

Crosslinking of Self-Assembled Protein–Polymer Conjugates with Divanillin
Zihao Li A , Yanyan Jiang B , Kilian Wüst A , Manuela Callari A and Martina H. Stenzel A C
A C
A Centre for Advanced Macromolecular Design (CAMD), School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia.
B School of Materials Science and Engineering, Shandong University, Jinan 250061, China.
C Corresponding author. Email: M.Stenzel@unsw.edu.au
Australian Journal of Chemistry 73(10) 1034-1041 https://doi.org/10.1071/CH19617
Submitted: 29 November 2019 Accepted: 31 March 2020 Published: 19 June 2020
Journal Compilation © CSIRO 2020 Open Access CC BY
Abstract
Protein-based materials are widely used in biomedical applications. Often the proteins need to be crosslinked in order to be stable for application. Here, we explored the use of 5,5′-bisvanillin as a potentially non-toxic crosslinker that can react with lysine residues on proteins. To demonstrate the success of the crosslinking reaction, polymer–protein conjugates based on bovine serum albumin (BSA) and poly(N-isopropyl acrylamide) (PNIPAM) were employed. The BSA-PNIPAM conjugate is water soluble at room temperature, but heated above the cloud point, BSA-PNIPAM forms nanoparticles of around 70 nm that can again disassemble at lower temperatures. Reaction with 5,5′-bisvanillin prevented disassembly resulting in stable BSA nanoparticles of 50 nm in size. The formed nanoparticles were observed to be rather stable and were not easily cleaved in acidic conditions. The crosslinker 5,5′-bisvanillin was measured to have lower toxicity against A2780 lung cancer cell lines compared with the commonly applied crosslinker glutaraldehyde.
Introduction
Biomaterials based on proteins have widely been used, ranging from protein capsules for drug delivery[1] to fibres, films, and hydrogels.[2] Protein-based materials however succumb to low structural integrity and are prone to disintegration if not stabilized by crosslinking. Crosslinking of proteins to stabilize their interaction is now routinely applied in biomaterial research. The motivation for protein crosslinking can be found in the desire to create better protein-based biomaterials, but it is also widely used as a tool to understand the function of proteins. Monitoring the ubiquitous protein interaction, identifying the function of each protein, and illuminating the action along the way have been the focus of many recent efforts.[3] Crosslinking of proteins is then used to capture the interactions of proteins allowing analysis of the resulting fixed structure using techniques such as high performance liquid chromatography (HPLC)[4] or mass spectrometry.[5] The main demand on these crosslinkers is different to the ones in material design as the crosslinker needs to be permanent, the reaction needs to be efficient, and the length between two crosslinking points needs to be adjusted to suit the special requirements to capture protein interaction. Crosslinkers used for biomaterials such as for drug delivery or tissue engineering have, in contrast, different needs regarding their design. Although the reaction should still be efficient, other considerations are paramount, such as biocompatibility. Researchers are usually faced with a choice of crosslinking reactions as proteins possess a variety of functional groups that can be used including thiols, hydroxides, carboxylic acids, and amines.[2,6] Many of these reactions result in the formation of permanent bonds with the exception of the reaction between amino acids such as lysine and aldehyde that can be degraded under acidic conditions.[7] Glutaraldehyde is by far the most commonly used protein crosslinker and has been used to fix samples for histology and microscopy. It acts as a chemosterilizer as it reacts with the lysine groups of proteins and it is therefore used to preserve tissue.[8] However, there are some concerns with regard to the safety of glutaraldehyde and it is therefore not suitable for human consumption.[9]
Recently, vanillin-derived functional building blocks have been explored for the design of biopolymers based on renewable resources.[10] Among them, divanillin, specifically 5,5′-bisvanillin, with its two aldehyde functionalities (Scheme 1) can be used to form polymers using a Schiff base reaction with diamines,[11] via electrochemical reductive polymerization,[12] or via Biginelli multicomponent polymerization.[13] Divanillin, which is found naturally in vanilla pods, is used as a flavour enhancer and is considered safe for use as a food supplement.[14]

|
Inspired by the low toxicity of 5,5′-bisvanillin and the ability to form acid degradable Schiff bases with amines, we propose the use of 5,5′-bisvanillin as a pH-responsive crosslinker for protein-based materials. To test the imine bond formation and therefore the successful crosslinking, polymer–protein conjugates based on bovine serum albumin (BSA) and poly(N-isopropyl acrylamide) (PNIPAM), a well studied system,[15] were self-assembled into micellar structures above the lower critical solution temperature (LCST) of the polymer. Subsequent crosslinking captures the structure preventing disassembly at lower temperatures (Scheme 1).
Experimental
Unless otherwise specified, all chemicals were reagent grade and used as supplied. Butylamine, BSA, NH4OH, lithium bromide, and 4-methoxyphenol were purchased from Sigma–Aldrich. N,N-Dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) were purchased from Chem-Supply. Acetonitrile and trifluoroacetic acid were purchased from VWR chemicals. Deuterated NMR solvents (CDCl3, d6-DMSO, and D2O) were purchased from Cambridge Isotope Laboratories. Unless otherwise specified, all chemicals and materials used for cell work were purchased from Sigma–Aldrich.
The synthesis of the crosslinker 5,5′-bisvanillin[16] and furan-protected-maleimide terminated poly(N-isopropylacrylamide) (fpMAL-PNIPAM)[17] are described elsewhere as referenced.
Imine Formation Study of Divanillin with Butylamine
Divanillin (20.2 mg, 0.0668 mmol, 1.0 equiv.) and n-butylamine (19.5 µL, 0.197 mmol, 3.0 equiv.) were mixed in anhydrous acetonitrile (2.0 mL) and stirred at 40°C for 1 h. After removing solvent by reduced pressure, the resulting solid was analysed by 1H NMR spectroscopy. δH (400 MHz, DMSO-d6) 0.91 (m, 6H, CH2CH2CH2CH3), 1.35 (sextet, 4H, CH2CH2CH2CH3), 1.60 (quintet, 4H, CH2CH2CH2CH3), 3.52 (t, 4H, CH2CH2CH2CH3), 3.75 (m, 6H, OCH3), 7.31 (m, 4H, ArH), 8.22 (s, 1.68H, CHN), 9.63 (s, 0.18H, CHO) revealing a conversion of aldehyde into imine of 90.3 %.
Synthesis of BSA-PNIPAM Conjugates
The maleimide group of fpMAL-PNIPAM was first deprotected by leaving the sample in a vacuum oven overnight at 95°C to yield maleimide-terminated poly(N-isopropylacrylamide) (MAL-PNIPAM). MAL-PNIPAM (12.5 mg, 5.0 × 10−4 mmol 1.0 equiv.) was dissolved in DMSO (3 mL) and added dropwise into BSA (33.1 mg, 5.0 × 10−4 mmol, 1.0 equiv.) solution with phosphate buffered solution (7 mL). The solutions were stirred for 60 h, dialyzed against cold deionized water (MWCO = 50 kDa) for 3 days, and freeze-dried. The conjugates synthesized were analysed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE, 10 % acrylamide) and aqueous phase gel permeation chromatography (GPC).
Crosslinking BSA-PNIPAM Micelles with Divanillin
A BSA-PNIPAM solution (0.5 mg mL−1, 300 µL, 1.6 × 10−6 mmol, 1.0 equiv.) in pH 8.0 phosphate buffer was pre-heated at 40°C for 5 min, followed by addition of 5,5′-bisvanillin solution (DMSO, 0.20 mg mL−1, 24.9 µL, 1.6 × 10−5 mmol, 10.0 equiv.) and 10 h slow stirring at 40°C to form crosslinked micelles. The hydrodynamic diameter of the resulting product was recorded by dynamic light scattering (DLS) at 25 and 40°C. The transmission electron microscopy (TEM) images of the crosslinked micelles were taken after staining with 2 % aqueous uranyl acetate.
Cell Culture
The human lung cancer (A-549) cell lines were grown in a ventilated tissue culture flask T-75 using Roswell Park Memorial Institute (RPMI-1640) media containing 10 % fetal bovine serum (FBS) and antibiotics. The cells were incubated at 37°C under a 5 % CO2 humidified atmosphere and passaged when monolayers of 80 % confluence were formed. The cytotoxicity was measured by a standard sulforhodamine B colourimetric proliferation assay (SRB assay). The SRB assay was established by the USA National Cancer Institute for rapid, sensitive, and inexpensive screening of antitumour drugs in microtiter plates. The cells were seeded at a density of 4000 cells per well in 96-well plates containing 200 µL of growth medium per well and incubated for 24 h. The medium was then replaced with fresh medium (200 µL) containing various concentrations of the material being tested. After 72 h incubation, the culture medium was discarded and the cells were fixed with 100 µL of cold 10 % w/v trichloroacetic acid (TCA), incubated at 4°C for 30 min, and then washed five times with reverse osmosis water to remove TCA, growth medium, and low molecular weight metabolites. TCA-fixed cells were stained for 30 min with 0.4 % (w/v) SRB dissolved in 1 % acetic acid. At the end of the staining period, SRB was removed and cultures were quickly rinsed five times with 1 % acetic acid to remove the unbound dye. The cultures were then air-dried until no conspicuous moisture was visible. The bound dye was dissolved with 200 µL of 10 mM Tris buffer and the plates were analysed using a microtiter plate reader scanning spectrophotometer BioTek’s PowerWave HT Microplate Reader and KC4 software. The plates were shaken for 30 s and read at an absorbance of 490 nm. All experiments were repeated three times.
Analysis
1H NMR Spectroscopy
All NMR measurements were performed on a Bruker DPX-400 with a 1H/X inverse broadband z gradient BBI probe at 400 MHz frequency and 16 scans as default. Samples were dissolved and analysed in deuterated chloroform (CDCl3), unless otherwise specified.
DMF GPC
DMF GPC was performed with a Shimadzu modular system consisting of a DGU-12A degasser, LC-10AT pump, SIL-10AD auto-injector, CTO-10A column oven (50°C), a guard column, three Phenomexex 5.0 µm bead-size columns (105, 104, and 103 Å), and an RID-10A refractive index detector. DMF containing 0.1 % LiBr and 0.04 % 4-methoxyphenol was used as the mobile phase (flow rate: 1 mL min−1). The instrument was calibrated with commercially available linear poly(methyl methacrylate) (PMMA) standards (Polymer Laboratories).
Aqueous Phase GPC
Aqueous phase GPC (MilliQ water containing 0.02 % w/v NaN3) was performed using a Shimadzu modular system comprising a DGU-12A solvent degasser, an LC-10AT pump, a CTO-10A column oven, and an RID-10A refractive index detector (flow rate: 0.8 mL min−1). The system was equipped with a Polymer Laboratories 5.0 mm bead-size guard column (50 × 7.8 mm2) followed by three 300 × 7.8 mm PL columns (30, 40, and 50 respectively in type). Calibration was conducted with poly(ethylene oxide) (PEO) standards ranging from 500 to 500 000 g mol−1.
SDS–PAGE
SDS–PAGE was performed using a premixed electrophoresis buffer which contained 25 mM Tris, 192 mM glycine, and 0.1 % SDS (Tris/glycine/SDS buffer) to determine the conjugation efficiency between the BSA and the PNIPAM. A mixture of eight proteins (6.5–200 kDa) (Bio-Rad) was used as molecular weight standards. Samples and protein molecular weight markers were diluted by a pre-made Laemmli sample buffer with reducing agent and then heated at 95°C for 5 min to denature the protein. Commercially available 4–20 % precast polyacrylamide gel, 8.6 × 6.8 cm (W × L) was then loaded with the protein samples and ran at a constant 120 V for 70 min. The samples were stained using Coomassie Brilliant Blue R-250 Staining Solution (Bio-Rad) for 2 h and washed with premixed eluent (ethanol/water/acetic acid = 5 : 4 : 1 (v/v/v)). Gel images were recorded using a Bio-Rad GS-800 calibrated densitometer. Native BSA was also used as a control.
DLS
The particle size distribution and zeta potential of polyion micelles were measured using DLS. The concentration of albumin in relevant samples was 1 mg mL−1, and the concentration of oligonucleotide was 0.2 mg mL−1. The data were obtained using a Malvern Nano-ZS as particle size analyser (laser, 4 mW, λ = 632 nm; measurement angle 12.8° and 175°). Samples were run at least three times at 25°C.
TEM
TEM analysis was performed to investigate the morphology and distribution of the nanoparticles with a Philips CM200 microscope operating at 200 kV. Samples were prepared by placing a drop of solution on carbon-coated copper grids and draining the excess with filter paper. Samples were stained with uranyl acetate (2 % aqueous solution) and then air-dried for 2 h.
HPLC
HPLC analysis was performed on a Shimadzu modular system comprised of a LC-20AD pump, DGU-20A degasser, CTO-20A oven, and SPD-20A UV-vis detector. A Resteck C18 column (5.0 μm bead sizes, 250 × 4.60 mm) was used in all measurements. The mobile phase consisted of a mixture of deionized water with 0.1 % NH4OH and 10 % acetonitrile, operating at a flow rate of 1 mL min−1. The column was kept at 30°C, and elution was monitored at 275 nm.
Results and Discussion
The maleimide terminated PNIPAM was prepared by RAFT polymerization using established procedures. The resulting polymer had a number average molar weight (Mn) of 25 kDa and a dispersity Ð of 1.12. Removal of the furan protecting group was confirmed using NMR analysis and led to a reactive polymer with similar molecular weights (Mn = 25 kDa, Ð of 1.12) (Fig. 1). The resulting polymer had a cloud point of 33°C, close to the LCST values of PNIPAM reported in the literature (32°C).
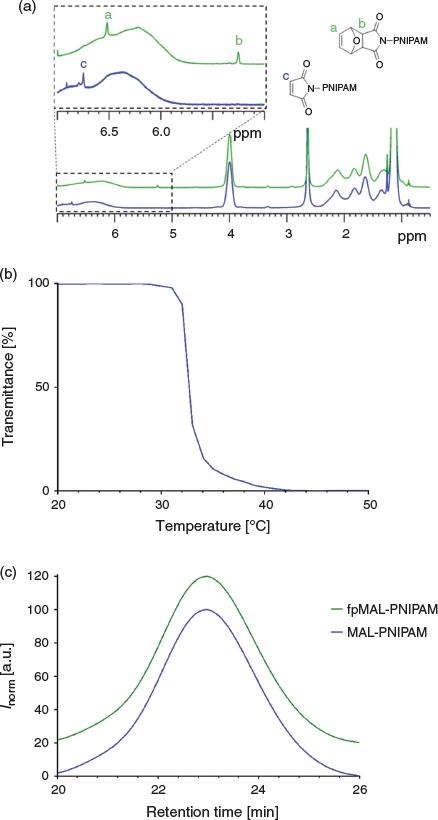
|
The polymers were subsequently conjugated to BSA at an equimolar ratio of BSA and polymer. As commercially available BSA samples may not have all the thiols on the cysteine residues in the reduced form due to dimerization, the amount of available thiol had to be determined using Ellman’s reaction. The fraction of reactive BSA was measured to be 60 %. The successful conjugation was determined using SDS–PAGE, which showed a new band appearing at around 90 kDa, which is in agreement with the expected molecular weight of the polymer–protein conjugate. Aqueous GPC analysis did not reveal a third peak in the higher molecular weight region, possibly due to the molecular weight of the polymer–protein conjugate being over the measuring range of the GPC columns. However, the small peak overlapping with the MAL-PNIPAM sample indicated most of the unreacted PNIPAM polymer was successfully removed by dialysis (Fig. 2).

|
Meanwhile, 5,5′-bisvanillin was synthesized using a mixture of vanillin, Na2S2O8, and FeSO4 in water, which was stirred at 50°C for 5 days.[16] It is also possible to obtain this crosslinker by enzymatic preparation.[18] To evaluate the activity of the compound, model reactions were carried out using 5,5′-divanillin and n-butyl amine. 5,5′-Bisvanillin (0.33 M) was stirred with a slight excess of n-butyl amine (3 equiv.) without the addition of any catalyst at 40°C for 1 h. The almost complete disappearance of the aldehyde signal at 9.8 ppm in the 1H NMR spectrum and the simultaneous appearance of the signal at 8.2 ppm belonging to the newly formed imine functionality confirmed the successful reaction (Fig. 3).

|
5,5′-Bisvanillin can be used to crosslink BSA directly, but here we use the thermo-responsive BSA-PNIPAM as it facilitates monitoring of the crosslinking process. The polymer–protein conjugate, which is fully water soluble at room temperature, will self-assemble into nanoparticles upon heating of the solution. Cooling of the solution results again in the formation of fully water soluble polymers, thus disassembly occurs. Crosslinking of BSA will ensure the structural integrity of the nanoparticle and stable nano-sized BSA-PNIPAM gel particles are created.
The solution of PNIPAM-BSA conjugates in cold water was absent of any nanoparticles due to the solubility of both PNIPAM and BSA in water (Fig. 4). The measured hydrodynamic diameter of around 7–8 nm was indicative of the soluble conjugate. The aqueous solution was subsequently heated above the cloud point of PNIPAM, which led to the formation of amphiphilic BSA–polymer conjugates. Ultimately, nanoparticles with hydrodynamic diameters of around 60–70 nm were formed. Repeated cooling and heating cycles led to disassembly and assembly of the micelle.
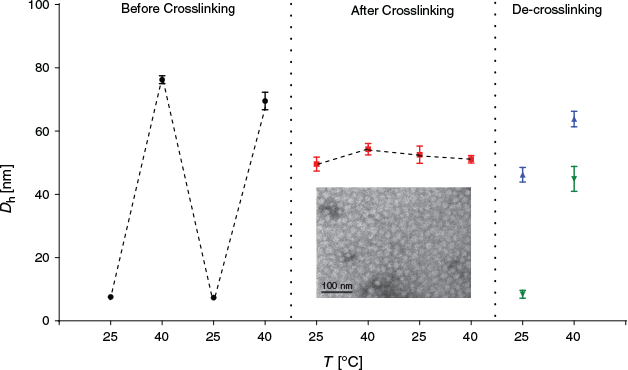
|
The subsequent crosslinking reaction was carried out in phosphate buffer solution at pH 8. However, 5,5′-bisvanillin seems to be barely soluble in aqueous solution and had to be added as a solution in DMSO. This can potentially lead to partial precipitation of the crosslinker. The reaction conditions therefore had to be optimized, which included changes in reaction time, amount of DMSO, molar ratio between crosslinker and BSA, and stirring. The latter was deemed important as the low crosslinker solubility may have led to a dispersion, with the crosslinker slowly settling out from the solution. Successful crosslinking was confirmed using DLS as the crosslinked product should not disassemble upon cooling. Summarized in Table 1, it was found that stirring is indeed necessary. Important is also a relatively high amount of DMSO in solution to ensure crosslinker solubility. Higher amounts of crosslinker were also required for efficient crosslinking. We hypothesize that it is not only the actual ratio that drives the reaction to higher yields, but more the fact that the crosslinker has a low solubility in water. With the addition of more crosslinker and more DMSO that can dissolve the crosslinker, the yield increased. Altogether, it seems that parameters that ensure good contact of the crosslinker with BSA such as stirring and better solubility results in better outcomes.

|
To learn more about the reaction rate, samples were taken from the solution at various time intervals to evaluate the consumption of the 5,5′-bisvanillin crosslinker (Fig. 5). In the absence of stirring, no crosslinker was consumed while stirring led to the reaction of around 40 % of all available crosslinker within 100 min. Considering the crosslinker property of 5,5′-bisvanillin and the 10 to 1 molar ratio of it with BSA, around 4 to 8 crosslinkers are attached per BSA molecule. However, it should be noted that not all 5,5′-bisvanillin molecules will act as crosslinkers as successful bridging between two large BSA molecules has certain solvent accessibility and special requirements for lysine-amines.
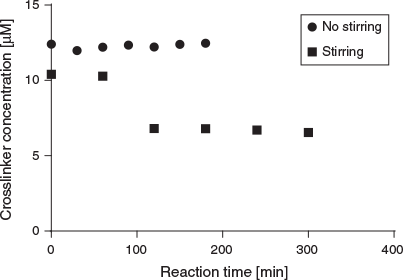
|
Upon crosslinking above the LCST, hydrodynamic diameters of around 50 nm were measured indicating that the crosslinking process may have led to a tighter packing of the polymers. Disassembly of the nanoparticle when cooling below the LCST was now prevented. However, the enhanced solubility of PNIPAM at low temperatures may result in a water-swollen core, thus an increase in measured size.[19] This is not clearly visible during temperature cycling (Fig. 4) suggesting that crosslinking may result in rather stiff structures. The inserted TEM image in Fig. 4 clearly revealed the nanoparticle structure that is stable at low temperatures. The size is around 30 nm, which is significantly lower than that measured by DLS, which is the result of the particle dehydration during the measurement in the dry state. It should be noted here that the micelle like structure as it is depicted in Scheme 1 is unlikely to exist as depicted. The structures obtained are probably nanoparticles with a surface enrichment of BSA while some BSA may also be in the core.
The formed imine functionality may be susceptible to acid catalysed degradation. This feature is often used to trigger the release of payload in an acidic environment such as cancer tissue or the endosomes inside cells.[20] Incubation of the crosslinked albumin nanoparticle in acidic conditions (pH 5.3) revealed however that the particles are stable (Fig. 4). Traces of free BSA-PNIPAM were measured, but this was in some samples not reproducible suggesting that the formed bond is rather stable under these conditions. The used 5,5′-bisvanillin crosslinker is therefore suitable to stabilize protein-based systems against premature disassembly, however probably not for the design of pH responsive nanocarriers.[21]
Finally, it is important to prove the hypothesis that the crosslinker is indeed less toxic than the commonly used glutaraldehyde. As shown in Fig. 6, glutaraldehyde displays toxicity at significantly lower concentrations than 5,5′-bisvanillin for lung cancer cell line A2780. Some toxicity was observed at the highest concentration used after the non-toxic BSA-PNIPAM micelle was crosslinked.

|
Conclusions
We have shown here that 5,5′-bisvanillin is a suitable alternative crosslinker for albumin. It was able to react with amino groups such as that found in lysine and was therefore able to stabilize the self-assembled BSA-PNIPAM conjugate against disassembly. The challenge was however the low solubility of 5,5′-bisvanillin in water. DMSO (7.7 wt-%) was required to ensure sufficient solubility. The need for organic solvents may be the limiting factor of this approach as many proteins are prone to denaturing in the presence of large amounts of DMSO. The resulting imine (Schiff base) was found to be surprisingly stable even under slightly acidic conditions.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgement
The authors thank the Australian Research Council (ARC) for funding.
References
[1] (a) U. Shimanovich, G. J. L. Bernardes, T. P. J. Knowles, A. Cavaco-Paulo, Chem. Soc. Rev. 2014, 43, 1361.| Crossref | GoogleScholarGoogle Scholar | 24336689PubMed |
(b) Y. Jiang, M. Stenzel, Macromol. Biosci. 2016, 16, 791.
| Crossref | GoogleScholarGoogle Scholar |
[2] N. Reddy, R. Reddy, Q. Jiang, Trends Biotechnol. 2015, 33, 362.
| Crossref | GoogleScholarGoogle Scholar | 25887334PubMed |
[3] (a) J. E. Bruce, Proteomics 2012, 12, 1565.
| Crossref | GoogleScholarGoogle Scholar | 22610688PubMed |
(b) G. W. Preston, A. J. Wilson, Chem. Soc. Rev. 2013, 42, 3289.
| Crossref | GoogleScholarGoogle Scholar |
[4] R. Kluger, A. Alagic, Bioorg. Chem. 2004, 32, 451.
| Crossref | GoogleScholarGoogle Scholar | 15530987PubMed |
[5] A. Leitner, T. Walzthoeni, R. Aebersold, Nat. Protoc. 2014, 9, 120.
| Crossref | GoogleScholarGoogle Scholar | 24356771PubMed |
[6] H. M. C. Azeredo, K. W. Waldron, Trends Food Sci. Technol. 2016, 52, 109.
| Crossref | GoogleScholarGoogle Scholar |
[7] S. Binauld, M. H. Stenzel, Chem. Commun. 2013, 49, 2082.
| Crossref | GoogleScholarGoogle Scholar |
[8] I. Migneault, C. Dartiguenave, M. J. Bertrand, K. C. Waldron, Biotechniques 2004, 37, 790.
| Crossref | GoogleScholarGoogle Scholar | 15560135PubMed |
[9] T. Takigawa, Y. Endo, J. Occup. Health 2006, 48, 75.
| Crossref | GoogleScholarGoogle Scholar | 16612035PubMed |
[10] (a) C. Pang, J. Zhang, G. Wu, Y. Wang, H. Gao, J. Ma, Polym. Chem. 2014, 5, 2843.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. Firdaus, M. A. R. Meier, Eur. Polym. J. 2013, 49, 156.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Fache, E. Darroman, V. Besse, R. Auvergne, S. Caillol, B. Boutevin, Green Chem. 2014, 16, 1987.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. Llevot, E. Grau, S. Carlotti, S. Grelier, H. Cramail, Polym. Chem. 2015, 6, 6058.
| Crossref | GoogleScholarGoogle Scholar |
(e) B. G. Harvey, A. J. Guenthner, H. A. Meylemans, S. R. L. Haines, K. R. Lamison, T. J. Groshens, L. R. Cambrea, M. C. Davis, W. W. Lai, Green Chem. 2015, 17, 1249.
| Crossref | GoogleScholarGoogle Scholar |
[11] A. S. Amarasekara, A. Razzaq, ISRN Polym Sci. 2012, 2012, 532171.
| Crossref | GoogleScholarGoogle Scholar |
[12] A. S. Amarasekara, B. Wiredu, A. Razzaq, Green Chem. 2012, 14, 2395.
| Crossref | GoogleScholarGoogle Scholar |
[13] A. C. Boukis, A. Llevot, M. A. R. Meier, Macromol. Rapid Commun. 2016, 37, 643.
| Crossref | GoogleScholarGoogle Scholar | 26800511PubMed |
[14] U. Krings, V. Esparan, R. G. Berger, Flavour Fragrance J. 2015, 30, 362.
| Crossref | GoogleScholarGoogle Scholar |
[15] (a) C. Boyer, J. Liu, L. Wong, M. Tippett, V. Bulmus, T. P. Davis, J. Polym. Sci. A Polym. Chem. 2008, 46, 7207.
| Crossref | GoogleScholarGoogle Scholar |
(b) V. Vazquez-Dorbatt, Z. P. Tolstyka, H. D. Maynard, Macromolecules 2009, 42, 7650.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Li, P. De, S. R. Gondi, B. S. Sumerlin, Macromol. Rapid Commun. 2008, 29, 1172.
| Crossref | GoogleScholarGoogle Scholar |
(d) Y. Fukui, D. Sakai, K. Fujimoto, Colloids Surf. B Biointerfaces 2016, 148, 503.
| Crossref | GoogleScholarGoogle Scholar |
(e) X. Liu, P. Zhou, Y. Huang, M. Li, X. Huang, S. Mann, Angew. Chem. Int. Ed. 2016, 55, 7095.
| Crossref | GoogleScholarGoogle Scholar |
[16] M. Delomenede, F. Bedos-Belval, H. Duran, C. Vindis, M. Baltas, A. Negre-Salvayre, J. Med. Chem. 2008, 51, 3171.
| Crossref | GoogleScholarGoogle Scholar | 18465848PubMed |
[17] K. N. R. Wuest, V. Trouillet, A. S. Goldmann, M. H. Stenzel, C. Barner-Kowollik, Macromolecules 2016, 49, 1712.
| Crossref | GoogleScholarGoogle Scholar |
[18] R. T. Nishimura, C. H. Giammanco, D. A. Vosburg, J. Chem. Educ. 2010, 87, 526.
| Crossref | GoogleScholarGoogle Scholar |
[19] L. Zhang, J. Bernard, T. P. Davis, C. Barner-Kowollik, M. H. Stenzel, Macromol. Rapid Commun. 2008, 29, 123.
| Crossref | GoogleScholarGoogle Scholar |
[20] S. Binauld, M. H. Stenzel, Chem. Commun. 2013, 49, 2082.
| Crossref | GoogleScholarGoogle Scholar |
[21] (a) H. X. Lu, L. Noorani, Y. Y. Jiang, A. W. Du, M. H. Stenzel, J. Mater. Chem. B Mater. Biol. Med. 2017, 5, 9591.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. Taguchi, H. Lu, Y. Jiang, T. T. Hung, M. H. Stenzel, J. Mater. Chem. B Mater. Biol. Med. 2018, 6, 6278.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Jiang, H. Lu, A. Dag, G. Hart-Smith, M. H. Stenzel, J. Mater. Chem. B Mater. Biol. Med. 2016, 4, 2017.
| Crossref | GoogleScholarGoogle Scholar |