS | 10698679PubMed |
[20] B. D. Kocar, T. Borch, S. Fendorf, Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite. Geochim. Cosmochim. Acta 2010, 74, 980.
| Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1SlsbrN&md5=94f50fdca1a25eda55126631fd7ade89CAS |
[21] P. A. O’Day, D. Vlassopoulos, R. Root, N. Rivera, The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 13 703.
| The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXotVygtrk%3D&md5=ed0e21739bc198e411ffdfdb7ae42a0eCAS |
[22] P. M. Fox, J. A. Davis, R. Kukkadapu, D. M. Singer, J. R. Bargar, K. H. Williams, Abiotic UVI reduction by sorbed FeII on natural sediments. Geochim. Cosmochim. Acta 2013, 117, 266.
| Abiotic UVI reduction by sorbed FeII on natural sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXht1Whs7fF&md5=a073b5d04f0707b5fa474ef40085e9a2CAS |
[23] D. E. Latta, M. I. Boyanov, K. M. Kemner, E. J. O'Loughlin, M. M. Scherer, Abiotic reduction of uranium by FeII in soil. Appl. Geochem. 2012, 27, 1512.
| Abiotic reduction of uranium by FeII in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xmt1OhtLs%3D&md5=b27e0e9f788a9e9fac61a6c8e4018b7cCAS |
[24] B. Hua, B. Deng, Reductive immobilization of uranium(VI) by amorphous iron sulfide. Environ. Sci. Technol. 2008, 42, 8703.
| Reductive immobilization of uranium(VI) by amorphous iron sulfide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlSqtbvJ&md5=5013eba36ebe973eb6614437230f7105CAS | 19192785PubMed |
[25] B. Hua, H. Xu, J. Terry, B. Deng, Kinetics of uranium(VI) reduction by hydrogen sulfide in anoxic aqueous systems. Environ. Sci. Technol. 2006, 40, 4666.
| Kinetics of uranium(VI) reduction by hydrogen sulfide in anoxic aqueous systems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmtFSgsrY%3D&md5=c963814ced6db8625dfedbf9008608b3CAS | 16913122PubMed |
[26] S. P. Hyun, J. A. Davis, K. Sun, K. F. Hayes, Uranium(VI) reduction by iron(II) monosulfide mackinawite. Environ. Sci. Technol. 2012, 46, 3369.
| Uranium(VI) reduction by iron(II) monosulfide mackinawite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVSntbc%3D&md5=703baafd467a0cb6fdaadbaf894c7e3dCAS | 22316012PubMed |
[27] H. Veeramani, A. C. Scheinost, N. Monsegue, Abiotic reductive immobilization of UVI by biogenic mackinawite. Sci.Technol. 2013, 47, 2361.
| Abiotic reductive immobilization of UVI by biogenic mackinawite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFaltrk%3D&md5=a3a39dbf5020dd5b1d5177ada6dd2a16CAS |
[28] D. R. Lovley, E. J. P. Phillips, Y. A. Gorby, E. R. Landa, Microbial reduction of uranium. Nature 1991, 350, 413.
| Microbial reduction of uranium.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXitVegsb8%3D&md5=16c536a2ec75545b90678ed32a2088e9CAS |
[29] D. R. Lovley, E. Phillips, Reduction of uranium by Desulfovibrio desulfuricans. Appl. Environ. Microbiol. 1992, 58, 850.
| 1:CAS:528:DyaK38Xhs12rtbo%3D&md5=dfa234eee38f39a777e56de1970632a5CAS | 1575486PubMed |
[30] D. E. Cummings, F. Caccavo, S. Fendorf, R. F. Rosenzweig, Arsenic mobilization by the dissimilatory FeIII-reducing bacterium Shewanella alga BrY. Environ. Sci. Technol. 1999, 33, 723.
| Arsenic mobilization by the dissimilatory FeIII-reducing bacterium Shewanella alga BrY.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFSguw%3D%3D&md5=f3a16d14eeccc2884b60026eec607035CAS |
[31] V. K. Stucker, K. H. Williams, M. J. Robbins, J. F. Ranville, Arsenic geochemistry in a biostimulated aquifer: an aqueous speciation study. Environ. Toxicol. Chem. 2013, 32, 1216.
| Arsenic geochemistry in a biostimulated aquifer: an aqueous speciation study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXntVCgu7k%3D&md5=23ca4f4bf805c243a1160ff124a7fe72CAS | 23401165PubMed |
[32] R. Oremland, J. Stolz, The ecology of arsenic. Science 2003, 300, 939.
| The ecology of arsenic.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVyjsLs%3D&md5=840e38f53bf5d9201af72c47691ceb50CAS | 12738852PubMed |
[33] S. Fendorf, P. S. Nico, B. D. Kocar, Y. Masue, K. J. Tufano, Arsenic chemistry in soils and sediments. Dev. Soil Sci. 2010, 34, 357.
| Arsenic chemistry in soils and sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotVOqu7s%3D&md5=ae829247042a65b238335bd65b94c614CAS |
[34] W. R. Cullen, K. J. Reimer, Arsenic speciation in the environment. Science 1989, 89, 713.
| 1:CAS:528:DyaL1MXktVaitbg%3D&md5=4f62255a7fe1f0f64d6429815eef729dCAS |
[35] B. D. Kocar, S. E. Fendorf, Thermodynamic constraints on reductive reactions influencing the biogeochemistry of arsenic in soils and sediments. Environ. Sci. Technol. 2009, 43, 4871.
| Thermodynamic constraints on reductive reactions influencing the biogeochemistry of arsenic in soils and sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtFaktLo%3D&md5=a856a9b30a972537ff629f30b93de87dCAS | 19673278PubMed |
[36] W. A. Gezahegne, C. Hennig, S. Tsushima, B. Planer-Friedrich, A. C. Scheinost, B. J. Merkel, EXAFS and DFT investigations of uranyl arsenate complexes in aqueous solution. Environ. Sci. Technol. 2012, 46, 2228.
| EXAFS and DFT investigations of uranyl arsenate complexes in aqueous solution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xks1Wqsw%3D%3D&md5=6c150542b042369eb0478f1049d45c07CAS | 22229913PubMed |
[37] Y. Tang, R. J. Reeder, Uranyl and arsenate cosorption on aluminum oxide surface. Geochim. Cosmochim. Acta 2009, 73, 2727.
| Uranyl and arsenate cosorption on aluminum oxide surface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltVals7k%3D&md5=7659ae6caf4097b704e01c466b706f70CAS |
[38] R. Donahue, M. J. Hendry, Geochemistry of arsenic in uranium mine mill tailings, Saskatchewan, Canada. Appl. Geochem. 2003, 18, 1733.
| Geochemistry of arsenic in uranium mine mill tailings, Saskatchewan, Canada.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXmtVSlurc%3D&md5=c52e035d26a10d55a262fd7a7dc11546CAS |
[39] A. Abdelouas, W. Lutze, E. Nuttall, Chemical reactions of uranium in ground water at a mill tailings site. J. Contam. Hydrol. 1998, 34, 343.
| Chemical reactions of uranium in ground water at a mill tailings site.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXnslKju7k%3D&md5=d35cb44a5c2b2262f0b0fe4ddd108a49CAS |
[40] K. M. Campbell, R. K. Kukkadapu, N. P. Qafoku, A. D. Peacock, E. Lesher, K. H. Williams, J. R. Bargar, M. J. Wilkins, L. Figueroa, J. Ranville, J. A. Davis, P. E. Long, Geochemical, mineralogical and microbiological characteristics of sediment from a naturally reduced zone in a uranium-contaminated aquifer. Appl. Geochem. 2012, 27, 1499.
| Geochemical, mineralogical and microbiological characteristics of sediment from a naturally reduced zone in a uranium-contaminated aquifer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotlOnsLg%3D&md5=cb4e1b2f2df34f1cbe33826c5dacf8ccCAS |
[41] T. Ohnuki, N. Kozai, M. Samadfam, R. Yasuda, S. Yamamoto, K. Narumi, H. Naramoto, T. Murakami, The formation of autunite (Ca(UO2)2(PO4)2nH2O) within the leached layer of dissolving apatite: incorporation mechanism of uranium by apatite. Chem. Geol. 2004, 211, 1.
| The formation of autunite (Ca(UO2)2(PO4)2nH2O) within the leached layer of dissolving apatite: incorporation mechanism of uranium by apatite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXns1Cgsb0%3D&md5=60423810629b33f1d7f8752b2c912519CAS |
[42] K. R. Salome, S. J. Green, M. J. Beazley, S. M. Webb, J. E. Kostka, M. Taillefert, The role of anaerobic respiration in the immobilization of uranium through biomineralization of phosphate minerals. Geochim. Cosmochim. Acta 2013, 106, 344.
| The role of anaerobic respiration in the immobilization of uranium through biomineralization of phosphate minerals.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjtVejsbo%3D&md5=38d42f87b80fc9f658e85bff27f08aa2CAS |
[43] R. Donahue, M. J. Hendry, P. Landine, Distribution of arsenic and nickel in uranium mill tailings, Rabbit Lake, Saskatchewan, Canada. Appl. Geochem. 2000, 15, 1097.
| Distribution of arsenic and nickel in uranium mill tailings, Rabbit Lake, Saskatchewan, Canada.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisFalu7c%3D&md5=1dfa23e876e38dada77db544d39e417eCAS |
[44] J. Essilfie-Dughan, M. J. Hendry, J. Warner, T. Kotzer, Arsenic and iron speciation in uranium mine tailings using X-ray absorption spectroscopy. Appl. Geochem. 2013, 28, 11.
| Arsenic and iron speciation in uranium mine tailings using X-ray absorption spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsleisbnK&md5=52da8b6c7d14c8889356e64114ade1c8CAS |
[45] B. J. Moldovan, D. T. Jiang, M. J. Hendry, Mineralogical characterization of arsenic in uranium mine tailings precipitated from iron-rich hydrometallurgical solutions. Environ. Sci. Technol. 2003, 37, 873.
| Mineralogical characterization of arsenic in uranium mine tailings precipitated from iron-rich hydrometallurgical solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXptV2lsA%3D%3D&md5=bbf0241983f34028d3d5d7be79fef80eCAS | 12666915PubMed |
[46] J. L. Domingo, Reproductive and developmental toxicity of natural and depleted uranium: a review. Reprod. Toxicol. 2001, 15, 603.
| Reproductive and developmental toxicity of natural and depleted uranium: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXovVChurk%3D&md5=0826851742d9beb32a3a6fd5cec0981cCAS | 11738513PubMed |
[47] E. S. Craft, A. W. Abu-Qare, M. M. Flaherty, M. C. Garofolo, H. L. Rincavage, M. B. Abou-Donia, Depleted and natural uranium: chemistry and toxicological effects. J. Toxicol. Env. Health B 2004, 7, 297.
| 1:CAS:528:DC%2BD2cXkvFGgsrc%3D&md5=34309d7c6ab652d8cac4738fb36b87c6CAS |
[48] G. W. Kunze, J. B. Dixon, Pretreatment for mineralogical analysis, in Methods of Soil Analysis: Part 1. Physical and Mineralogical Methods (Ed. A. Klute) 1986, pp. 91–100 (Soil Science Society of America: Madison, WI ).
[49] K. H. Tan, Methods of soil chemical analysis, in Soil Sampling, Preparation, and Analysis, 2nd edn 2005, pp. 82–98 (Taylor & Francis Group: Boca Raton, FL).
[50] L. J. Poppe, V. F. Paskevich, J. C. Hathaway, D. S. Blackwood, Separation of the silt and clay fractions for x-ray powder diffraction by centrifugation, in A Laboratory Manual for X-Ray Powder Diffraction, US Geological Survey Open-File Report 01-041 2001 (Woods Hole, MA). Available at http://pubs.usgs.gov/of/2001/of01-041/htmldocs/methods/centrifu.htm [Verified 1 November 2013].
[51] L. D. Whittig, W. R. Allardice, X-Ray diffraction techniques, in Methods of Soil Analysis: Part 1. Physical and Mineralogical Methods (Ed. A. Klute) 1986, pp. 331–362 (Soil Science Society of America: Madison, WI).
[52] W. Harris, G. N. White, X-Ray diffraction techniques for soil mineral identification, in Methods of Soil Analysis: Part 5. Mineralogical Methods (Eds A. L. Ulery, L. R. Drees) 2008, pp. 81–115 (Soil Science Society of America: Madison, WI).
[53] P. N. Soltanpour, J. Benton Jones, S. M. Workman, Optical emission spectroscopy, in Methods of Soil Analysis: Part 2. Chemical and Microbiological Properties, 2nd edn (Ed. A. L. Page) 1982, pp. 55–57 (Soil Science Society of America: Madison, WI).
[54] J.-H. Huang, R. Kretzschmar, Sequential extraction method for speciation of arsenate and arsenite in mineral soils. Anal. Chem. 2010, 82, 5534.
| Sequential extraction method for speciation of arsenate and arsenite in mineral soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXntVSjsrg%3D&md5=6b7f3783c80f1d9f894f1a39c66df5bfCAS | 20524641PubMed |
[55] M. J. Beazley, R. J. Martinez, S. M. Webb, P. A. Sobecky, M. Taillefert, The effect of pH and natural microbial phosphatase activity on the speciation of uranium in subsurface soils. Geochim. Cosmochim. Acta 2011, 75, 5648.
| The effect of pH and natural microbial phosphatase activity on the speciation of uranium in subsurface soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFWltLvK&md5=5f190d2039c479e763884cf2c3ef935dCAS |
[56] A. Tessier, P. Campbell, M. Bisson, Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 1979, 51, 844.
| Sequential extraction procedure for the speciation of particulate trace metals.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXitV2rtr4%3D&md5=8f350c2b3f8f0286c5489ff0ff6c6df7CAS |
[57] T. Borch, Y. Masue-Slowey, R. K. Kukkadapu, S. E. Fendorf, Phosphate imposed limitations on biological reduction and alteration of ferrihydrite. Environ. Sci. Technol. 2007, 41, 166.
| Phosphate imposed limitations on biological reduction and alteration of ferrihydrite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtlCit7jF&md5=dd278d5f98a3a92c098186d98d028d56CAS | 17265943PubMed |
[58] K. Amstaetter, T. Borch, P. Larese-Casanova, A. Kappler, Redox transformation of arsenic by FeII-activated goethite (α-FeOOH). Environ. Sci. Technol. 2010, 44, 102.
| Redox transformation of arsenic by FeII-activated goethite (α-FeOOH).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1artbrI&md5=094665af2b901d73dd8f585e995a4fabCAS | 20039739PubMed |
[59] B. Ravel, M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537.
| ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXltlCntLo%3D&md5=38393bb2fbbd8e2e2db909c600fda025CAS | 15968136PubMed |
[60] E. J. Schofield, H. Veeramani, J. O. Sharp, E. Suvorova, R. Bernier-Latmani, A. Mehta, J. Stahlman, S. M. Webb, D. L. Clark, S. D. Conradson, E. S. Ilton, J. R. Bargar, Structure of biogenic uraninite produced by Shewanella oneidensis strain MR-1. Environ. Sci. Technol. 2008, 42, 7898.
| Structure of biogenic uraninite produced by Shewanella oneidensis strain MR-1.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFGksbfF&md5=29346c8ab8d6dc2529324f25bb263957CAS | 19031878PubMed |
[61] P. A. O'day, N. Rivera, R. Root, S. Carroll, X-Ray absorption spectroscopic study of Fe reference compounds for the analysis of natural sediments. Am. Mineral. 2004, 89, 572.
| 1:CAS:528:DC%2BD2cXivFCrtLo%3D&md5=30c43a25ee71aa17eb14496fa6803c9aCAS |
[62] R. M. Cornell, U. Schwertmann, Formation, in The Iron Oxides 2006, pp. 345–364 (Wiley-VCH: Weinheim, Germany).
[63] J. J. Moran, M. K. Newburn, M. L. Alexander, R. L. Sams, J. F. Kelly, H. W. Kreuzer, Laser ablation isotope ratio mass spectrometry for enhanced sensitivity and spatial resolution in stable isotope analysis. Rapid Commun. Mass Spectrom. 2011, 25, 1282.
| Laser ablation isotope ratio mass spectrometry for enhanced sensitivity and spatial resolution in stable isotope analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXks1ajtrc%3D&md5=ca7d87344ca505d1c0f2d499722cbb3bCAS | 21488126PubMed |
[64] J. D. Wall, L. R. Krumholz, Uranium reduction. Annu. Rev. Microbiol. 2006, 60, 149.
| Uranium reduction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1WhtbzM&md5=72737ed375d2903c406d7d0a5920c3feCAS | 16704344PubMed |
[65] J. K. Fredrickson, J. M. Zachara, D. W. Kennedy, M. C. Duff, Y. A. Gorby, S. W. Li, K. M. Krupka, Reduction of UVI in goethite (α-FeOOH) suspensions by a dissimilatory metal-reducing bacterium. Geochim. Cosmochim. Acta 2000, 64, 3085.
| Reduction of UVI in goethite (α-FeOOH) suspensions by a dissimilatory metal-reducing bacterium.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXnt1yju7c%3D&md5=39761bebf5f968f8f63397ec280e5657CAS |
[66] D. Langmuir, Carbonate chemistry, in Aqueous Environmental Geochemistry 1997, pp. 193–230 (Prentice Hall: Upper Saddle River, NJ).
[67] R. J. Reeder, M. Nugent, C. D. Tait, D. E. Morris, S. M. Heald, K. M. Beck, W. P. Hess, A. Lanzirotti, Coprecipitation of uranium(VI) with calcite: XAFS, micro-XAS, and luminescence characterization. Geochim. Cosmochim. Acta 2001, 65, 3491.
| Coprecipitation of uranium(VI) with calcite: XAFS, micro-XAS, and luminescence characterization.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXotVOrurY%3D&md5=8c68801d452c73f8e5cdaa3a38c8174cCAS |
[68] Z. Zheng, T. K. Tokunaga, J. Wan, Influence of calcium carbonate on UVI sorption to soils. Environ. Sci. Technol. 2003, 37, 5603.
| Influence of calcium carbonate on UVI sorption to soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXos1Whs70%3D&md5=be6d5ad29cd70b5516fe3dbb31f4433aCAS | 14717170PubMed |
[69] J. D. C. Begg, I. T. Burke, J. R. Lloyd, C. Boothman, S. Shaw, J. M. Charnock, K. Morris, Bioreduction behavior of UVI sorbed to sediments. Geomicrobiol. J. 2011, 28, 160.
| Bioreduction behavior of UVI sorbed to sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXisVajtLk%3D&md5=3444a9e56635e7425383ceffbcb34167CAS |
[70] P. Smedley, D. Kinniburgh, A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517.
| A review of the source, behaviour and distribution of arsenic in natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhvVSmur0%3D&md5=e91f95bcfc6b5336d0e64af1fca2a6c1CAS |
[71] B. A. Manning, S. Goldberg, Adsorption and stability of arsenic(III) at the clay mineral – water interface. Environ. Sci. Technol. 1997, 31, 2005.
| Adsorption and stability of arsenic(III) at the clay mineral – water interface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjtlGisbk%3D&md5=64d28d19b15213fd25c6f011a4b33818CAS |
[72] M. F. Lengke, C. Sanpawanitchakit, R. N. Tempel, The oxidation and dissolution of arsenic-bearing sulfides. Can. Mineral. 2009, 47, 593.
| The oxidation and dissolution of arsenic-bearing sulfides.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVKmt7bI&md5=6746bf232019fe7dc23aee984303f9c9CAS |
[73] M. F. Lengke, R. N. Tempel, Reaction rates of natural orpiment oxidation at 25 to 40 °C and pH 68 to 82 and comparison with amorphous As2S3 oxidation. Geochim. Cosmochim. Acta 2002, 66, 3281.
| Reaction rates of natural orpiment oxidation at 25 to 40 °C and pH 68 to 82 and comparison with amorphous As2S3 oxidation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XmslKkurw%3D&md5=afdef53077a2f7c5dbc303c2320a59feCAS |
[74] D. K. Newman, T. J. Beveridge, F. Morel, Precipitation of arsenic trisulfide by Desulfotomaculum auripigmentum. Appl. Environ. Microbiol. 1997, 63, 2022.
| 1:CAS:528:DyaK2sXjtFGmtbo%3D&md5=7aa57146e378e8a5d391168d03890f42CAS | 16535611PubMed |
[75] B. C. Bostick, Arsenite sorption on troilite (FeS) and pyrite (FeS2). Geochim. Cosmochim. Acta 2003, 67, 909.
| Arsenite sorption on troilite (FeS) and pyrite (FeS2).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXht1WgtLw%3D&md5=319a74cb9ee0668002d66cd1d5c8af02CAS |
[76] Agency for Toxic Substances and Disease Registry, Minimal risk levels (MRLs), in Toxicological Profile for Uranium 2013, pp. 20–21 (US Department of Health and Human Services, Public Health Service: Atlanta, GA).
[77] S. Kelly, D. Hesterberg, B. Ravel, Analysis of soils and minerals using X-ray absorption spectroscopy, in Methods of Soil Analysis: Part 5. Mineralogical Methods (Eds A. L. Ulery, L. R. Drees), 2008, pp. 387–463 (Soil Science Society of America: Madison, WI).