

Polarity Inversion Catalysis by the 1,4-Addition of N-Heterocyclic Carbenes
Xuan B. Nguyen A , Yuji Nakano A and David W. Lupton A B
A B
A School of Chemistry, Monash University, Clayton, Vic. 3800, Australia.
B Corresponding author. Email: david.lupton@monash.edu

Xuan B. Nguyen completed a Bachelor of Biotechnology majoring in chemistry and pharmacology before graduating with Honours (first class) in chemistry under the supervision of Professor David W. Lupton at Monash University in 2017. In 2018, he commenced studies towards a Doctorate of Philosophy in the same group. His studies are focused on the discovery of polarity reversal of conjugate acceptors enabled by NHC catalysis. |

Yuji Nakano completed a Bachelor of Science (Science Scholar Program) at Monash University in 2012 and a Ph.D. in 2017 at Monash University under the supervision of Professor David W. Lupton, where he worked on enantioselective N-heterocyclic carbene catalysis. From there, Yuji moved to Princeton University as an Endeavour Postdoctoral Fellow to conduct post-doctoral research with Professor Todd K. Hyster, investigating photoenzymatic catalyzed processes. In mid-2019, Yuji returned to Melbourne and took up a position as a post-doctoral fellow with Professor Jonathan B. Baell, where he is undertaking medicinal chemistry research at the Monash Institute of Pharmaceutical Sciences. |

David W. Lupton graduated with a Bachelor of Science (Honours, first class) in 2001 (University of Adelaide) before being awarded a Doctorate of Philosophy for studies under the supervision of Professor Martin G. Banwell (Australian National University) in 2005. Dr Lupton then undertook a post-doctoral fellowship with Professor Barry M. Trost (Stanford University) as a Sir Keith Murdoch fellow of the American Australian Association. In 2007, he returned to Australia to take up an academic appointment at Monash University in Melbourne, receiving an Australian Research Council Future Fellowship in 2011, and promotion to Professor in 2018. In 2010, he received the Athel Beckwith Lectureship of the Royal Australian Chemical Institute (RACI) while in 2013 he received the Rennie Medal of the RACI. Studies under David’s supervision are focused on the capacity of catalysis to enable discoveries in chemical synthesis. |
Australian Journal of Chemistry 73(1) 1-8 https://doi.org/10.1071/CH19550
Submitted: 25 October 2019 Accepted: 27 November 2019 Published: 3 February 2020
Abstract
Polarity inversion is the hallmark of N-heterocyclic carbene (NHC) organocatalysis, with the generation and reaction of acyl anion equivalents known for more than 70 years. In contrast, polarity inversion through 1,4-addition of NHCs to conjugate acceptors was first applied in a catalytic reaction in 2006. This sub-field of NHC-organocatalysis has developed steadily over the subsequent years, enabling novel coupling reactions, enantioselective cycloisomerizations, polymerizations, and other reactions. In this review, this emerging area of NHC-organocatalysis is discussed with comprehensive coverage. In addition, notes regarding the use of other Lewis base catalysts for related reactions, and comments regarding NHC selection for this type of catalysis, are provided.
Introduction
Although catalysis via polarity-inverted intermediates can be achieved with various catalysts and substrates,[1] reactions of acyl anions formed using N-heterocyclic carbenes (NHCs) are some of the most common.[2] Such chemistry was discovered more than 70 years ago by Ukai,[3] with mechanistic clarity introduced by Breslow in 1958.[4] Specifically, a nucleophilic carbene 1 adds to an aldehyde and the resultant alkoxide tautomerizes to give the acyl anion equivalent 2, colloquially referred to as the Breslow intermediate. Subsequent coupling to an electrophile is followed by elimination of the NHC to complete the catalytic cycle (Scheme 1a). Before the turn of this century, these events were largely limited to the benzoin condensation and Stetter reaction. More recently, Breslow intermediates derived from more sophisticated aldehydes have been accessed, thereby providing access to a range of secondary reactive intermediates, and fuelling the rapid growth in NHC-organocatalysis.[2c]
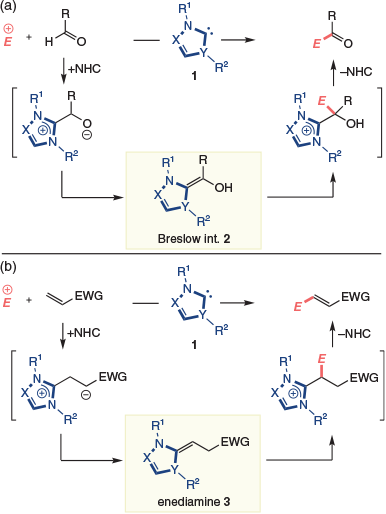
|
In little more than the last decade, a parallel field of NHC-organocatalysis has emerged in which the polarity of conjugate acceptors is inverted by 1,4-addition of the NHC. As with the formation of the Breslow intermediate, polarity inversion occurs by addition of the NHC and tautomerization of the resultant enolate, resulting in reactive intermediate 3. Enediamine 3 (also referred to as the β-azolium ylide or deoxy-Breslow intermediate[5]) couples to an electrophile and, after elimination of the NHC, gives a β-alkylated product (Scheme 1b). In the present review, we will examine the development of this chemistry in a comprehensive fashion. In the section Phosphine Catalysis, related catalysis achieved with phosphines is discussed. In the section Stoichiometric Access to the Enediamine, stoichiometric routes to the enediamine are discussed, with the section NHC-Catalysis Involving Single C–C Bond Formation detailing catalytic reactions involving single C–C bond-forming events. Finally, multiple C–C bond formation is discussed in the section NHC-Catalysis Involving Multiple C–C Bond Formation, while comments on catalyst selection and concluding remarks are made in the sections Miscellaneous Reactions of the Enediamine and Catalyst Selection.
Phosphine Catalysis
In 1962, Takashina and Price reported the triphenyl phosphine-catalyzed hexamerization of acrylonitrile (Scheme 2).[6] In contrast to phosphine-catalyzed reactions of the β-phosphonium enolate (i.e. 4) – a species that would attract significant attention over the following years[6b,7] – this transformation was achieved in the presence of ethanol as a cosolvent. It was postulated that the alcoholic additive allows tautomerization of enolate 4 to give the β-phosphonium ylide 5 that then couples with a second acrylonitrile to give 6. Four subsequent Michael additions and elimination of the catalyst then gives 7. Although the utility of such products is not clear, this pioneering report demonstrates the potential of catalysis exploiting polarity inversion of conjugate acceptors.
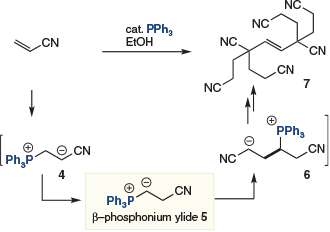
|
Related phosphine-catalyzed dimerizations of electron-poor olefins have been examined sporadically in the years following this report,[8] often in the context of non-electrochemical routes to adiponitrile. Most recently, Han and coworkers reported the dimerization of vinylphosphonate 8 to give 9 in 48 % yield in the presence of a catalytic trialkyl phosphine (Scheme 3).[8c]
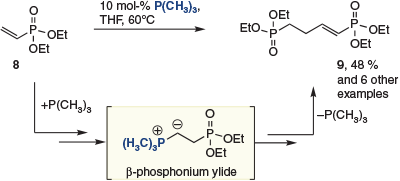
|
Although these reports demonstrate the viability of phosphine-mediated polarity inversion, the development of reactions beyond commodity chemical dimerization and oligomerization is yet to be demonstrated. The variability and abundance of phosphine catalysts in the literature suggest that this subfield could, in the future, expand into a profitable area of organocatalysis.
Stoichiometric Access to the Enediamine
Enders and coworkers reported the stoichiometric preparation of an enediamine in 1995.[9] Like the β-phosphonium ylide introduced in the previous section, the formation of enediamine 10 is thought to occur by 1,4-addition of NHC 11 to an electron-poor olefin and subsequent tautomerization (Scheme 4). Further reactions of the enediamine were not explored; however, this report introduced NHCs and substrates that could subsequently be applied in catalytic reactions.
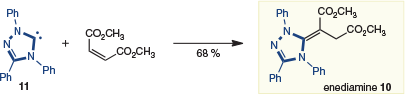
|
Stoichiometric formation of related enediamines was reported by Chen and coworkers in 2012[10] in their studies of NHC-mediated polymerization reactions. Specifically, they found that the combination of IMes 12 and methyl methacrylate (13) afforded enediamine 14, which was characterized by X-ray crystallography (Scheme 5). Whether enediamine formation (i.e. 14), dimerization, or polymerization occurs with methylmethacrylate (13) was found to be sensitive to the nature of the azolium.
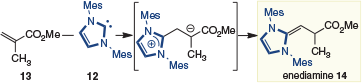
|
More recently, Biju and coworkers reported the stoichiometric formation of enediamine 15 when chalcone 16 is exposed to various imidazolium-derived NHCs (i.e. 12) in the presence of air (Scheme 6).[5e] In this reaction, 1,4-addition provides enolate 17, which is oxidized to the 1,2-dicarbonyl 18, which is then deprotonated to give enediamine 15.
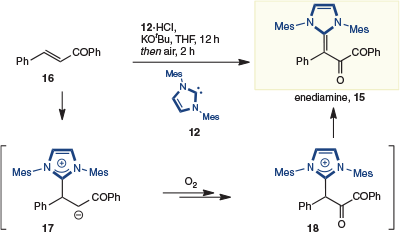
|
NHC-Catalysis Involving Single C–C Bond Formation
The first catalytic reaction implicating the enediamine was reported in 2006 by Fu and coworkers. In studies examining a nickel-catalyzed Mizoroki-Heck reaction, it was discovered that the cyclization of 19 to give 20 (Scheme 7a) could be catalyzed by NHC ligand 21 in the absence of a transition metal.[11] The observed reaction was rationalized as occurring by 1,4-addition of the Lewis base catalyst to give enolate 22, which then undergoes tautomerization to enediamine 23. Subsequent displacement of the tethered bromide provides the cyclized material, with elimination of the catalyst giving the observed product 20. What is striking in this reaction is that enolate 22 does not undergo direct cyclization to give cyclohexene 24 (Ph = OEt). In contrast, with phosphines – Lewis base catalysts also capable of polarity inversion (see above) – cyclization via the enolate is observed to provide the six-membered product 24 (Scheme 7b).[12]
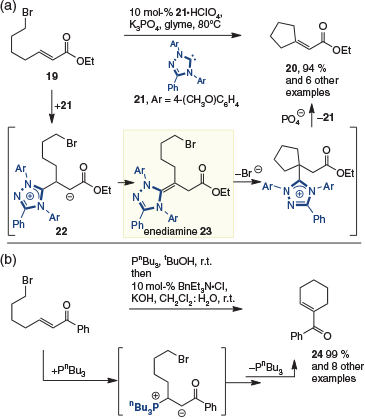
|
Computational investigations by Wang and coworkers examined the divergent reactivity of NHC and phosphine catalysts, and concluded that formation of enediamine 23 was thermodynamically favoured over enolate 22, something that was not the case for the analogous β-phosphonium ylide.[13]
This intriguing report from Fu introduced a new reactive intermediate to the field and demonstrated that NHC-organocatalysis by 1,4-addition is not limited to enolate reactions.[14] Despite this advance, it was some 5 years before the next report of catalysis via the enediamine emerged. In 2011, Matsuoka and coworkers reported the tail-to-tail dimerization of acrylates effected by triazolium-derived NHC 11 (Scheme 8a).[15] The proposed mechanism for this reaction involves addition of the NHC to provide enolate 25, which then tautomerizes to give the enediamine 26. Nucleophilic addition to another equivalent of the starting material then installs the new C–C bond in 27, with elimination of the catalyst by an E1cB mechanism providing the observed product 28. Independent studies reported concurrently by Glorius and coworkers introduced a similar transformation.[16] In their work, it was also possible to achieve the cross-coupling of acrylates with vinylphosphonates in the presence of triazolium-derived NHC 11 to give, for example, 29 (Scheme 8b). Presumably the dual activation within the vinyl phosphonate allows chemoselective addition of the NHC to this partner.
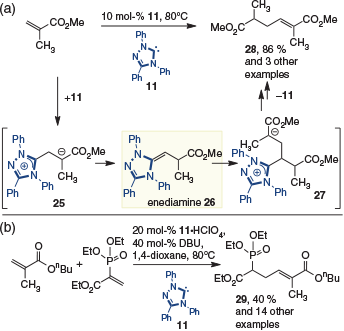
|
Subsequently, Matsuoka and coworkers expanded on these initial discoveries to include α,β-unsaturated nitrile 30 as a precursor to enediamine 31. This transformation proved sensitive to H-bond donor additives, with favourable results obtained in the presence of propan-2-ol and 2-naphthol (Scheme 9).[17]
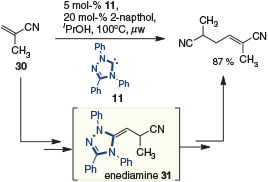
|
The first enantioselective reaction to exploit the enediamine intermediate was introduced by Nakano and Lupton in 2016. In this work, the cycloisomerization of bis-Michael acceptor 32 was achieved to allow access to enantioenriched aryl propiolate 33 (Scheme 10).[18] Specifically, the reaction starts with polarity inversion of the more electrophilic β-unsubstituted vinyl ketone using the nucleophilic p-methoxyphenyl NHC 34 to give enediamine 35. Subsequent addition into the second Michael acceptor gives rise to enolate 36. Diastereoselective protonation, to establish the only stereogenic centre retained in the product, is then followed by elimination of the catalyst and keto-enol tautomerism to give 33. The reaction occurred with moderate levels of enantioselectivity, which could be augmented using hexafluoroisopropanol (HFIP) as an additive. This is likely due to the HFIP serving as a bulky acid for the diastereoselective protonation.
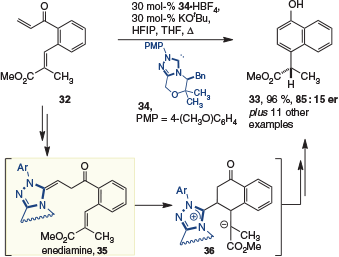
|
Although enantioselectivity was possible with the previous design, an alternative cycloisomerization was developed by the same group to access more highly enantioenriched materials (Scheme 11).[19] In this transformation, the cyclization sets a stereocentre that is retained in the product. As a consequence, this design was more robust, allowing a wide array of cyclopentanes (i.e. 37) to be prepared from bis-Michael acceptors (i.e. 38) with high levels of enantioselectivity.
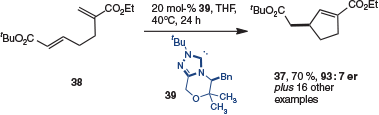
|
Recently, polarity inversion of aryl acrylamide 40 has enabled the preparation of 2-quinolones 41 (Scheme 12).[20] In this report, Tobisu and coworkers exploit bespoke high-nucleophilicity NHCs (i.e. 42) to give enediamine 43, which then undergoes a concerted aromatic substitution reaction (CSNAr, supported by computational studies) to give 44 and ultimately the quinolone 41. This study is a rare example of the enediamine adding to an electrophile that is not a Michael acceptor.
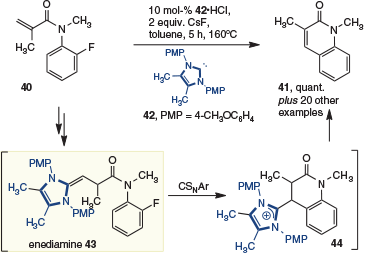
|
In addition to small-molecule synthesis, the enediamine has been exploited in polymerization studies.[21] For example, Chen and coworkers reported linear chain propagation of dimethacrylate 45 by tail-to-tail dimerization catalyzed by 11. This reaction furnished polyesters of high atomic weight and narrow dispersity (Scheme 13).
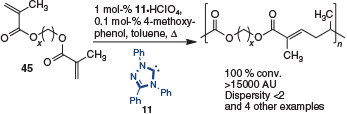
|
NHC-Catalysis Involving Multiple C–C Bond Formation
Formation of the enediamine proceeds via initial formation of an enolate – the key intermediate for the Morita–Baylis–Hillman and Rauhut–Currier reactions.[7] In addition, mechanistic investigations have revealed how the enediamine can reform following a bond-forming reaction (see below). Thus, reactions can be designed that exploit multiple C–C bond-forming events involving either intermediate.
In 2015, Berkessel and coworkers reported studies on the dimerization of dimethyl acrylamide 46 with the Enders triphenyltriazolium (TPT) catalyst 11. Analogous to adducts formed with acrylates and phosphonates, the tail-to-tail dimer 47 formed, although this was also accompanied by a small amount of trimer 48 (Scheme 14).[22] Mechanistically, it would appear that formation of the first enediamine (i.e. 49) is followed by coupling to an acrylamide to give enolate 50. This can then either eliminate the NHC to give the dimer 47, or tautomerize to the second enediamine (i.e. 51). The latter pathway then allows a third acrylamide to be incorporated, to give 52, which then eliminates the NHC to give trimer 48.

|
Repeated formation of the enediamine can also be inferred from deuterium labelling studies reported by Matsuoka (Scheme 15).[23] Specifically, when acrylate 53 is dimerized in the presence of deuterated methanol, isotope incorporation is observed in product 54 that is consistent with formation of a second enediamine (i.e. 55) before elimination of the NHC.
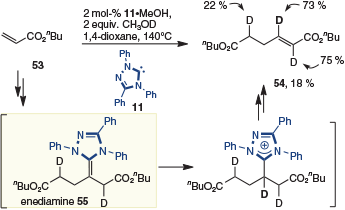
|
The potential utility of multiple enediamine formation was recently exploited with the development of dimerizing/cycloisomerization, and cycloisomerization, reactions by Lupton and coworkers (Scheme 16).[24] The former reaction design was possible using homochiral NHC 56, allowing several cyclohexanones (i.e. 57) to be prepared (Scheme 16a) with high levels of enantioselectivity. The second reaction design was achieved with achiral catalysts 58. A lack of reactivity with chiral NHCs was attributed to the decreased nucleophilicity of these catalysts,[25] thus making them unsuitable for substrates in which the three electrophiles are linked through the α-position of the first conjugate acceptor (Scheme 16b). Mechanistically, these reactions are related. If we consider the latter reaction it likely commences by 1,4-addition and tautomerization to provide enediamine 59 (Scheme 16c). This species then undergoes an intramolecular conjugate addition to give enolate 60, which tautomerizes to give the second enediamine 61. This species undergoes the second cyclization to give 62, which following elimination of the catalyst, provides the bicyclic product 63.
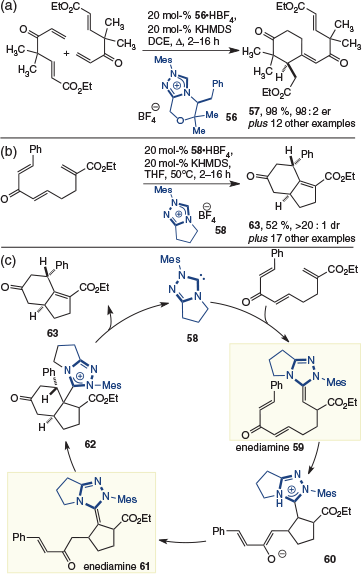
|
The fine lines between dimerization, oligomerization, and polymerization as outcomes in reactions involving the enediamine were exemplified in studies by Matsuoka reported in 2013. Using methyl acrylate in the presence of imidazolium-derived NHC 12, the tetramer 64 was produced (Scheme 17a).[26] A mechanism was postulated in which enolate 65 is initially formed and undergoes a Michael addition, followed by tautomerization to yield enediamine 66 (Scheme 17b). Its addition into a third acrylate then gives enolate 67, which adds to a fourth acrylate in a second Michael addition to give 68. Finally, tautomerization to give 69, followed by Dieckmann reaction and elimination of the NHC, gives cyclopentenone 64.

|
A related trimerization exploiting both enolate and enediamine intermediates was reported by Taton and coworkers when methyl methacrylate was exposed to an equivalent of NHC 70 (Scheme 18).[27] In this case, the ylide 71 was isolated in 51 % yield.
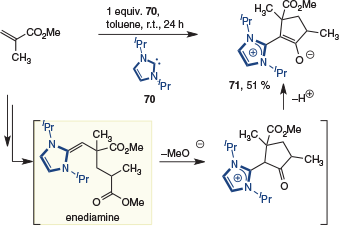
|
Miscellaneous Reactions of the Enediamine
The intermediacy of the enediamine was exploited by Scheidt and coworkers in the rearrangement of 1,1-disulfone 72 to 1,2-disulfone 73, within the context of a [3 + 2] annulation.[28] Crossover experiments confirmed the formation of the enediamine intermediate 74, followed by ejection of the sulfonate and recombination to give 75. Finally, elimination of NHC 76 provided the substrate for the [3 + 2]-dipolar cycloaddition with nitrone 77 to give 78 (Scheme 19).
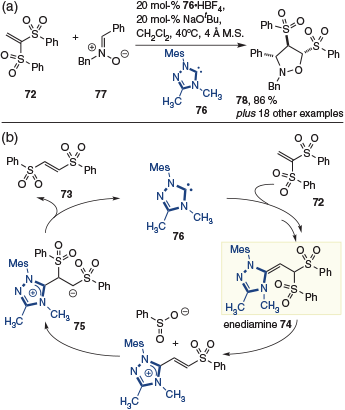
|
Arylogous acrylonitrile 79 has been shown to undergo tail-to-tail dimerization reactions to give dinitrile product 80 (Scheme 20). The groups of Glorius[29] and Matsuoka,[23] using conditions similar to those already reported (Scheme 8), were able to execute this coupling reaction with good yields.
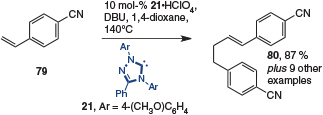
|
Catalyst Selection
As is common in studies on NHC-organocatalysis, a broad array of catalysts have been reported. In some cases, it can be seen that a systematic investigation into the NHC suited to the reaction design has been undertaken, although this is not always the case. Limiting factors such as accessibility can influence catalyst selection. With this said, a few general comments to guide NHC selection for this type of reaction can be made. First, the use of triazolium-derived NHCs bearing electron-withdrawing N-substituents, such as N-C6F5 or N-2,4,6-Cl3C6H2, has not been reported. These are common NHCs used in the polarity inversion of aldehydes. Their absence is likely related to a higher degree of nucleophilicity required for the 1,4-addition. In our studies, we have examined a range of NHCs in all enantioselective reaction designs and found that in most, but not all, designs, higher-nucleophilicity carbenes are ideal.
Another feature evident in the studies presented herein is that β-unsubstituted Michael acceptors are most common, with only a limited number of reactions with a β-substituent reported. In these cases, more nucleophilic triazolium (or indeed imidazolium)-derived NHCs or forcing conditions are required. Presumably, this correlates with a decrease in electrophilicity of substrate bearing β-substituents.[30]
Conclusions
Catalysis via polarity inversion of conjugate acceptors was reported little more than a decade ago and, after an induction period, is beginning to receive attention within the community. Trends in catalyst selection and substrate suitability are becoming clear, allowing new reaction designs to be established. There are still several limitations in this field, with only tentative forays into enantioselective catalysis reported, and a limited number of reaction designs that exploit electrophilic partners that are not a Michael acceptor. The next stage of development for this type of reaction is likely to involve an expansion of the range of electrophilic partners, and the creation of more general solutions to enantioselectivity.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors acknowledge financial support from the Australian Research Council through the Discovery Program.
References
[1] (a) For an introduction, see: G. Wittig, P. Davis, G. Koenig, Chem. Ber. 1951, 84, 627.| Crossref | GoogleScholarGoogle Scholar |
(b) D. Seebach, Angew. Chem. Int. Ed. Engl. 1979, 18, 239.
| Crossref | GoogleScholarGoogle Scholar |
(c) For metal-catalyzed umpolung, see: G. Zanoni, A. Pontiroli, A. Marchetti, G. Vidari, Eur. J. Org. Chem. 2007, 3599.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. Streuff, Synthesis 2013, 281.
| Crossref | GoogleScholarGoogle Scholar |
(e) For organocatalytic umpolung, see: X. Bugaut, F. Glorius, Chem. Soc. Rev. 2012, 41, 3511.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. Waser, J. Novacek, Angew. Chem. Int. Ed. 2015, 54, 14228.
| Crossref | GoogleScholarGoogle Scholar |
(g) For radical ion umpolung, see: M. A. Ischay, T. P. Yoon, Eur. J. Org. Chem. 2012, 3359.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) For general NHC catalysis, see: D. Enders, O. Niemeier, A. Henseler, Chem. Rev. 2007, 107, 5606.
| Crossref | GoogleScholarGoogle Scholar | 17956132PubMed |
(b) M. N. Hopkinson, C. Richter, M. Schedler, F. Glorius, Nature 2014, 510, 485.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. M. Flanigan, F. Romanov-Michailidis, N. A. White, T. Rovis, Chem. Rev. 2015, 115, 9307.
| Crossref | GoogleScholarGoogle Scholar |
(d) For acyl azolium enolates, see: J. Douglas, G. Churchill, A. D. Smith, Synthesis 2012, 2295.
| Crossref | GoogleScholarGoogle Scholar |
(e) For cascade catalysis, see: A. Grossmann, D. Enders, Angew. Chem. Int. Ed. 2012, 51, 314.
| Crossref | GoogleScholarGoogle Scholar |
(f) For acyl anion free catalysis, see: S. J. Ryan, L. Candish, D. W. Lupton, Chem. Soc. Rev. 2013, 42, 4906.
| Crossref | GoogleScholarGoogle Scholar |
(g) For acyl azolium catalysis: S. De Sarkar, A. Biswas, R. C. Samanta, A. Studer, Chem. – Eur. J. 2013, 19, 4664.
| Crossref | GoogleScholarGoogle Scholar |
(h) C. Zhang, J. F. Hooper, D. W. Lupton, ACS Catal. 2017, 7, 2583.
| Crossref | GoogleScholarGoogle Scholar |
(i) S. Mondal, S. R. Yetra, S. Mukherjee, A. T. Biju, Acc. Chem. Res. 2019, 52, 425.
| Crossref | GoogleScholarGoogle Scholar |
(j) For cooperative catalysis: M. H. Wang, K. A. Scheidt, Angew. Chem. Int. Ed. 2016, 55, 14912.
| Crossref | GoogleScholarGoogle Scholar |
[3] T. Ukai, R. Tanaka, T. Dokawa, J. Pharm. Soc. Jpn. 1943, 63, 296.
| Crossref | GoogleScholarGoogle Scholar |
[4] R. Breslow, J. Am. Chem. Soc. 1958, 80, 3719.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) Enediamine 3 is a type of deoxy-Breslow intermediate, a name used to describe compounds with a double bond from the NHC to a carbon lacking an oxygen substituent. In this review, the term ‘enediamine’ is used to describe all intermediates forming from 1,4-addition and tautomerization. For other types of deoxy-Breslow intermediates, see: C. E. I. Knappke, J.-M. Neudörfl, A. J. von Wangelin, Org. Biomol. Chem. 2010, 8, 1695.
| Crossref | GoogleScholarGoogle Scholar |
(b) C. E. I. Knappke, A. J. Arduengo, H. Jiao, J.-M. Neudörfl, A. J. von Wangelin, Synthesis 2011, 3784.
(c) R. N. Reddi, P. K. Prasad, A. Sudalai, Angew. Chem. Int. Ed. 2015, 54, 14150.
| Crossref | GoogleScholarGoogle Scholar |
(d) M. Schedler, N. E. Wurz, C. G. Daniliuc, F. Glorius, Org. Lett. 2014, 16, 3134.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. Bhunia, S. Thorat, R. G. Gonnade, A. T. Biju, Chem. Commun. 2015, 13690.
| Crossref | GoogleScholarGoogle Scholar |
(f) For studies examining the properties of deoxy-Breslow intermediates, see: A. Berkessel, S. Elfert, Adv. Synth. Catal. 2014, 356, 571.
| Crossref | GoogleScholarGoogle Scholar |
(g) B. Maji, M. Horn, H. Mayr, Angew. Chem. Int. Ed. 2012, 51, 6231.
| Crossref | GoogleScholarGoogle Scholar |
(h) B. Maji, H. Mayr, Angew. Chem. Int. Ed. 2012, 51, 10408.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) N. Takashina, C. C. Price, J. Am. Chem. Soc. 1962, 84, 489.
| Crossref | GoogleScholarGoogle Scholar |
(b) For a review covering this work: H. Guo, Y. C. Fan, Z. Sun, Y. Wu, O. Kwon, Chem. Rev. 2018, 118, 10049.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) M. M. Rauhut, H. Currier, U.S. Patent 3074999 1963 [Chem. Abstr. 1963, 58, 66109].
(b) For selected reviews, see: C. E. Aroyan, A. Dermenci, S. J. Miller, Tetrahedron 2009, 65, 4069.
| Crossref | GoogleScholarGoogle Scholar |
(c) W. Li, J. Zhang, Chem. Soc. Rev. 2016, 45, 1657.
| Crossref | GoogleScholarGoogle Scholar |
(d) H. Ni, W.-L. Chan, Y. Lu, Chem. Rev. 2018, 118, 9344.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) For examples, see: C. D. Hall, N. Lowther, B. R. Tweedy, A. C. Hall, G. Shaw, J. Chem. Soc. Perkin Trans. 2 1998, 2, 2047.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. N. Khong, Y. S. Tran, O. Kwon, Tetrahedron 2010, 66, 4760.
| Crossref | GoogleScholarGoogle Scholar |
(c) X.-B. Wang, Y. Saga, R. Shen, H. Fujino, M. Goto, L.-B. Han, RSC Adv. 2012, 2, 5935.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Takizawa, K. Kishi, Y. Yoshida, S. Mader, F. A. Arteaga, S. Lee, M. Hoshino, M. Rueping, M. Fujita, H. Sasai, Angew. Chem. Int. Ed. 2015, 54, 15511.
| Crossref | GoogleScholarGoogle Scholar |
[9] D. Enders, K. Breuer, G. Raabe, J. Runsink, J. H. Teles, J.-P. Melder, K. Ebel, S. Brode, Angew. Chem. Int. Ed. Engl. 1995, 34, 1021.
| Crossref | GoogleScholarGoogle Scholar |
[10] Y. Zhang, E. Y.-X. Chen, Angew. Chem. Int. Ed. 2012, 51, 2465.
| Crossref | GoogleScholarGoogle Scholar |
[11] C. Fischer, S. W. Smith, D. A. Powell, G. C. Fu, J. Am. Chem. Soc. 2006, 128, 1472.
| Crossref | GoogleScholarGoogle Scholar | 16448117PubMed |
[12] (a) M. E. Krafft, T. F. N. Hazell, J. Am. Chem. Soc. 2005, 127, 10168.
| Crossref | GoogleScholarGoogle Scholar | 16028918PubMed |
(b) M. E. Krafft, K. A. Seibert, T. F. N. Hazell, C. Hirosawa, Chem. Commun. 2005, 5772.
| Crossref | GoogleScholarGoogle Scholar |
[13] L. Zhao, X. Y. Chen, S. Ye, Z.-X. Wang, J. Org. Chem. 2011, 76, 2733.
| Crossref | GoogleScholarGoogle Scholar | 21375260PubMed |
[14] (a) For NHC-mediated enolate reactions, see: L. He, T.-Y. Jian, S. Ye, J. Org. Chem. 2007, 72, 7466.
| Crossref | GoogleScholarGoogle Scholar | 17705547PubMed |
(b) P. Goswami, S. Sharma, G. Singh, R. V. Anand, J. Org. Chem. 2018, 83, 4213.
| Crossref | GoogleScholarGoogle Scholar |
(c) S.-i. Matsuoka, N. Awano, M. Nakazawa, M. Suzuki, Tetrahedron Lett. 2016, 57, 5707.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. Bhunia, S. R. Yetra, R. G. Gonnadec, A. T. Biju, Org. Biomol. Chem. 2016, 14, 5612.
| Crossref | GoogleScholarGoogle Scholar |
(e) R. L. Atienza, K. A. Scheidt, Aust. J. Chem. 2011, 64, 1158.
| Crossref | GoogleScholarGoogle Scholar |
(f) X. Wu, L. Zhou, R. Maiti, C. Mou, L. Pan, Y. R. Chi, Angew. Chem. Int. Ed. 2019, 58, 477.
| Crossref | GoogleScholarGoogle Scholar |
(g) S. Bae, C. Zhang, R. M. Gillard, D. W. Lupton, Angew. Chem. Int. Ed. 2019, 58, 13370.
| Crossref | GoogleScholarGoogle Scholar |
[15] S.-i. Matsuoka, Y. Ota, A. Washio, A. Katada, K. Ichioka, K. Takagi, M. Suzuki, Org. Lett. 2011, 13, 3722.
| Crossref | GoogleScholarGoogle Scholar |
[16] A. T. Biju, M. Padmanaban, N. E. Wurz, F. Glorius, Angew. Chem. Int. Ed. 2011, 50, 8412.
| Crossref | GoogleScholarGoogle Scholar |
[17] T. Kato, S.-i. Matsuoka, M. Suzuki, J. Org. Chem. 2014, 79, 4484.
| Crossref | GoogleScholarGoogle Scholar | 24773333PubMed |
[18] Y. Nakano, D. W. Lupton, Angew. Chem. Int. Ed. 2016, 55, 3135.
| Crossref | GoogleScholarGoogle Scholar |
[19] L. Scott, Y. Nakano, C. Zhang, D. W. Lupton, Angew. Chem. Int. Ed. 2018, 57, 10299.
| Crossref | GoogleScholarGoogle Scholar |
[20] K. Yasui, M. Kamitani, M. Tobisu, Angew. Chem. Int. Ed. 2019, 58, 14157.
| Crossref | GoogleScholarGoogle Scholar |
[21] (a) M. Hong, X. Tang, L. Falivene, L. Caporaso, L. Cavallo, E. Y.-X. Chen, J. Am. Chem. Soc. 2016, 138, 2021.
| Crossref | GoogleScholarGoogle Scholar | 26779897PubMed |
(b) M. Hong, E. Y.-X. Chen, Angew. Chem. Int. Ed. 2014, 53, 11900.
| Crossref | GoogleScholarGoogle Scholar |
[22] O.-a. Rajachan, M. Paul, V. R. Yatham, J.-M. Neudörfl, K. Kanokmedhakul, S. Kanokmedhakul, A. Berkessel, Tetrahedron Lett. 2015, 56, 6537.
| Crossref | GoogleScholarGoogle Scholar |
[23] S.-i. Matsuoka, M. Nakazawa, M. Suzuki, Bull. Chem. Soc. Jpn. 2015, 88, 1093.
| Crossref | GoogleScholarGoogle Scholar |
[24] X. B. Nguyen, Y. Nakano, N. M. Duggan, L. Scott, D. W. Lupton, Angew. Chem. Int. Ed. 2019, 58, 11483.
| Crossref | GoogleScholarGoogle Scholar |
[25] (a) For the impact of substituent on nucleophilicity of the NHC, see: B. Maji, M. Breugst, H. Mayr, Angew. Chem. Int. Ed. 2011, 50, 6915.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. Levens, F. An, M. Breugst, H. Mayr, D. W. Lupton, Org. Lett. 2016, 18, 3566.
| Crossref | GoogleScholarGoogle Scholar |
[26] S.-i. Matsuoka, S. Namera, A. Washio, K. Takagi, M. Suzuki, Org. Lett. 2013, 15, 5916.
| Crossref | GoogleScholarGoogle Scholar |
[27] W. N. Ottou, D. Bourichon, J. Vignolle, A.-L. Wirotius, F. Robert, Y. Landais, J.-M. Sotiropoulos, K. Miqueu, D. Taton, Chem. – Eur. J. 2014, 20, 3989.
| Crossref | GoogleScholarGoogle Scholar |
[28] R. L. Atienza, H. S. Roth, K. A. Scheidt, Chem. Sci. 2011, 2, 1772.
| Crossref | GoogleScholarGoogle Scholar | 22448316PubMed |
[29] M. Schedler, N. E. Wurz, C. G. Daniliuc, F. Glorius, Org. Lett. 2014, 16, 3134.
| Crossref | GoogleScholarGoogle Scholar | 24824735PubMed |
[30] D. S. Allgäuer, H. Jangra, H. Asahara, Z. Li, Q. Chen, H. Zipse, A. R. Ofial, H. Mayr, J. Am. Chem. Soc. 2017, 139, 13318.
| Crossref | GoogleScholarGoogle Scholar | 28921959PubMed |