

Elimination of Ethene from 1,2-Diiodoethane Induced by N-Heterocyclic Carbene Halogen Bonding*
Tiffany B. Poynder A B , Dharmeshkumar P. Savaliya A B , Andrew Molino A B , David J. D. Wilson A C and Jason L. Dutton A CA Department of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Vic. 3086, Australia.
B These authors contributed equally to this work.
C Corresponding authors. Email: david.wilson@latrobe.edu.au; j.dutton@latrobe.edu.au
Australian Journal of Chemistry 72(8) 614-619 https://doi.org/10.1071/CH19237
Submitted: 27 May 2019 Accepted: 27 June 2019 Published: 24 July 2019
Abstract
The attempted synthesis of N-heterocyclic carbene (NHC)-stabilised dicarbon (C2) fragments via nucleophilic substitution at 1,2-diiodoethane is reported. Rather than the expected SN2 pathway, clean elimination of ethene and formation of an iodoimidazolium cation was observed. The resistance towards nucleophilic substitution piqued interest, and subsequent investigation determined NHC-halogen bonding as the source. This is in contrast to reactions between NHCs and other alkyl halides, where substitution or elimination pathways are reported. A detailed theoretical study between these cases highlights the importance of iodine as a halogen bond donor compared with other halogens, and shows that NHCs are excellent halogen bond acceptors. This reactivity suggests potential for application of the halogen bonding interaction between NHCs and organic compounds.
Introduction
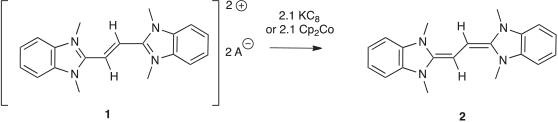
The isolation of molecules with C2 fragments in a zero oxidation state is a topic of current interest.[1–3] Our laboratory is particularly interested in the coordination chemistry of carbene ligands with C2 fragments.[4–6] Recently, we discovered that reduction of dication 1 generated a bis-N-heterocyclic olefin 2 (Scheme 1),[7] which demonstrated unique reactivity, such as the first example of atmospheric O2 cleaving a C=C bond.[8] It is of interest to extend the chemistry of this system, however, reported methods to access the starting compounds are limited to benzimidazolium fused N-heterocyclic carbene (NHC) derivatives, as the syntheses start with 1,2-phenylenediamine to generate benzimidazoles via two subsequent condensations with maleic anhydride.[9] The reported method is also a rather onerous synthesis, with the second condensation using polyphosphoric acid as a solvent at high temperatures.
|
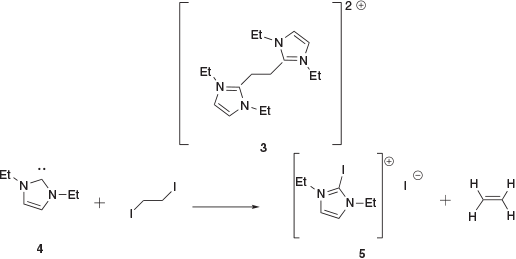
To find a more efficient route, while also expanding the classes of NHCs that could potentially be used, we intended to access NHC-stabilised C2 compounds via direct reaction of the free carbene with a suitable C2 precursor. As an initial attempt we intended to form dication 3 via an SN2 reaction between Et2NHC (the nucleophile) and 1,2-diiodoethane (Scheme 2). Dication 3 could then simply be deprotonated, furnishing a derivative of 2 in only two steps. However, rather than the desired SN2 reactivity, we discovered that the NHC reacts cleanly with the alkyl halide to eliminate ethene.
|
Here we report a combined synthetic and theoretical investigation of the unexpected reactivity of NHCs with alkyl halides, in order to characterise and assess the wider applicability of this new reactivity. In particular, reactivity based on a consideration of halogen bonding offers the potential for new perspectives in the interaction of NHCs with organic compounds.
Results and Discussion
The reaction of NHC 4 (generated in situ from the imidizaolium and potassium bis(trimethylsilyl)amide (KHMDS)) and 1,2-diiodoethane in C6D6 solvent in a 1 : 1 stoichiometric ratio at room temperature resulted in the immediate visible evolution of a gas and precipitation of an orange solid. 1H and 13C NMR studies of the in situ reaction mixture revealed the presence of a signal at 5.26 ppm in the 1H NMR spectrum. This was consistent with the presence of ethylene, which was confirmed in the 13C NMR spectrum by a resonance at 123 ppm. A sample of the solid that precipitated from the reaction was dissolved in CDCl3, for which the 1H NMR spectrum showed a single NHC-containing compound, missing the imidazolium proton. The ESI mass spectrum gave signals at m/z 251 and 127.1 in positive and negative ion mode, respectively, consistent with the iodoimidazolium cation being present as an iodide salt (5). The isolated yield of the salt was 87 %, indicating the reaction primarily generates 3 and ethylene. The products, while not the goal of the reaction, caught our attention as it was not the expected result of a reaction between these two species. In effect, the product requires a reduction of the 1,2-diiodoethane and oxidation of the NHC.
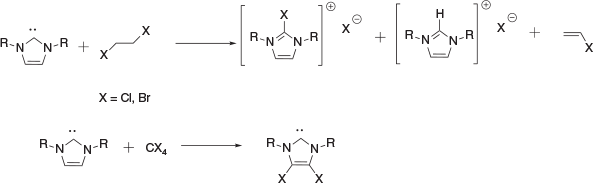
Denk and co-workers have described the reduction of haloalkanes with folates and imidazolidine derivatives,[10,11] but not with free carbenes. Jones and Junk have reported reactions between NHC and 1,2-dibromoethane to give a mixture of protonated NHC, bromoimidazolium cations, and vinyl bromide as the primary fate of the bromoethane.[12] Reactions of NHCs with 1,2-dichloroethane reported by Kuhn gives chloroimidazolium;[13] however, Jones and Junk indicate in their study that the reaction gives a substantial fraction of protonated carbene and vinyl chloride from an elimination pathway as well (Scheme 3).
|
Early reports from Arduengo shortly after the isolation of the first N-heterocyclic carbene involving C1 perchloroalkanes indicate that the reactions with NHCs primarily result in halogenation of the NHC backbone,[14] with the same observed by Jones and Junk in reactions of NHC with CBr4.[12]
The elimination of ethene and intermediacy of [Ph3P–I]+ has been recently reported in a version of the Appel reaction between triphenylphosphine and 1,2-diiodoethane at elevated temperatures (60–100°C, 2 h), giving alkyl iodides from alcohols. Direct attack on the I by PPh3 was hypothesised but not analysed in detail, nor was the [Ph3P–I]+ intermediate observed or isolated.[15]
In our system, nucleophilic substitution reactions should give a product with NHC bound to the C2 fragment, whereas elimination pathways should generate protonated carbene as the main NHC-containing product together with vinyl iodide. In light of the report from Jones and Junk discussed above, the exclusive and clean formation of 3 and ethylene thus merits attention. We hypothesised that the mechanism might be direct attack by the NHC onto an iodine atom via a halogen bonding mechanism. Halogen bonding is a topic of increasing interest, primarily for supramolecular applications, but also in organic synthesis.[16] NHCs are considered good halogen bond acceptors,[17] however, a transformation of this type induced by halogen bonding with NHCs has not been proposed before.
The energy profiles of two possible pathways, one involving direct interaction with iodine and the other a standard SN2 attack on the carbon atom were modelled at the B3LYP-D3(BJ)/def2-TZVP (SMD,MeCN)//DSDPBEP86/def2-TZVPP (SMD,MeCN) level of theory, using an N-Me substituted NHC as a model system. The reaction energy profile is plotted in Fig. 1, including the expected SN2 reaction, direct attack of the NHC on iodine via a halogen-bonding mechanism, and a reaction between Me2NHC and 1,2-diiodoethane.

|
In the typical SN2 pathway, the initial transition state barrier is calculated to be 110.4 kJ mol−1, with the products 206.5 kJ mol−1 below that of the reactants. To reach the final observed products, attack on the carbenic carbon by iodide is required together with rupture of the NHC C–C bond to yield the ethylene and iodoimidazolium products. This second step is calculated to be endergonic by ~100 kJ mol−1. This suggests the reaction should stop after formation of the cation arising from displacement of iodide if one equivalent of NHC is used and the mechanism is of a classic SN2 reaction.
Deprotonation and elimination for 1,2-diiodoethane gives a similarly high barrier at 106 kJ mol−1. However, direct attack of the NHC on the iodine atom, breaking the C–I bonds and forming ethene in a single step, has a much lower transition state barrier of 65.5 kJ mol−1, leading directly to the observed products, and consistent with the rapid room temperature reaction. The requirement for elevated temperatures in the analogous reaction with PPh3 and 1,2-diiodoethane[15] suggests that NHCs have a superior propensity for halogen bonding to alkyl iodides than do phosphines.
An examination of the lowest unoccupied molecular orbital (LUMO) of 1,2-diiodoethane is consistent with the proposed halogen bond induced pathway (Fig. 2). The LUMO is of σ antibonding character with respect to the I–C bonds, but π bonding with respect to the C–C bond. Population of the LUMO on 1,2-diiodoethane by the lone pair of electrons from the electron-donating NHC along the I–C bond axis would then promote the observed transformation involving rupture of the I–C bonds, formation of the C=C bond, and release of ethene.

|
Examination of the analogous reaction for 1,2-dibromoethane and NHC (Fig. 3) yields a barrier for halogen bonding attack on the bromine atom that is much higher at 123 kJ mol−1. That is consistent with a C–Br bond being a worse halogen bond acceptor than a C–I bond. The calculated energy of the LUMO for 1,2-dibromoethane is 0.4 eV higher in energy than the LUMO for 1,2-diiodoethane and is thus less accessible for halogen bonding. For 1,2-dibromoethane the pathway with the lowest barrier is the elimination reaction (113 kJ mol−1), consistent with Jones’ observations.[12] The barrier for direct attack on the bromine is very similar in energy (123 kJ mol−1), accounting for the observation of bromoimidazolium as a competing product in Jones’ work.

|
Pyridine derivatives have been reported to undergo SN2 reactions with 1,2-haloalkanes, rather than elimination or the halogen bonding based attack observed for phosphine/NHC.[18] Modelling of this process using 4-DMAP (4-dimethylaminopyridine) as the pyridine indicates that the transition state barrier for the SN2 is significantly lowered in comparison with NHCs, with a barrier of 73 kJ mol−1 for 4-DMAP (Fig. 4). For the pathway associated with halogen bonding attack on iodine, a transition state was not able to be located, however the overall reaction is thermodynamically unfavourable. The inability to locate a transition state may also indicate that pyridines are less prone to engaging in halogen bonding interactions than NHCs. The lack of experimental observation of elimination/deprotonation as a pathway can be rationalised by the lowered basicity of pyridines as compared with NHCs.

|
Conclusions
The attempted synthesis of NHC-stabilised dicarbon fragments via nucleophilic substitution at 1,2-diiodoethane resulted in a clean reaction via an NHC–halogen bonding interaction, eliminating ethene. The resistance to the substitution pathway inspired an in-depth theoretical study comparing the reaction pathways between other nucleophiles and alkyl halides. The results highlight the importance of the halogen bonding capacity between NHCs and iodine compared with other systems, suggesting potential applications for halogen bonding interactions between NHCs and organic compounds.
Experimental
1,3-Diethylimidazolium iodide was synthesised according to the literature procedure.[19] Reactions were carried out in a glovebox under an N2 atmosphere. Solvents were dried over CaH2, distilled, and stored over 3 Å molecular sieves in the glovebox.
1,3-Diethylimidazolium iodide (91 mg, 0.36 mmol) was suspended in 2 mL of C6D6 with stirring. KHMDS (76 mg, 0.38 mmol) was added in one portion to the solution and was left to stir. After 1 h, the reaction mixture was centrifuged and 1,3-diethylimidazolylidene 4 was collected in the supernatant.
1,2-Diiodoethane (102 mg, 0.36 mmol) was added to a solution of 1,3-diethylimidazolylidene (45 mg, 0.36 mmol) in C6D6 with stirring. There was an immediate precipitation of an orange solid and the evolution of a gas was observed. The reaction was left to stir for 5 min. The solid was filtered, washed with diethyl ether (3 × 10 mL), and dried under vacuum giving 5 (119 mg, 87 %). δH (CDCl3, 400 MHz) 7.34 (s, 2 H), 4.28 (q, 4 H), 1.54 (t, 6 H). m/z (ESI) 251.0 [M]+, 127.1 [M]−.
All calculations were performed using Gaussian 16 revision A.03 unless noted.[20] Geometry optimisations were performed using the B3LYP[21] density functional with the def2-TZVP basis set,[22] inclusive of solvent effects using the polarisable continuum model (IEF-PCM) with Truhlar’s SMD model with parameters for acetonitrile (ϵ = 35.688).[23] Grimme’s D3 dispersion with Becke–Johnson damping was included, labelled D3(BJ).[24] Harmonic vibrational frequencies were computed analytically at the same level of theory in order to characterise the stationary points as minima, representing equilibrium structures, or transition states (TSs). For all TSs, intrinsic reaction coordinate (IRC) calculations were carried out to ensure connectivity between the local minima along the reaction path. Vibrational frequencies were also utilised to determine corresponding thermochemical data (within the harmonic limit and determined at 1 atm and 298 K). In order to provide more accurate energetics, single-point calculations at the optimised geometries were carried out at the DSDPBEP86[25] level of theory with the def2-TZVPP basis set, inclusive of solvent effects using the polarisable continuum model (IEF-PCM) with Truhlar’s SMD model with parameters for acetonitrile (ϵ = 35.688). Reported free energies were determined by adding the thermal corrections determined at the B3LYP-D3(BJ)/def2-TZVP level of theory to the DSDPBEP86/def2-TZVPP solvent-corrected single point energies, which is labelled DSDPBEP86/def2-TZVPP//B3LYP-D3(BJ)/def2-TZVP (SMD, MeCN).
Supplementary Material
NMR and mass spectra as well as calculated Cartesian coordinates are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank The La Trobe Institute for Molecular Science for their generous funding of this project. This work was also supported by an ARC Future Fellowship (JLD, FT16010007). Generous allocation of computing resources from National Computational Infrastructure (NCI), Intersect, and La Trobe University are acknowledged.
References
[1] L. Jin, M. Melaimi, L. Liu, G. Bertrand, Org. Chem. Front. 2014, 1, 351.| Crossref | GoogleScholarGoogle Scholar |
[2] Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley, H. W. Roesky, Angew. Chem. Int. Ed. 2014, 53, 4168.
| Crossref | GoogleScholarGoogle Scholar |
[3] D. Wu, Y. Li, R. Ganguly, R. Kinjo, Chem. Commun. 2014, 50, 12378.
| Crossref | GoogleScholarGoogle Scholar |
[4] D. C. Georgiou, B. D. Stringer, N. Holzmann, C. F. Hogan, P. J. Barnard, D. J. D. Wilson, G. Frenking, J. L. Dutton, Chem. – Eur. J. 2015, 21, 3377.
| Crossref | GoogleScholarGoogle Scholar | 25588368PubMed |
[5] J. L. Dutton, D. J. D. Wilson, Angew. Chem. Int. Ed. 2012, 51, 1477.
| Crossref | GoogleScholarGoogle Scholar |
[6] V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt, S. Inoue, Chem. Rev. 2018, 118, 9678.
| Crossref | GoogleScholarGoogle Scholar | 29969239PubMed |
[7] M. M. D. Roy, E. Rivard, Acc. Chem. Res. 2017, 50, 2017.
| Crossref | GoogleScholarGoogle Scholar |
[8] D. C. Georgiou, L. Zhao, D. J. D. Wilson, G. Frenking, J. L. Dutton, Chem. – Eur. J. 2017, 23, 2926.
| Crossref | GoogleScholarGoogle Scholar | 27935139PubMed |
[9] D. Bhabagrahi, E. K. Dora, C. S. Panda, Indian J. Chem. Sect. B 1982, 21, 697.
[10] M. K. Denk, N. S. Milutinovic, K. M. Marczenko, N. M. Sadowski, A. Paschos, Chem. Sci. 2017, 8, 1883.
| Crossref | GoogleScholarGoogle Scholar | 28553478PubMed |
[11] M. K. Denk, N. Milutinovic, K. Marczenko, U. S. Patent, Ed. 2017.
[12] M. L. Cole, C. Jones, P. C. Junk, New J. Chem. 2002, 26, 1296.
| Crossref | GoogleScholarGoogle Scholar |
[13] N. Kuhn, J. Fahl, R. Fawzi, C. Maichle-Mossmer, M. Steimann, Z. Naturforsch. B 1998, 53, 720.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. J. I. Arduengo, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall, T. K. Prakasha, J. Am. Chem. Soc. 1997, 119, 12742.
| Crossref | GoogleScholarGoogle Scholar |
[15] J. Chen, J. Lin, J. Xiao, Org. Lett. 2018, 20, 3061.
| Crossref | GoogleScholarGoogle Scholar | 29741387PubMed |
[16] D. Bulfield, S. M. Huber, Chem. – Eur. J. 2016, 22, 14434.
| Crossref | GoogleScholarGoogle Scholar | 27465662PubMed |
[17] B. Pinter, N. Nagels, W. A. Herrebout, F. De Proft, Chem. – Eur. J. 2013, 19, 519.
| Crossref | GoogleScholarGoogle Scholar | 23169478PubMed |
[18] H. Kubota, Y. Takahashi, J. Harada, T. Inabe, Cryst. Growth Des. 2014, 14, 5575.
| Crossref | GoogleScholarGoogle Scholar |
[19] C. Schmidt, B. Karge, R. Misgeld, A. Prokop, R. Franke, M. Bronstrup, I. Ott, Chem. – Eur. J. 2017, 23, 1869.
| Crossref | GoogleScholarGoogle Scholar | 27865002PubMed |
[20] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian 16, Revision A.03 2016 (Gaussian, Inc.: Wallingford, CT).
[21] A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
| Crossref | GoogleScholarGoogle Scholar |
[22] F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 2005, 7, 3297.
| Crossref | GoogleScholarGoogle Scholar | 16240044PubMed |
[23] A. V. Marenich, C. J. Cramer, D. G. Truhlar, J. Phys. Chem. B 2009, 113, 6378.
| Crossref | GoogleScholarGoogle Scholar | 19366259PubMed |
[24] S. Grimme, S. Ehrlich, L. Goerigk, J. Comput. Chem. 2011, 32, 1456.
| Crossref | GoogleScholarGoogle Scholar | 21370243PubMed |
[25] S. Kozuch, J. M. L. Martin, J. Comput. Chem. 2013, 34, 2327.
| 23983204PubMed |
* Jason Dutton is the winner of the 2018 RACI Organometallic Award.