

Generation and Rearrangement of (1-Hydroxycyclopropyl)- and (1-Hydroxycyclobutyl)carbene
Joseph D. DeAngelo A , Sayaka Hatano B C and Dasan M. Thamattoor A C
A , Sayaka Hatano B C and Dasan M. Thamattoor A C
A Department of Chemistry, Colby College, Waterville, ME 04901, USA.
B Department of Chemistry, Graduate School of Science, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, Hiroshima 739-8526, Japan.
C Corresponding authors. Email: sa-hatano@hiroshima-u.ac.jp; dmthamat@colby.edu
Australian Journal of Chemistry 72(11) 890-893 https://doi.org/10.1071/CH19379
Submitted: 5 August 2019 Accepted: 11 September 2019 Published: 10 October 2019
Abstract
Photolysis of exo-1-(1a,9b-dihydro-1H-cyclopropa[l]phenanthren-1-yl)cyclopropan-1-ol and exo-1-(1a,9b-dihydro-1H-cyclopropa[l]phenanthren-1-yl)cyclobutan-1-ol in benzene-d6 produces (1-hydroxycyclopropyl)- and (1-hydroxycyclobutyl)carbene respectively. It was observed that (1-hydroxycyclopropyl)carbene rearranges to cyclobutanone whereas (1-hydroxycyclobutyl)carbene forms cyclopentanone. Formation of both ketones is attributed to tautomerization of the corresponding enols that arise from ring expansion of the carbenes. Products assignable to intramolecular C–H insertions were not detected in the photolysates.
Introduction
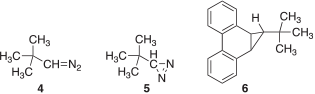
The rearrangement of tert-butylcarbene (1), generated by the thermal decomposition of various precursors, gives 1,1-dimethylcylopropane (2) as the overwhelmingly major product, with only minor amounts of 2-methyl-2-butene (3) produced (Eqn 1).[1] These observations suggest a marked preference for 1 to undergo insertion into the γ C–H bond rather than a 1,2-methyl migration. However, the photolysis of the diazo compound 4 and diazirine 5 produces 2 and 3 in nearly equal amounts.[1f] These peculiar results were attributed to the incursion of excited-state chemistry of the nitrogenous carbene sources used in the study.[1i,2] In subsequent work, when 1 was generated by photolyzing the non-nitrogenous precursor 6 at room temperature, compounds 2 and 3 were formed in a 9 : 1 ratio, which is comparable with the results obtained from the thermal experiments (Chart 1).[1i] It is also of interest to note that the generation of 1 at −78°C has been reported to give 2 as the sole rearrangement product.[1h,1i]
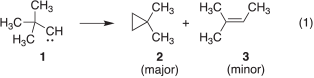
|
|
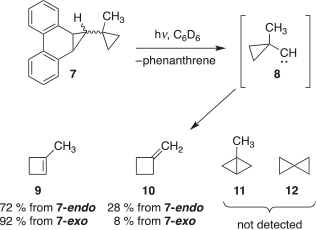
In related work, our laboratory has reported that photolysis of isomeric (endo and exo) phenanthrene-based precursors 7 produces carbene 8, which may be thought of as a cyclic version of 1 with two of its methyl groups ‘tied back’ into a cyclopropyl ring.[3] As shown in Scheme 1, 8 undergoes ring expansion to give 1-methylcyclobutene (9). Minor amounts of methylenecylobutane (10) were also found in the photolysate, presumably from the isomerization of 9. Notably, the intramolecular C–H insertion products 1-methylbicyclo[1.1.0]butane (11) and spiropentane (12) were not observed. In this case, perhaps strain issues have diverted the rearrangement of 8 to favour carbon migration over C–H insertion.
|
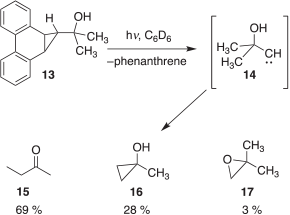
We have also reported that the solution photolysis of precursor 13 (Scheme 2) generates 2-hydroxy-2-methylpropylidene (14).[4] Carbene 14 is formally related to 1 by replacement of one of the methyl groups in the latter by a hydroxy substituent. In sharp contrast to 1, however, 14 is more inclined to undergo a 1,2-methyl shift to produce 2-butanone (15), via the corresponding enol, as the major product. Insertion into a C–H bond to form 1-methylcyclopropanol (16) and the O–H bond to form 2,2-dimethyloxirane (17) also occurs but these are now minor products.
|
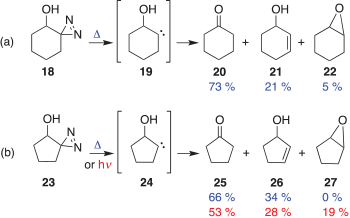
The behaviour of 14 was rationalized in terms of the bystander effect of the β-hydroxy group, which facilitates the migration of the methyl group. Interestingly, when diazirine 18 was thermally decomposed, the putative carbene 19 only gave products arising from hydrogen insertions such as 20, 21, and 22 (Scheme 3a).[5] Likewise the related diazirine 23, a source of carbene 24, only gave the hydrogen insertion products 25, 26, and 27 (Scheme 3b).[6] No ring-contraction product, indicating a carbon shift, was observed for either 19 or 24.
|
In the present work, we report our results on the behaviour of (1-hydroxycyclopropyl)carbene (28) and (1-hydroxycyclobutyl)carbene (29) in both of which the divalent carbon is exocylic to the ring bearing the β-hydroxy group (Fig. 1).

|
Results and Discussion
Synthesis of Precursors
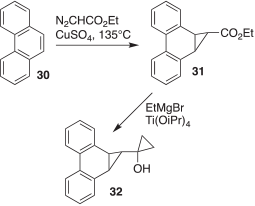
The synthesis of compound 32, the desired photochemical source of carbene 28, was achieved in two straightforward steps, as depicted in Scheme 4. In the first step, ethyl diazoacetate was added dropwise to molten phenanthrene (30) containing catalytic amounts of CuSO4 to obtain the cyclopropyl ester 31.[7] Then, the Kulinkovich reaction of 31 with ethylmagnesium bromide, in the presence of catalytic titanium isopropoxide, delivered 32.[8]
|
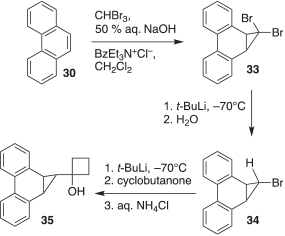
Compound 35, the targeted precursor to carbene 29, was obtained in three steps as shown in Scheme 5. The first step involved a phase-transfer-catalyzed addition of dibromocarbene to 30 to afford 33 following our previously reported procedure.[9] Subsequent treatment of 33 with t-BuLi followed by quenching with water provided the monobromo derivative 34.[10] Finally, 34 was reacted with t-BuLi followed by the sequential addition of cyclobutanone and aqueous ammonium chloride to obtain 35.
|
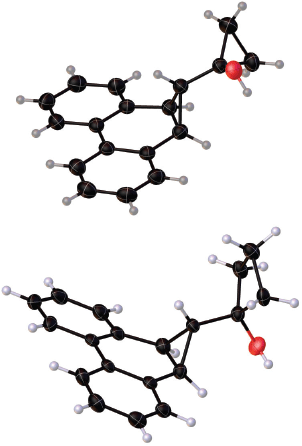
Crystals of precursors 32 and 35 suitable for X-ray diffraction experiments were grown and their solid-state structures determined.[11] These are shown in Fig. 2.
|
Photolysis Experiments
A solution of precursor 32, dissolved in benzene-d6, was taken up in a glass NMR tube. The tube was placed in a Rayonet reactor and photolyzed (~315–400 nm) at ambient temperature. Progress of the photolysis was monitored periodically by 1H NMR spectroscopy until the starting material was consumed. Analysis of the photolysate indicated a clean reaction that produced phenanthrene (30) and cyclobutanone (37) as the only product isomeric with carbene 28 (Scheme 6). The most likely mechanism for the formation of 37 is the ring expansion of 28 to the enol 36, which subsequently undergoes tautomerization. Notably, neither the O–H insertion product 38 nor the C–H insertion product 39 were observed in the reaction mixture, indicating that these pathways are not competitive with the alkyl shift.
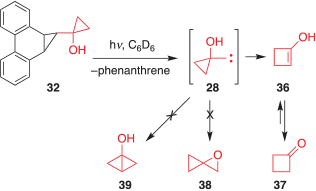
|
The photolysis of 35 was performed in an analogous manner and provided similar results, which are summarized in Scheme 7. Only phenanthrene (30) and cyclopentanone (41) were observed in the photolysate. The O–H and C–H insertion products, 42 and 43 respectively, were not detected.
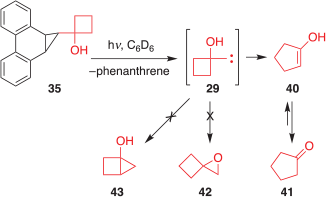
|
Thus, it is evident that carbene 29, like 28, shows an overwhelming preference for ring expansion to the enol 40, which then tautomerizes to 41.[12]
Conclusions
Two cyclopropanated phenanthrene derivatives, 32 and 35, were synthesized and photolyzed in solution to produce β-hydroxycarbenes 28 and 29 respectively. Like the parent cyclopropylcarbene,[13] and its 1-methyl derivative 8,[3] carbene 28 shows a strong preference to ring-expand, as evident from the formation of cyclobutanone (37) via the enol 36. No C–H or O–H insertion products were observed in the photolysate. Increasing the ring size in 28, as in 29, has no apparent effect in the overall rearrangement chemistry. Carbene 29 also undergoes ring expansion as the exclusive rearrangement pathway and no products attributable to C–H or O–H insertions were found post photolysis.
Experimental
General Remarks
Tetrahydrofuran was dried by passage through two columns (60 × 10 cm) of activated alumina. All other solvents and reagents were used as obtained from commercial sources. All reactions were carried out under an argon atmosphere in oven-dried glassware. Flash chromatography was performed on prepacked silica gel columns (70–230 mesh). NMR spectra were recorded at 500 MHz for 1H and 125 MHz for 13C in CDCl3 or C6D6. The shifts (δ) are reported in parts per million and referenced to either tetramethylsilane (TMS) or the residual proton signal from the solvent. Fourier-transform (FT)-IR data were acquired on neat solids using an attenuated total reflectance (ATR) accessory. Gas chromatography–mass spectrometry (GC/MS) data were obtained with a capillary gas chromatograph interfaced with a quadrupole triple-axis mass-selective detector operating in electron impact (EI) mode. Single-crystal X-ray diffraction data were collected at 173 K on a diffractometer employing graphite-monochromated Mo Kα radiation (λ 0.71073 Å), and equipped with a complementary metal oxide semiconductor (CMOS) or charge-coupled device (CCD) detector.
Synthesis of exo-1-(1a,9b-Dihydro-1H-cyclopropa[l]phenanthren-1-yl)cyclopropan-1-ol (32)
Ethylmagnesium bromide (3.0 mL, 3.0 M in Et2O, 9 mmol) was added dropwise by syringe to a magnetically stirred solution of the exo ester 31[7] (1.06 g, 4.0 mmol) and titanium tetraisopropoxide (237 μL, 0.8 mmol, 20 mol-%) in anhydrous THF (20 mL). The temperature was maintained between 22 and 30°C with a water bath during addition. Stirring was continued at room temperature for an additional 30 min after the completion of addition. The dark green reaction mixture was then cooled to −5°C and quenched by the dropwise addition of 10 % aqueous sulfuric acid (40 mL), producing a white precipitate. During the addition, the temperature was maintained between −10 and 0°C. The reaction mixture was stirred at 0°C for an additional 1 h, maintaining temperature with an ice bath. After warming to room temperature, the organic and aqueous layers were separated, and the latter was extracted with diethyl ether (2 × 20 mL). The organic layers were combined, washed with saturated sodium bicarbonate (2 × 20 mL), and freed of solvent at the rotary evaporator. The final product, a white solid, was recrystallized from hexanes and ethyl acetate. Yield: 63 %. δH (500 MHz, CDCl3) 8.01–7.95 (m, 2H), 7.40 (dd, J 5.0, 2.2, 2H), 7.31–7.22 (m, 4H), 2.50 (d, J 4.4, 2H), 1.99 (s, 1H), 0.92–0.86 (m, 2H), 0.77–0.72 (m, 1H), 0.67–0.60 (m, 2H). δC (126 MHz, CDCl3) 134.9, 129.4, 128.8, 127.7, 126.1, 123.1, 56.0, 30.9, 24.5, 12.7.
Synthesis of exo-1-(1a,9b-Dihydro-1H-cyclopropa[l]phenanthren-1-yl)cyclobutan-1-ol (35)
The monobromo derivative 34 (2.71 g, 10 mmol) was dissolved in 100 mL dry THF and cooled to −70°C. Then, t-butyllithium (1.7 M in pentane, 13 mL, 22 mmol) was added dropwise over 10 min, keeping the temperature below −65°C. The resulting dark green reaction mixture was left stirring at −70°C for 1 h. Cyclobutanone (0.9 mL, 13.7 mmol) was added to the reaction mixture, which was left stirring for an additional 45 min at −70°C. The solution slowly warmed to room temperature, and was quenched with the addition of saturated aqueous NH4Cl (25 mL). The resulting mixture was poured into a separatory funnel and the reaction flask was rinsed with Et2O (10 mL) and H2O (10 mL). The rinses were added to the separatory funnel and the aqueous layer was separated from the organic suspension of the product. The aqueous layer was then extracted with Et2O (2 × 25 mL) and the extracts were combined with the organic suspension. The suspension was washed sequentially with H2O (2 × 25 mL) and brine (1 × 25 mL). The solvent was removed to obtain a crude yellow product, which was recrystallized from hexanes and ethyl acetate. Yield: 68 %. δH (500 MHz, C6D6) 7.87–7.79 (m, 2H), 7.27–7.20 (m, 2H), 7.15–7.04 (m, 4H), 2.49 (d, J 4.5, 2H), 1.88–1.74 (m, 4H), 1.46 (dtd, J 11.5, 9.4, 4.6, 1H), 1.19 (dp, J 11.3, 8.8, 1H), 1.02 (s, 1H), 0.46 (m, 1H). δC (126 MHz, C6D6) 128.8, 127.9, 127.8, 127.6, 127.4, 125.9, 123.2, 73.0, 35.9, 22.8, 12.0.
Photolysis Experiments
Photolysis was performed at room temperature in a Rayonet photochemical reactor equipped with 16 30-cm 8-W lamps with output centred at ~350 nm (range ~315 to 400 nm). In a typical photolysis experiment, the carbene precursor (~10–15 mg) was weighed out and dissolved in 0.6 to 0.8 mL of benzene-d6. The resulting solution was then placed in an NMR tube (glass) for the photolysis and initial 1H and 13C spectra were taken. The photolysis was monitored by 1H NMR, until it was deemed that all precursor was consumed.
Supplementary Material
Spectral data for precursors 32 and 35 and photolysis results are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
D.M.T. acknowledges funding from the United States National Science Foundation (grant number CHE-1665278) for support of this work.
References
[1] (a) L. Friedman, J. G. Berger, J. Am. Chem. Soc. 1961, 83, 500.| Crossref | GoogleScholarGoogle Scholar |
(b) W. Kirmse, B. V. Wedel, Justus Liebigs Ann. Chem. 1963, 666, 1.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. J. Goldstein, W. R. Dolbier, J. Am. Chem. Soc. 1965, 87, 2293.
| Crossref | GoogleScholarGoogle Scholar |
(d) H. M. Frey, I. D. R. Stevens, J. Chem. Soc. 1965, 3101.
| Crossref | GoogleScholarGoogle Scholar |
(e) H. M. Frey, Adv. Photochem. 1966, 4, 225.
(f) K. T. Chang, H. Shechter, J. Am. Chem. Soc. 1979, 101, 5082.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. Fukushima, M. Jones, U. H. Brinker, Tetrahedron Lett. 1982, 23, 3211.
| Crossref | GoogleScholarGoogle Scholar |
(h) B. M. Armstrong, M. L. McKee, P. B. Shevlin, J. Am. Chem. Soc. 1995, 117, 3685.
| Crossref | GoogleScholarGoogle Scholar |
(i) H. C. Glick, I. R. Likhovorik, M. Jones, Tetrahedron Lett. 1995, 36, 5715.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) R. Bonneau, M. T. H. Liu, in Advances in Carbene Chemistry (Ed. U. H. Brinker) 1998, Vol. 2, pp. 1–28 (JAI Press: Stamford, CT).
(b) M. S. Platz, in Advances in Carbene Chemistry (Ed. U. H. Brinker) 1998, Vol. 2, pp. 133–174 (JAI Press: Stamford, CT).
[3] D. M. Thamattoor, J. R. Snoonian, H. M. Sulzbach, C. M. Hadad, J. Org. Chem. 1999, 64, 5886.
| Crossref | GoogleScholarGoogle Scholar |
[4] R. A. Farlow, D. M. Thamattoor, R. B. Sunoj, C. M. Hadad, J. Org. Chem. 2002, 67, 3257.
| Crossref | GoogleScholarGoogle Scholar | 12003533PubMed |
[5] E. Schmitz, A. Stark, C. Hoerig, Chem. Ber. 1965, 98, 2509.
| Crossref | GoogleScholarGoogle Scholar |
[6] K. M. Morgan, M. J. O’Connor, J. L. Humphrey, K. E. Buschman, J. Org. Chem. 2001, 66, 1600.
| Crossref | GoogleScholarGoogle Scholar | 11262102PubMed |
[7] (a) N. L. Drake, T. R. Sweeney, J. Org. Chem. 1946, 11, 67.
| Crossref | GoogleScholarGoogle Scholar | 21013437PubMed |
(b) W. T. Ford, M. Newcomb, J. Am. Chem. Soc. 1973, 95, 6277.
| Crossref | GoogleScholarGoogle Scholar |
[8] O. G. Kulinkovich, S. V. Sviridov, D. A. Vasilevski, Synthesis 1991, 234.
| Crossref | GoogleScholarGoogle Scholar |
[9] J. M. Nguyen, D. M. Thamattoor, Synthesis 2007, 2093.
[10] K. S. Graves, D. M. Thamattoor, P. R. Rablen, J. Org. Chem. 2011, 76, 1584.
| Crossref | GoogleScholarGoogle Scholar | 21338136PubMed |
[11] CIF files for the two precursors have been deposited at the Cambridge Crystallographic Data Centre (CCDC). CCDC deposition numbers are 1945138 for 32 and 1945137 for 35.
[12] For a recent theoretical discussion of the structure and chemistry of cyclobutylcarbenes, see: M. G. Rosenberg, U. H. Brinker, Tetrahedron Lett. 2018, 59, 645.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) L. Friedman, H. Shechter, J. Am. Chem. Soc. 1960, 82, 1002.
| Crossref | GoogleScholarGoogle Scholar |
(b) W. W. Schoeller, J. Org. Chem. 1980, 45, 2161.
| Crossref | GoogleScholarGoogle Scholar |