

Redox-Initiated Reversible Addition–Fragmentation Chain Transfer (RAFT) Polymerization
Amin Reyhani A , Thomas G. McKenzie A , Qiang Fu A and Greg G. Qiao A BA Polymer Science Group, Department of Chemical Engineering, The University of Melbourne, Parkville, Vic. 3010, Australia.
B Corresponding author. Email: gregghq@unimelb.edu.au

Amin Reyhani earned his undergraduate degree in chemical engineering from Sahand University of Technology, Iran, in 2010. He graduated from Sharif University of Technology, Iran, with his Master’s degree in chemical engineering in 2012. He completed his Ph.D. studies in chemical engineering at The University of Melbourne, Australia, under the supervision of Professor Greg Qiao in 2019. His Ph.D. research was focused on harnessing the power of Fenton chemistry in the RAFT polymerization technique. |

Dr Thomas (Tom) McKenzie is originally from New Zealand, having completed his undergraduate degree at Victoria University of Wellington. After a brief stint in industry, he then went on to complete his doctoral studies under the supervision of Professors Greg Qiao and Dave Dunstan at the University of Melbourne, where he is now a Research Fellow specializing in novel strategies for externally regulated controlled polymer synthesis. |

Dr Qiang Fu received his B.E. in chemical engineering from Shanghai Jiao Tong University in 2004. He completed his Ph.D. degree in polymer chemistry at Fudan University in 2009 before working as a postdoctoral fellow with Professor Greg Qiao at the University of Melbourne. He was awarded an ARC Super Science Fellowship in 2011 and an ARC Future Fellowship in 2018. His research interests range from fundamental studies to applied sciences and encompass the fields of two-dimensional polymers, macromolecular engineering and self-assembly, and membrane materials for separations. |

Professor Greg Qiao received his Ph.D. degree from the University of Queensland in 1996. He joined the University of Melbourne in 1996 and became a full professor in 2009. He was the Australian Research Council’s professorial Future Fellow (2012–2015) and the Chair of the Polymer Division of the Royal Australian Chemical Institute (RACI) (2015–2016). He received the Applied Research Award in 2017, ExxonMobil Award in 2015, RACI’s Polymer Division Citation in 2011, and Freehills Award in 2010. He has published more than 250 journal papers and is a co-inventor in more than 20 patents. His key research interests are in polymeric architectures, new activation methods for RAFT, peptide polymers, tissue scaffolds, and gas membranes. |
Australian Journal of Chemistry 72(7) 479-489 https://doi.org/10.1071/CH19109
Submitted: 6 March 2019 Accepted: 11 May 2019 Published: 12 June 2019
Abstract
Reversible addition–fragmentation chain transfer (RAFT) polymerization initiated by a radical-forming redox reaction between a reducing and an oxidizing agent (i.e. ‘redox RAFT’) represents a simple, versatile, and highly useful platform for controlled polymer synthesis. Herein, the potency of a wide range of redox initiation systems including enzyme-mediated redox reactions, the Fenton reaction, peroxide-based reactions, and metal-catalyzed redox reactions, and their application in initiating RAFT polymerization, are reviewed. These redox-RAFT polymerization methods have been widely studied for synthesizing a broad range of homo- and co-polymers with tailored molecular weights, compositions, and (macro)molecular structures. It has been demonstrated that redox-RAFT polymerization holds particular promise due to its excellent performance under mild conditions, typically operating at room temperature. Redox-RAFT polymerization is therefore an important and core part of the RAFT methodology handbook and may be of particular importance going forward for the fabrication of polymeric biomaterials under biologically relevant conditions or in biological systems, in which naturally occurring redox reactions are prevalent.
Introduction
Reversible deactivation radical polymerization (RDRP) methods such as atom transfer radical polymerization (ATRP),[1] nitroxide-mediated polymerization (NMP),[2] and reversible addition–fragmentation chain transfer (RAFT) polymerization[3] have played a significant role in the paradigm shift towards the synthesis of tailor-made polymers with pre-determined size, composition, and architecture. The speed with which polymers can be synthesized in a controlled manner via RDRP has become an interesting scientific discussion.[4] Among the RDRP methods, the RAFT technique has been of great interest to polymer chemists due to its compatibility with a diverse range of reagents (i.e. monomers, catalysts, etc.), and its versatility towards various operating conditions.[5] A typical RAFT reaction involves the use of a radical initiator to induce a chain growth process which is mediated by participating in a degenerative chain transfer for rapid ‘deactivation’ of the propagating radical with a highly efficient chain transfer agent (CTA) – typically a thiocarbonylthio-containing compound, that is referred to in this role as a ‘RAFT agent’ (Scheme 1a).[3] RAFT agents have been demonstrated to be versatile[6] and highly efficient chain transfer agents for such purposes[7] while maintaining excellent tolerance towards a wide range of chemical functionalities.[5c,8] It is known that RAFT polymerization is the process, as the RAFT agent is used in the free radical polymerization to achieve well controlled polymers.[7] The general mechanism of RAFT is shown in Scheme 1b, where an initiation step results in the addition of monomer (M) to the active radical and subsequent propagating polymer chain (Pn•) with rate constant kp. The degenerative chain transfer process occurs across the thiocarbonyl group of the RAFT CTA, securing a radical intermediate that may fragment to liberate the R group from the RAFT agent. The released R group radical then re-initiates the polymerization, producing another actively propagating polymer chain (Pm•). Although thiocarbonylthio-bound polymer chains (Pn and Pm) are dormant – i.e. they do not react directly with available monomers – they participate actively in the dynamic equilibrium with propagating radical chains. Therefore, while only propagating polymers (Pn• and Pm•) would actively participate in polymerization reactions, they may transfer the radical to a dormant chain, thereby becoming dormant themselves while activating previously dormant thiocarbonylthio compounds. The equilibrium between active propagating and dormant chains provides a distributed opportunity for all chains to grow evenly, resulting in polymers with well controlled or low dispersity (Ð) molecular weight distributions.[3a,9]

|
RAFT polymerization has been initiated by a broad range of physical and chemical stimuli including thermally activated azo-initiators (e.g. azobisisobutyronitrile (AIBN)[3a,5a] or 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044)[5b,5c]), enzyme-mediated reactions,[10] and photo-induced processes.[6,11] Nevertheless, researchers are always keen to look for new approaches that can address various drawbacks of these systems. For instance, the rate of visible light-induced RAFT polymerization is very slow in the absence of photo catalysts.[12] Although the rate of RAFT polymerization catalyzed by photoredox catalysts is relatively fast, such photo catalysts (e.g. iridium and ruthenium-based ones[11d,13]) are quite costly. Likewise, photo-mediated RAFT polymerizations may face difficulties in terms of the equipment required to perform photo-initiation in a scaled-up process.[5b] With the use of azo-initiators, elevated temperatures are required, limiting the potential for in situ RAFT polymerization in biological systems where heat may denature biological components.[14] The employment of such thermally activated initiators will confine the flexibility of the RAFT process when different reaction temperatures are necessary.[9]
Redox-initiated RAFT polymerization (‘redox-RAFT’) techniques can allow for a simple ambient temperature initiation without the need for external stimuli, and therefore offers an attractive alternative to thermal and photo-based techniques.[5b,5d,15] Such redox-RAFT polymerization systems have many advantages over other RAFT processes including low needed activation energies, facile control over the polymerization rate at low temperatures, high elimination of the side reactions, and very short induction periods.[15c,15d,16] From an industrial standpoint, the use of a redox initiation system at ambient temperature is highly practical and easy to perform as simple, cheap, and effective redox pairs can be employed.[5b]
Redox Pairs for RAFT Initiation
Bai and co-workers[15b] reported a non-aqueous redox-RAFT polymerization of methyl acrylate (MA), methyl methacrylate (MMA), and styrene (St) in bulk (or tetrahydrofuran (THF) solution) in the presence of the CTAs benzyl 1H-imidazole-1-carbodithioate (BICDT) and 2-cyanoprop-2-yldithiobenzoate (CPDB) by using a traditional redox system consisting of benzoyl peroxide (BPO)/N,N-dimethylaniline (DMAn), performed at room temperature. In both bulk and THF solution they accomplished well controlled polymers with Ð values below 1.2 and experimental molecular weights matched well with their theoretical values.[15b] Bai and his group later published an application of the BPO/DMAn redox couple in initiating the RAFT polymerization of a vinyl azide monomer (i.e. 4-azidophenyl methacrylate, APM), along with BICDT and CPDB as CTAs, in dimethylformamide (DMF) as solvent at room temperature.[15f] The obtained experimental results showed that the redox-RAFT polymerization bore all the hallmarks of a living radical polymerization, and the azide-based polymeric products possessed controlled molecular weight and low Ð values. RAFT co-polymerizations of APM with MA, MMA, and St were also successfully conducted.
Various redox initiation systems have also been employed for the aqueous RAFT polymerization of temperature-sensitive monomers such as N-isopropylacrylamide (NIPAM)[15d] and N-vinyl pyrrolidone (VP).[15e] For instance, Bai and co-workers[15d] have demonstrated the use of an inexpensive and effective redox system consisting of potassium persulfate (KPS, K2S2O8) and sodium thiosulfate (STS, Na2S2O3) for the aqueous RAFT polymerization of NIPAM and acrylamide (AM) in the presence of 2-(1-carboxy-1-methyl-ethylsulfanylthiocarbonyl-sulfanyl)-2-methyl-propionic acid (CMP) as RAFT agent at room temperature. High molecular weight linear PNIPAM and PAM with low Ð values (< 1.15 for NIPAM and < 1.40 for PAM) were achieved. Control of the RAFT polymerization and its ‘livingness’ (i.e. retention of the thiocarbonylthio moiety) was demonstrated by kinetic studies and chain extension experiments. Another redox pair consisting of tert-butylhydroperoxide (TBHP)/ascorbic acid (AsAc) was used for the initiation of an aqueous RAFT polymerization of VP at ambient conditions in the presence of a xanthate CTA (i.e. O-ethyl-S-(1-methoxycarbonyl)ethyl dithiocarbonate, XA) without any observable side reactions.[15e] High monomer conversions (> 90 %) and high molecular weight polymers with low Ð values (< 1.3) were successfully obtained. High chain-end fidelity of the RAFT agent in polymer chains was demonstrated by synthesizing poly(acrylamide) (PAM)-PVP diblock copolymers and matrix-assisted laser desorption ionization time-of-flight (MALDI-ToF) analysis.[15e] The TBHP/AsAc redox pair has been also taken to initiate RAFT polymerization of N-vinylcaprolactam with XA in water/ethanol solvent mixtures at ambient conditions. Monomer conversion values increased with an increase in water volume fraction in the solvent mixture because of an enhanced polymer solvation.[17] Junkers and co-workers introduced an acid-induced redox initiation system (i.e. cyclohexanone (CH)/TBHP) to aqueous RAFT polymerization of n-butyl acrylate with 2-(dodecylthiocarbonothioylthio) propionic acid (DoPAT) at room temperature. An acid (p-toluenesulfonic acid) was used in conjugation with the CH/TBHP redox pair as catalytic co-initiator.[15a]
Destarac and co-workers reported aqueous RAFT polymerization of VP with a xanthate CTA initiated by the TBHP/Na2SO3 as redox pair.[18] In this work, they replaced the AsAc used in their previous study[15e] with Na2SO3 because it is a reducing agent which allows for a faster reaction under basic conditions, therefore minimizing the formation of N-(α-hydroxyethyl)pyrrolidone – the main by-product of VP polymerization conducted under acidic conditions. This redox-RAFT process was ultimately used for the aqueous synthesis of PVP-based hydrophilic diblock copolymers including PAM-b-PVP and poly(acrylic acid)-b-PVP through the VP polymerization at room temperature using several hydrophilic macro-RAFT initiators, leading to well controlled copolymers. Later, Perrier and co-workers[5b] employed the TBHP/AsAc redox pair for the aqueous room temperature polymerization of multiblock copolymers (up to seven blocks) consisting of different polymer blocks such as poly(4-acryloylmorpholine) (PNAM), PNIPAM, and poly(2-hydroxyethyl acrylate) (PHEA), with the final Ð < 1.35. Scheme 2a shows the structures of all reagents used in this work. As an example, Scheme 2b illustrates the strategy for the synthesis of the acrylate/acrylamide heptablock copolymers. The high livingness/end group fidelity is seemingly retained, highlighted by the successive shifts in molecular weight distribution after each block extension. The main benefit of employing this redox-RAFT polymerization is the possibility to incorporate thermo-responsive blocks and to limit side reactions that are sometimes observed in the polymerization of acrylate-type monomers at high temperatures.[5b] Table 1 summarizes the reagents employed for synthesizing linear polymers in some redox-initiated RAFT processes.

|

|
Using the same redox pair (TBHP/AsAc), Kempe and co-workers reported the synthesis of novel, thermo-responsive comb polymers from alternating N-acylated poly(aminoester)s via a combination of spontaneous zwitterionic copolymerization and redox-initiated RAFT polymerization.[19] On the basis of the above, Kempe’s group has successfully prepared a series of functional comb polymers from multi-stimuli responsive poly(aminoester) macromonomers.[20] Therefore, it can be concluded that the redox-initiating system was especially appealing to the polymerization of thermoresponsive (macro)monomers. More recently, Perrier’s group[21] investigated the performance of three different redox initiating systems, TBHP/AsAc, TBHP/Brüggolit FF7 (Brug7), and ammonium persulfate (APS)/SFS by following the kinetics of a model RAFT polymerization. It was found that the TBHP/Brug7 redox pair displayed the fastest polymerization rate with excellent control over molecular weight and polydispersity compared with other redox initiating systems.[21] Armes and co-workers reported the preparation of pyrrolidone-based homo- or block-polymers via RAFT aqueous polymerization using the KPS/AsAc redox pair at 30°C.[22]
In all the above-mentioned studies the redox initiation systems employed for room temperature RAFT polymerization require the simultaneous addition of both reducing and oxidizing agents. An amine-containing vinylic monomer (2-vinylpyridine (2VP)) has been reported to participate in the initiation stage of an aqueous RAFT process at room temperature, as shown by Xin and co-workers.[15c] In other words, redox-RAFT of 2VP was carried out in the presence of BPO as oxidizing agent without any reducing agent. It was claimed that the amine-based 2VP together with BPO generated benzoyloxyl radicals via a redox reaction, providing initiating radicals to stimulate the RAFT polymerization. This was proved further by demonstrating that St (an amine-free monomer) could not polymerize through this process. Although well defined P2VPs with low Ð values (< 1.2) were achieved, the monomer conversion after 24 h was less than 50 % indicating the rate of polymerization in this redox-RAFT system was very low.[15c]
RAFT Polymerization Initiated by Ternary Enzymatic-Redox Systems
Enzymes, the proteins that catalyze biological reactions, have been recently employed to effectively initiate various RAFT polymerizations. Among them, horseradish peroxide (HRP) and glucose oxidase (GOx) have attracted special attention due to their ability to generate initiating radical species and consume oxygen, respectively. An and co-workers[23] harnessed the ability of a HRP/H2O2/acetylacetone (ACAC) ternary initiating system for the RAFT polymerization of N,N-dimethylacrylamide (DMA), HEA, VP, poly(ethylene glycol) methyl ether acrylate (PEGA), and poly(ethylene glycol) methyl ether methacrylate (PEGMA) using a trithiocarbonate (2-ethylsulfanylthiocarbonylsulfanyl-propionic acid methyl ester) and a xanthate (2-ethoxythiocarbonylsulfanyl-2-methyl-propionic acid 2-hydroxy-ethyl ester) as CTAs at mild, biocompatible, and ambient conditions. HRP can catalyze the oxidation of ACAC (with the co-substrate H2O2) to generate ACAC radicals capable of initiating the RAFT polymerization (Scheme 3). In more detail, the HRP facilitates an electron transfer by converting a two-electron oxidation of ferriprotoporphyrin units by H2O2 into two subsequent single electron transfer steps to generate two ACAC radicals (step I) which then initiate polymerization of DMA (step II).[24] Well defined and high molecular weight homopolymers and di-/tri-block copolymers with low Ð values and high monomer conversions were readily achieved in aqueous buffer solution (pH 7) at low HRP concentrations via this ternary enzymatic redox-RAFT polymerization.[23]
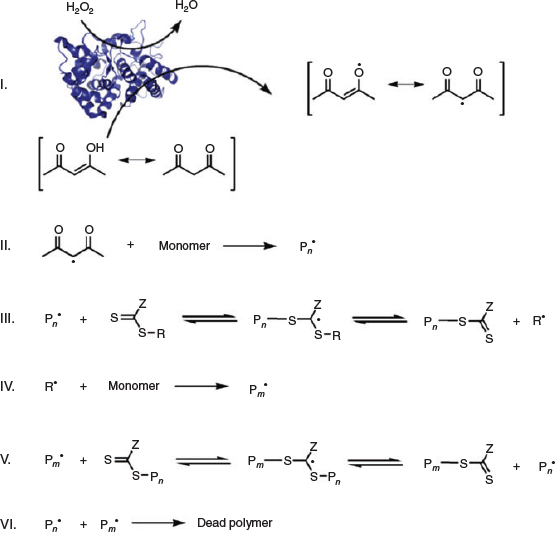
|
Independently, Konkolewicz and co-workers[25] also demonstrated the use of this enzymatic redox initiating system for the RAFT polymerization of DMA, NIPAM, and oligo(ethylene oxide) methyl ether acrylate (OEOA) in the presence of (2-propionic acid)yl ethyl trithiocarbonate (PAETC) as CTA in a buffer solution (pH 5.5) at room temperature. Although high monomer conversion values were achieved, the Ð values were above 1.25. This HRP-initiated RAFT was used to create block copolymers and protein–polymer conjugates. Recently, Konkolewicz and co-workers provided more detail about the mechanism of this HRP-mediated RAFT polymerization.[26] These papers introduced a burgeoning interest in the use of biocatalysts for RAFT (and other controlled) polymerizations.[23,24] The use of biocatalysts such as enzymes in RAFT polymerization is promising for a more ‘green’ synthesis of well defined polymers. The promising results demonstrated to date may pave the way for the fabrication of bio-based polymers under biologically relevant conditions.
Another ternary enzymatic-redox initiating system that has been employed for the initiation of RAFT polymerization consists of glucose oxidase (GOx) and d-glucose, along with a reducing agent capable of reacting with the H2O2 produced by the GOx enzyme. GOx has been used extensively for dissolved oxygen removal in various RDRPs. It can catalyze the oxidation of d-glucose to d-gluconic acid (or 2-dehydro-d-glucose) in the presence of molecular oxygen, generating H2O2 as a by-product.[27] The GOx-generated H2O2 can react with an added reducing agent to produce highly reactive hydroxyl radicals (HO•).[28] An and co-workers utilized the formation of such hydroxyl radicals for initiating an aqueous RAFT polymerization of DMA at low temperatures in the presence of air (in either open or sealed reaction vessels) under different operating conditions (i.e. stirring speed, temperature (30 and 40°C), pre-polymerization incubation time, and concentrations of the reagents) by using AsAc as the reducing agent.[28] An and co-workers have also demonstrated the use of a dual-enzyme cascade methodology in which the GOx/d-glucose/O2 system is combined with the HRP/H2O2/ACAC system to generate the requisite initiating ACAC radicals (Scheme 4).[23] This GOx-HRP-mediated RAFT polymerization showed pseudo first-order kinetics regarding monomer conversions, well controlled polymers (i.e. PDMAs) with low Ð values (< 1.2), and experimental molecular weights well matched with theoretical values.

|
An and co-workers took the enzymatic cascade system in which GOx was replaced with pyranose oxide (P2Ox, a homotetrameric flavoprotein found in root fungi) due to its higher enzymatic efficiency to synthesize hydrophilic multiblock and ultra-high molecular weight (UHMW) polymers via the RAFT process in reaction vessels exposed to air (Scheme 5).[29] They successfully synthesized sequentially controlled multiblock copolymers up to 10 blocks with Mn ~100 kDa, and also formed controlled UHMW polymers with Mn ~2.3 million Da (MDa) with near-quantitative monomer conversion and low Ð values (< 1.5 for the copolymers and < 1.4 for the UHMW homopolymers).

|
RAFT Polymerization Initiated by Fenton Chemistry
Recently the power of Fenton chemistry has been used to initiate RAFT polymerization by Qiao and co-workers.[30] It was initially introduced for the initiation of a RAFT process by using ferrous ions, i.e. FeII, and H2O2 to speed up the polymerization rate, and termed as Fenton-RAFT polymerization (Scheme 6a).[30a] Several approaches to increase the rate of RAFT polymerization had previously been reported, such as the employment of microwave reactor technology,[31] the use of various photoinitiators,[32] or elevated reaction temperatures.[33] Likewise, some early flow reactor experiments appear promising for increasing the reaction kinetics.[34] Nevertheless, these works do not represent a step-change in the choice of initiation pathway, but rather an improvement of the traditional approaches. The aqueous Fenton-RAFT polymerization was demonstrated as an ultra-fast, easy to set-up, and oxygen tolerant process at ambient temperature. Polymerization of different water-soluble vinyl monomers (e.g. DMA, NAM, and HEA) was successfully accomplished with moderate monomer conversions (> 65 %) and low Ð (< 1.1) values, and experimental Mn matching theoretical values. The extremely rapid polymerization timescales (< 60 s) make this new technique of great interest in the development of the machine-programmable synthesis of polymers in high-throughput arrays. Afterwards, biological Fenton reagents (i.e. GOx and hemoglobin (Hb)) were also employed successfully for a Fenton RAFT polymerization in the presence of air – termed as BioRAFT.[30c] This BioRAFT polymerization led to well controlled polymers with near-quantitative conversion within 6 h. The resultant linear polymers showed very narrow dispersities (Ð < 1.1) and molecular weights in agreement with their theoretical values. The BioRAFT system, therefore, offered fascinating opportunities for in situ radical generation, and ‘blood-catalyzed’ reactions (in non-modified ovine blood) were demonstrated (Scheme 6b). In these reactions, it was hypothesized that the red blood cells could act directly as the catalyst for the Fenton reaction, via their iron-rich Hb enzymes, providing initiating radicals for RAFT polymerization when used in conjunction with the H2O2-generating GOx enzyme. Lysis of the red blood cells in reverse osmosis (RO) water (unbuffered water) was required for the reaction to proceed due to the release of Hb into the reaction solution increasing its availability for subsequent participation in the Fenton process. Taken together, obtained results indicated that the content of red blood cells could be exploited as precursors for the Fenton reaction-mediated radical polymerization, potentially even exploiting the natural production of H2O2 from certain biological responses to provide the peroxide required to complete the redox process.

|
Inspired by the dual catalyst system (i.e. GOx/Hb) in the BioRAFT technique that supplies hydroxyl radicals in a controlled manner, synthetic H2O2 was carefully and continuously dosed into the reaction solution by using a syringe pump, leading to the synthesis of well defined polymers with low Ð values and full monomer conversions, which wasn’t previously attainable without careful metering of the H2O2 concentration.[30b] By restricting the immediate flux of initiating radicals, metered H2O2 injection decreases the unwanted radical ‘wasting’ reaction (H2O2 + HO• → HO2• + H2O, k = 3.3 × 107 M−1 S−1) providing access to full conversions with high chain‐end functionality.[30b] The concept of a slow production of hydroxyl radicals led to the successful synthesis of unprecedently large polymers with UHMW values up to 20 MDa through a Bio-Fenton-RAFT process (Scheme 6c) where Hb was replaced by an inorganic source of FeII to expedite the reaction.[30e] To mimic the function of GOx synthetically, a syringe pump was employed to gradually add H2O2 into the reaction mixture, leading to UHMW polymers.
The residual of the employed catalysts (i.e. FeII salt and Hb) remain in the reaction solution at the end of polymerization, presenting a challenging issue for purification in all the above-mentioned Fenton-RAFT processes. To overcome this challenge, heterogeneous FeII-based metal–organic framework (MOF) particles were used to initiate the Fenton-RAFT polymerization – termed as MOF-Fenton-RAFT (Scheme 6d).[35] Well controlled polymers (Ð < 1.1) with near-quantitative monomer conversions were achieved, even in non-deoxygenated reaction solutions. It was shown that the heterogeneous MOF catalyst could be easily removed via centrifugation, recovered, and reused (five cycles of reuse shown). The MOF-Fenton-RAFT process may be important for biomedical systems where the presence of free metal catalysts is problematic.
RAFT Polymerization with Direct Activation of RAFT Agents
RAFT Polymerization Catalyzed by Transition Metal Catalysts
Transition metal catalysts have been widely used in RAFT polymerization at ambient temperature where a RAFT CTA is considered as an alkyl ‘pseudohalide’, in analogy with ATRP.[9,36] Use of these catalysts would overcome drawbacks of both conventional RAFT with azo-initiators and ATRP with alkyl halide initiators so that: (1) polymerization at ambient temperature may decrease the possibility of thermal self-initiation of monomer, thermal cross-linking, chain transfer, and other side reactions.[36f] Room temperature reactions would also stop the need for conventional thermal initiators which limit the flexibility of the RAFT process in terms of range of polymerization temperature;[9] (2) as shown in Scheme 1b, the initiating radicals are generated directly from the CTA, thus eliminating the formation of new chains resulting from the reaction of radical initiators with propagating polymer chains or monomer during traditional RAFT process;[9,36c,37] (3) using CTA as alkyl pseudohalides (R-X) removes the need for a minimal amount of deactivating high oxidation state metal complex while still retaining low Ð;[36b] (4) use of activators regenerated by electron transfer (ARGET) ATRP by employing a reducing agent such as AsAc allows the level of low oxidation state metal catalyst to be minimized to below ppm levels.[36a,37]
An example of this approach is the polymerization of the monomers glycidyl methacrylate (GMA), 4-vinylpyridine (4VP), butyl methacrylate (BMA), 2-(dimethylamino)ethyl methacrylate (DMAEMA), and poly(ethylene glycol)monomethyl ether methacrylate (PEGMA300) using Fe0 wire, together with 2-cyanoprop-2-yl 1-dithionaphthalate (CPDN) as reported by Zhu and co-workers.[38] Scheme 7 shows the proposed hybrid mechanism (RAFT and ATRP) for this Fe0-mediated polymerization in which Fe0 acts as an activator that breaks the C–S bond in CPDN, generating initiating radicals in the initial stage required for kicking off the polymerization. In this technique the RAFT agent plays two roles, as an alkyl pseudohalide initiator and a CTA. In the proposed mechanism, the RAFT process is thought to be the dominant mechanism for control due to the efficiency of radical chain transfer. Other zero-valent metals (e.g. cobalt, nickel, manganese, and zinc) were also found to be applicable for the polymerization of MMA,[38] in a similar manner.[39]

|
UHMW PMMA (Mn > 1 MDa) and high molecular weight PSt (Mn > 0.5 MDa) polymers were successfully synthesized for the first time within an RDRP system by using ppm levels of CuI complex catalyst and cumyl dithiobenzoate (CDB) via the ARGET ATRP process under atmospheric pressure by Matyjaszewski and co-workers.[37] AsAc was used as the reducing agent to regenerate the active, lower oxidation state catalyst. This approach provided excellent control over Mn, Ð, and chain-end fidelity. Moreover, a minimal concentration of deactivating CuII complex is not required for the retention of low Ð values, unlike typical ARGET ATRP techniques. With the same methodology, Zhang and co-workers employed inorganic compounds, cupric oxide (CuO) or cuprous oxide (Cu2O), in the polymerization of MMA with CPDN at room temperature, resulting in well defined polymers with low dispersity values (Ð < 1.25).[36a] These reactions using RAFT agents as alkyl pseudohalides could be a preferred technique from both environmental and industrial viewpoints. Some works have also reported the use of Cu-based organometallic catalysts, e.g. copper(ii) acetylacetonate (Cu(acac)2),[40] copper(ii) hexafluoroacetylacetonate (Cu(hfa)2),[40] and copper(ii) acetate (Cu(OAc)2),[41] for the RAFT polymerization of MMA and styrene (St) at high temperatures (80 and 120°C). It could be noticed that the polymerization rate at elevated temperatures was higher than that of processes at room temperature.
RAFT Polymerization in the Absence of Transition Metal Catalysts
The majority of the transition metals employed to catalyze the above-mentioned RAFT systems are considered toxic, with the need to remove them from the polymer post-synthesis – especially for biological and biomedical applications.[42] Moreover, these catalyst complexes increase the cost of production since the techniques used to purify the polymers are usually quite expensive.[43] Recently, Maxiniano et al.[43] have introduced a metal-free dissociative electron transfer (DET)-RAFT system at ambient temperature using sodium sulfite as the catalyst. These preliminary studies resulted in well controlled polymerizations for methyl acrylate (MA), St, and MMA monomers with Ð values < 1.2. Scheme 8 illustrates the proposed mechanism of the DET-RAFT system where the initial dissociation of dithionite anions into SO2•− is thermally driven, followed by the transfer of an electron to the CTA due to the difference in their redox potentials, resulting in a radical anion capable of fragmenting reversibly to activate the RAFT polymerization.
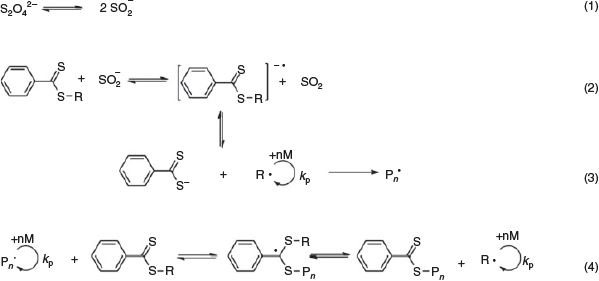
|
This mechanism is similar to that proposed by Boyer and colleagues for photo-redox catalyzed RAFT reactions via photo-induced electron/energy transfer (PET-RAFT).[11d] The RAFT polymerizations of various monomers can be achieved via a PET process by incorporating a suitable photoredox catalyst. Under irradiation, the employed photoredox catalyst is able to reduce the RAFT agent to generate an initiating carbon-centred radical (R•) and a resonance-stabilized anion specie (−S–C(=S)–Z), along with the photoredox catalyst in an elevated oxidation state. The resulting anion specie would back transfer an electron to the catalyst, leading to the generation of dormant species and the photocatalyst in its ground state. The PET-RAFT process has been demonstrated to be tolerant to oxygen due to the strong reductive capacity of the photocatalysts.[44] This robust technique was further combined with a high throughput (HT) approach to prepare a library of linear or star polymers in HT using 96- or 384-well plates.[45] For a comprehensive overview of the PET-RAFT polymerization the reader is referred to the following recent reviews.[13,46]
A newly emerging photo-initiated cationic RAFT polymerization of vinyl ethers in which the activation of a photocatalyst results in the direct activation of RAFT agent, has been recently reported by Fors and his group.[47] Inspired by these works, Fors and co-workers[48] activated the cationic RAFT polymerization of isobutyl vinyl ether by the electrochemical oxidation of a dithiocarbamate CTA, which led to well controlled polymers with experimental molecular weights well matched with theoretical values.
Conclusion
The capability and potency of redox-initiated RAFT polymerization has been illustrated in this review. From an industrial point of view, such redox-RAFT reactions may play a significant role in the fabrication of different types of polymers due to the simple control over the reaction at room temperature and the low activation energies. Examples of redox-RAFT polymerizations are given which show great potential for the synthesis of libraries of well-defined polymers with a wide range of tailored architectures via high throughput systems at room temperature, as well as in situ polymerization in bio-related environments, favourable for various applications, especially biological and biomedical systems.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
A.R. was supported by the Melbourne International Research Scholarship (MIRS) and the Melbourne International Fee Remission Scholarship (MIFRS).
References
[1] (a) J.-S. Wang, K. Matyjaszewski, Macromolecules 1995, 28, 7901.| Crossref | GoogleScholarGoogle Scholar |
(b) M. Kato, M. Kamigaito, M. Sawamoto, T. Higashimura, Macromolecules 1995, 28, 1721.
| Crossref | GoogleScholarGoogle Scholar |
[2] C. J. Hawker, A. W. Bosman, E. Harth, Chem. Rev. 2001, 101, 3661.
| Crossref | GoogleScholarGoogle Scholar | 11740918PubMed |
[3] (a) G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2012, 65, 985.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. Chiefari, Y. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. Le, R. T. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, Macromolecules 1998, 31, 5559.
| Crossref | GoogleScholarGoogle Scholar |
[4] M. Zhong, K. Matyjaszewski, Macromolecules 2011, 44, 2668.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) M. Kopeć, P. Krys, R. Yuan, K. Matyjaszewski, Macromolecules 2016, 49, 5877.
| Crossref | GoogleScholarGoogle Scholar |
(b) L. Martin, G. Gody, S. Perrier, Polym. Chem. 2015, 6, 4875.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. R. Hill, R. N. Carmean, B. S. Sumerlin, Macromolecules 2015, 48, 5459.
| Crossref | GoogleScholarGoogle Scholar |
(d) M. Roa-Luna, G. Jaramillo-Soto, P. V. Castañeda-Flores, E. Vivaldo-Lima, Chem. Eng. Technol. 2010, 33, 1893.
| Crossref | GoogleScholarGoogle Scholar |
(e) T. G. McKenzie, Q. Fu, E. H. Wong, D. E. Dunstan, G. G. Qiao, Macromolecules 2015, 48, 3864.
| Crossref | GoogleScholarGoogle Scholar |
(f) X. Tian, J. Ding, B. Zhang, F. Qiu, X. Zhuang, Y. Chen, Polymers 2018, 10, 318.
| Crossref | GoogleScholarGoogle Scholar |
[6] T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito, G. G. Qiao, Adv. Sci. 2016, 3, 1500394.
| Crossref | GoogleScholarGoogle Scholar |
[7] D. J. Keddie, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2012, 45, 5321.
| Crossref | GoogleScholarGoogle Scholar |
[8] G. Moad, E. Rizzardo, S. H. Thang, Chem. Asian J. 2013, 8, 1634.
| Crossref | GoogleScholarGoogle Scholar | 23606667PubMed |
[9] R. Nicolaÿ, Y. Kwak, Isr. J. Chem. 2012, 52, 288.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) S. Kobayashi, H. Uyama, S. Kimura, Chem. Rev. 2001, 101, 3793.
| Crossref | GoogleScholarGoogle Scholar | 11740921PubMed |
(b) S. R. Zavada, T. Battsengel, T. F. Scott, Int. J. Mol. Sci. 2016, 17, 195.
| Crossref | GoogleScholarGoogle Scholar |
[11] (a) H. Wang, Q. Li, J. Dai, F. Du, H. Zheng, R. Bai, Macromolecules 2013, 46, 2576.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. Y. Khan, M.-S. Cho, Y.-J. Kwark, Macromolecules 2014, 47, 1929.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Shanmugam, J. Xu, C. Boyer, J. Am. Chem. Soc. 2015, 137, 9174.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. Xu, K. Jung, A. Atme, S. Shanmugam, C. Boyer, J. Am. Chem. Soc. 2014, 136, 5508.
| Crossref | GoogleScholarGoogle Scholar |
(e) J. Xu, S. Shanmugam, H. Duong, C. Boyer, Polym. Chem. 2015, 6, 5615.
| Crossref | GoogleScholarGoogle Scholar |
(f) J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou, C. Boyer, J. Am. Chem. Soc. 2016, 138, 3094.
| Crossref | GoogleScholarGoogle Scholar |
[12] C. Ding, C. Fan, G. Jiang, X. Pan, Z. Zhang, J. Zhu, X. Zhu, Macromol. Rapid Commun. 2015, 36, 2181.
| Crossref | GoogleScholarGoogle Scholar | 26445213PubMed |
[13] M. Chen, M. Zhong, J. A. Johnson, Chem. Rev. 2016, 116, 10167.
| Crossref | GoogleScholarGoogle Scholar | 26978484PubMed |
[14] J. Niu, D. J. Lunn, A. Pusuluri, J. I. Yoo, M. A. O’Malley, S. Mitragotri, H. T. Soh, C. J. Hawker, Nat. Chem. 2017, 9, 537.
| Crossref | GoogleScholarGoogle Scholar | 28537595PubMed |
[15] (a) J. Vandenbergh, B. Schweitzer-Chaput, M. Klussmann, T. Junkers, Macromolecules 2016, 49, 4124.
| Crossref | GoogleScholarGoogle Scholar |
(b) H. Zheng, W. Bai, K. Hu, R. Bai, C. Pan, J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 2575.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Luo, M. Li, M. Xin, W. Sun, Macromol. Chem. Phys. 2015, 216, 1646.
| Crossref | GoogleScholarGoogle Scholar |
(d) W. Bai, L. Zhang, R. Bai, G. Zhang, Macromol. Rapid Commun. 2008, 29, 562.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. Guinaudeau, S. Mazières, D. J. Wilson, M. Destarac, Polym. Chem. 2012, 3, 81.
| Crossref | GoogleScholarGoogle Scholar |
(f) G. Li, H. Zheng, R. Bai, Macromol. Rapid Commun. 2009, 30, 442.
| Crossref | GoogleScholarGoogle Scholar |
[16] S. Şenel, I.-Y. Belma, O. Güven, Turk. J. Chem. 1996, 20, 62.
[17] I. Van Nieuwenhove, S. Maji, M. Dash, S. Van Vlierberghe, R. Hoogenboom, P. Dubruel, Polym. Chem. 2017, 8, 2433.
| Crossref | GoogleScholarGoogle Scholar |
[18] A. Guinaudeau, O. Coutelier, A. l. Sandeau, S. p. Mazières, H. D. Nguyen Thi, V. Le Drogo, D. J. Wilson, M. Destarac, Macromolecules 2014, 47, 41.
| Crossref | GoogleScholarGoogle Scholar |
[19] K. Kempe, P. A. De Jongh, A. Anastasaki, P. Wilson, D. M. Haddleton, Chem. Commun. 2015, 51, 16213.
| Crossref | GoogleScholarGoogle Scholar |
[20] (a) P. A. de Jongh, A. Mortiboy, G. S. Sulley, M. R. Bennett, A. Anastasaki, P. Wilson, D. M. Haddleton, K. Kempe, ACS Macro Lett. 2016, 5, 321.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. M. Mahmoud, A. Rajakanthan, K. Kempe, Polym. Chem. 2018, 9, 1562.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. A. de Jongh, M. R. Bennett, G. S. Sulley, P. Wilson, T. P. Davis, D. M. Haddleton, K. Kempe, Polym. Chem. 2016, 7, 6703.
| Crossref | GoogleScholarGoogle Scholar |
[21] L. Martin, G. Gody, S. Perrier, in Reversible Deactivation Radical Polymerization: Materials and Applications (Eds K. Matyjaszewski, H. Gao, B. S. Sumerlin, N. V. Tsarevsky) 2018, pp. 57–79 (American Chemical Society: Washington, D.C.).
[22] O. Deane, J. Lovett, O. Musa, A. Fernyhough, S. Armes, Macromolecules 2018, 51, 7756.
| Crossref | GoogleScholarGoogle Scholar | 30333669PubMed |
[23] B. Zhang, X. Wang, A. Zhu, K. Ma, Y. Lv, X. Wang, Z. An, Macromolecules 2015, 48, 7792.
| Crossref | GoogleScholarGoogle Scholar |
[24] S. Kobayashi, A. Makino, Chem. Rev. 2009, 109, 5288.
| Crossref | GoogleScholarGoogle Scholar | 19824647PubMed |
[25] A. P. Danielson, D. B. Van Kuren, M. E. Lucius, K. Makaroff, C. Williams, R. C. Page, J. A. Berberich, D. Konkolewicz, Macromol. Rapid Commun. 2016, 37, 362.
| Crossref | GoogleScholarGoogle Scholar | 26748786PubMed |
[26] A. Danielson, D. Van-Kuren, J. Bornstein, C. Kozuszek, J. Berberich, R. Page, D. Konkolewicz, Polymers 2018, 10, 741.
| Crossref | GoogleScholarGoogle Scholar |
[27] (a) R. Chapman, A. J. Gormley, K.-L. Herpoldt, M. M. Stevens, Macromolecules 2014, 47, 8541.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. Chapman, A. J. Gormley, M. H. Stenzel, M. M. Stevens, Angew. Chem. 2016, 55, 4500.
| Crossref | GoogleScholarGoogle Scholar |
[28] Y. Lv, Z. Liu, A. Zhu, Z. An, J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 164.
| Crossref | GoogleScholarGoogle Scholar |
[29] L. Brocken, P. D. Price, J. Whittaker, I. R. Baxendale, React. Chem. Eng. 2017, 2, 662.
| Crossref | GoogleScholarGoogle Scholar |
[30] (a) A. Reyhani, T. G. McKenzie, H. Ranji‐Burachaloo, Q. Fu, G. G. Qiao, Chem. – Eur. J. 2017, 23, 7221.
| Crossref | GoogleScholarGoogle Scholar | 28382790PubMed |
(b) M. D. Nothling, T. G. McKenzie, A. Reyhani, G. G. Qiao, Macromol. Rapid Commun. 2018, 39, 1800179.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Reyhani, M. N. Nothling, H. Ranji-Burachaloo, T. G. McKenzie, Q. Fu, S. Tan, G. Bryant, G. G. Qiao, Angew. Chem. Int. Ed. 2018, 57, 10288.
| Crossref | GoogleScholarGoogle Scholar |
(d) T. G. McKenzie, A. Reyhani, M. D. Nothling, G. G. Qiao, in Reversible Deactivation Radical Polymerization: Mechanisms and Synthetic Methodologies (Eds K. Matyjaszewski, H. Gao, B. S. Sumerlin, N. V. Tsarevsky) 2018, pp. 307–321 (American Chemical Society: Washington, D.C.).
(e) A. Reyhani, S. Allison-Logan, H. Ranji-Burachaloo, T. G. McKenzie, G. Bryant, G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem. 2019, in press
[31] D. Roy, A. Ullah, B. S. Sumerlin, Macromolecules 2009, 42, 7701.
| Crossref | GoogleScholarGoogle Scholar |
[32] Y. Shi, H. Gao, L. Lu, Y. Cai, Chem. Commun. 2009, 11, 1368.
| Crossref | GoogleScholarGoogle Scholar |
[33] G. Gody, R. Barbey, M. Danial, S. Perrier, Polym. Chem. 2015, 6, 1502.
| Crossref | GoogleScholarGoogle Scholar |
[34] (a) T. Junkers, B. Wenn, React. Chem. Eng. 2016, 1, 60.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. Wenn, T. Junkers, Macromolecules 2016, 49, 6888.
| Crossref | GoogleScholarGoogle Scholar |
[35] A. Reyhani, H. Ranji-Burachaloo, T. G. McKenzie, Q. Fu, G. G. Qiao, Macromolecules 2019, 52, 3278.
| Crossref | GoogleScholarGoogle Scholar |
[36] (a) Q. Zhang, Z. Zhang, W. Wang, J. Zhu, Z. Cheng, N. Zhou, W. Zhang, X. Zhu, J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 1424.
| Crossref | GoogleScholarGoogle Scholar |
(b) N. Haridharan, R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 1021.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Kwak, K. Matyjaszewski, Macromolecules 2008, 41, 6627.
| Crossref | GoogleScholarGoogle Scholar |
(d) Y. Kwak, R. Nicolaÿ, K. Matyjaszewski, Macromolecules 2008, 41, 6602.
| Crossref | GoogleScholarGoogle Scholar |
(e) N. Haridharan, K. Ponnusamy, R. Dhamodharan, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 5329.
| Crossref | GoogleScholarGoogle Scholar |
(f) S. Harihara Subramanian, R. Prakash Babu, R. Dhamodharan, Macromolecules 2008, 41, 262.
| Crossref | GoogleScholarGoogle Scholar |
(g) Z. Zhang, W. Wang, H. Xia, J. Zhu, W. Zhang, X. Zhu, Macromolecules 2009, 42, 7360.
| Crossref | GoogleScholarGoogle Scholar |
(h) R. Nicolaÿ, Y. Kwak, K. Matyjaszewski, Macromolecules 2008, 41, 4585.
| Crossref | GoogleScholarGoogle Scholar |
[37] R. Nicolay, Y. Kwak, K. Matyjaszewski, Angew. Chem. Int. Ed. 2010, 49, 541.
| Crossref | GoogleScholarGoogle Scholar |
[38] Y. Gu, J. Zhao, Q. Liu, X. Pan, W. Zhang, Z. Zhang, X. Zhu, Polym. Chem. 2015, 6, 359.
| Crossref | GoogleScholarGoogle Scholar |
[39] Z. Zhang, W. Wang, Z. Cheng, J. Zhu, N. Zhou, Y. Yang, Y. Tu, X. Zhu, Macromolecules 2010, 43, 7979.
| Crossref | GoogleScholarGoogle Scholar |
[40] Y. Zhang, K. Schröder, Y. Kwak, P. Krys, A. N. Morin, T. Pintauer, R. Poli, K. Matyjaszewski, Macromolecules 2013, 46, 5512.
| Crossref | GoogleScholarGoogle Scholar |
[41] H. Jiang, L. Zhang, X. Jiang, X. Bao, Z. Cheng, X. Zhu, Macromol. Rapid Commun. 2014, 35, 1332.
| Crossref | GoogleScholarGoogle Scholar | 24943002PubMed |
[42] J. Kreutzer, Y. Yagci, Polymers 2017, 10, 35.
| Crossref | GoogleScholarGoogle Scholar |
[43] P. Maximiano, P. V. Mendonça, J. R. Costa, N. L. Haworth, A. C. Serra, T. Guliashvili, M. L. Coote, J. F. Coelho, Macromolecules 2016, 49, 1597.
| Crossref | GoogleScholarGoogle Scholar |
[44] J. Yeow, R. Chapman, A. J. Gormley, C. Boyer, Chem. Soc. Rev. 2018, 47, 4357.
| Crossref | GoogleScholarGoogle Scholar | 29718038PubMed |
[45] (a) G. Ng, J. Yeow, R. Chapman, N. Isahak, E. Wolvetang, J. J. Cooper-White, C. Boyer, Macromolecules 2018, 51, 7600.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. J. Gormley, J. Yeow, G. Ng, Ó. Conway, C. Boyer, R. Chapman, Angew. Chem. Int. Ed. 2018, 57, 1557.
| Crossref | GoogleScholarGoogle Scholar |
[46] S. Shanmugam, J. Xu, C. Boyer, Macromol. Rapid Commun. 2017, 38, 1700143.
| Crossref | GoogleScholarGoogle Scholar | 28556363PubMed |
[47] (a) V. Kottisch, Q. Michaudel, B. P. Fors, J. Am. Chem. Soc. 2016, 138, 15535.
| Crossref | GoogleScholarGoogle Scholar | 27934022PubMed |
(b) V. Kottisch, Q. Michaudel, B. P. Fors, J. Am. Chem. Soc. 2017, 139, 10665.
| Crossref | GoogleScholarGoogle Scholar |
[48] B. M. Peterson, S. Lin, B. P. Fors, J. Am. Chem. Soc. 2018, 140, 2076.
| Crossref | GoogleScholarGoogle Scholar | 29385348PubMed |