

Radiolabelled Peptides: Optimal Candidates for Theranostic Application in Oncology
Andrew J. Hall A and Mohammad B. Haskali A B C
A B C
A Radiopharmaceutical Research Laboratory, The Peter MacCallum Cancer Centre, Melbourne, Vic. 3000, Australia.
B Sir Peter MacCallum Department of Oncology, The University of Melbourne, Melbourne, Vic. 3010, Australia.
C Corresponding author. Email: mo.haskali@petermac.org

Dr Mohammad Haskali is a radiopharmaceutical scientist (RPS) certified by the Australasian College of Physical Scientists and Engineers in Medicine in their A1 limited category. After receiving his PhD degree in the development of research radiopharmaceuticals and radiochemistry methodologies, he gained extensive experience in the radiopharmaceutical industry operating under the code of Good Manufacturing Practice (GMP) before joining the National Institute of Mental Health (NIMH) in the USA as a radiochemist. Currently, Dr Haskali is employed by the Peter MacCallum Cancer Centre in Melbourne where he is involved in radiopharmaceutical science research and the provision of radiopharmaceuticals for clinical use. |
Australian Journal of Chemistry 75(2) 34-54 https://doi.org/10.1071/CH21118
Submitted: 20 May 2021 Accepted: 21 September 2021 Published: 8 November 2021
Journal Compilation © CSIRO 2022 Open Access CC BY-NC-ND
Abstract
Theranostics are drugs suitable for use in both diagnostic and therapeutic applications, and have played an important role in the advancement of modern nuclear medicine. This review explains key elements that are common to successful theranostics and highlights significant developments in the field, including our own. Specific focus is given to peptides and those features that make them most suitable for theranostic application, as well as some key radioisotopes owing to their favourable properties and high clinical utility. This report provides an overview of the techniques at the researcher’s disposal, how they have been applied to current clinically significant targets, and how they might be used and improved upon for future targets.
Keywords: theranostic, radiopharmaceutical, radiolabelling, peptide, foldamers, oncology, RNT, PET, PRRT, radionuclide.
Introduction
Theranostic is a compound word of therapeutic and diagnostic, and describes a key component of nuclear medicine, which uses the same drug for both imaging and treatment. The field of theranostics in oncology has evolved from developments within molecular imaging and radionuclide therapy (RNT), and has had an increasing role in precision medicine owing to its outstanding capacity to identify the heterogeneity of cancers that exists between, and often within, patients.[1]
Molecular imaging describes a suite of non-invasive diagnostic techniques capable of accurately locating and staging various diseases with the goal of directing therapy. Positron emission tomography (PET) imaging is among its most popular techniques.[2] PET imaging utilises positron emitting radiopharmaceuticals (radiolabelled, biologically active drugs) that bind specifically to sites on diseased cells, enabling non-invasive whole body imaging and directing selection of patients suitable for RNT by showing a high uptake of the agent at all sites of disease.[2] Changing the diagnostic radionuclide to a therapeutic radionuclide produces a radiopharmaceutical able to effect RNT, enabling the targeted destruction of metastatic malignancies which cannot be treated efficiently by conventional therapies (Fig. 1).[3] A key benefit of theranostics is the ability to ‘see what you treat’ using the companion imaging agent.
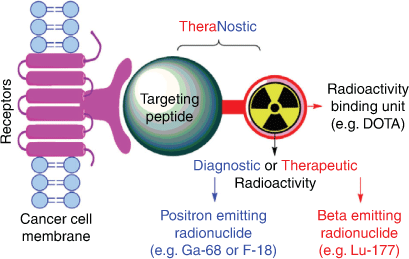
|
Theranostic radiopharmaceuticals are administered as an injection and selectively target tumour cells while minimising the impact on healthy tissue.[3] Successful theranostic agents have high specificity and binding affinity to the target of interest, exhibit good metabolic stability, and have good tumour targeting kinetics (i.e. high/rapid uptake and slow clearance in target tissue, with low uptake and rapid clearance from non-target tissue).[3] They must also have a safe immunogenicity and toxicity profile.[3]
Peptides: The Optimal Targeting Moieties for Theranostic Application
Peptides are compounds of two or more amino acids linked through amide bonds, and are a unique class of pharmaceuticals with distinct biochemical characteristics. They typically present high binding affinity, specificity, and low toxicity. In addition to their role as hormone analogues, peptides can effectively disrupt protein–protein interactions (PPIs), a fundamental process that underlies most cellular activity.[4] These unique characteristics make peptides of growing importance in medicinal chemistry, leading to a rapidly expanding role in the pharmaceutical industry with nearly 20 new peptide-based drugs entering clinical trials every year. A global industry analysis for peptide therapeutics estimated a compound annual growth of 9.1 % from 2016 to 2024.[5]
Peptide-based radiopharmaceuticals afford rapid target accumulation, fast clearance from background tissue, and exhibit good tissue penetration, making them ideal candidates for theranostic applications.[3,6,7] Moreover, peptide synthesis procedures such as solid-phase peptide synthesis are well established, reproducible, amenable to scale up, and enable efficient purification.[6,7]
While proteins such as antibodies can facilitate tight PPIs, they suffer from significant drawbacks including high production and purification costs, limited tissue penetration, obscure mode of action, and low resistance to denaturation, which often prove prohibitive impediments towards their therapeutic applications.[5] Moreover, their long circulation half-life is undesirable for theranostics development, as optimal target-to-background contrast is often only achieved many days after administration, resulting in inadvertent delivery of high doses to non-target tissue.[5]
Although the molecular weight cut-off for small-molecule drugs (< 900 Da) does not exclude small peptides, the recommendations that govern the development of small-molecule drugs can sometimes lead to inferior characteristics for radiopharmaceuticals. The Ghose filter extension on the Lipinski’s rule of five, for example, recommends moderate to high lipophilicity (i.e. a partition coefficient (Log P) of –0.4 to 5.6) as an essential parameter for small molecules to achieve a desirable drug profile.[8,9] While it is important to maintain moderate lipophilicity (i.e. Log P of 2 to 3.5) for radiotracers intended to cross the blood brain barrier for imaging of the central nervous system (CNS), this very same property contributes negatively to the biodistribution of theranostics intended for use in the periphery.[10,11] High lipophilicity affords extensive, undesired hepatobiliary excretion and high non-specific uptake in healthy tissue, which would result in a theranostic with limited imaging and therapeutic utility. Generally, the notion is to reduce the lipophilicity (i.e. a Log P < 0) of theranostics to enhance their tumour-to-background and tumour-to-organ ratios, this holds true for peptide-based radiopharmaceuticals (Table 1). As such, peptides occupy a ‘Goldilocks zone’ between antibodies and small molecules, making them ideal candidates for theranostic application.

|
While hydrophilicity is generally desirable, excessive hydrophilicity can reduce the overall retention of theranostics leading to low tumour accumulation.[22] Overly hydrophilic and highly charged peptides can also lead to high kidney uptake and retention by reabsorption in the proximal tubular cells, resulting in detrimental amounts of radiation being delivered to the kidney.[23] Hence it is important to strike a balance between hydro- and lipophilicity to maintain favourable biodistribution and pharmacokinetic profiles of theranostics. Nevertheless, high kidney uptake and retention of peptide-based radiopharmaceuticals can often be clinically managed by pre- or co-infusion of basic amino acids or plasma expander succinylated gelatin (Gelfusine) that competitively inhibit the renal transport mechanism (glomerular ultrafiltration) leading to lower kidney uptake and retention.[23]
Peptide-Based Theranostics Leading Clinical Work in Nuclear Medicine
Overexpression of a range of regulatory hormone receptors is common to many human cancers, hence investigation of peptide-based hormone analogues has been proven a successful method for discovering tumour targeting theranostics.[24,25] Using peptides to deliver a cytotoxic radiation dose to tumours via binding to overexpressed receptors is a form of RNT more specifically known as peptide receptor radionuclide therapy (PRRT).[26]
Interest in using peptides to deliver radionuclides to tumours was largely driven by the success of OctreoScan in the early 90s to visualise somatostatin receptor subtype 2 (SST-2R)-positive tumours.[27] Subsequent to this agent, many overexpressed receptors have been exploited to specifically target tumours using peptides. Below we briefly discuss three developments related to peptide-based theranostics targeting various receptors and enzymes overexpressed on cancer tissue.
Somatostatin Receptors in Neuroendocrine Tumours
Neuroendocrine tumours (NETs) are a family of cancers which arise from neuroendocrine cells that are dispersed all over the body and have traits similar to both endocrine cells (hormone producing) and nerve cells (neurons). Somatostatin receptors are frequently overexpressed in NETs, specifically SST-2R, enabling the development of theranostics that target SST-2R positive NETs.[27,28] Somatostatin receptors are a type of G-protein coupled receptor that bind the somatostatin hormone, specifically somatostatin-14 and somatostatin-28.[29] The activation of somatostatin receptors by its native peptides induce a variety of physiological responses, which includes inhibiting the secretion of many other hormones throughout the body. However, native somatostatin-14 has a low metabolic half-life (2–3 min) making it unsuitable for theranostic application, hence research focussed on developing more metabolically stable analogues.[29–31] Octreotide, an eight amino acid analogue of somatostatin-14, was the first long acting analogue in clinical use with high SST-2R affinity and selectivity.[27]
Octreotide was initially modified with diethylenetriaminepentaacetic acid chelator (DTPA) at the N-terminus, as the C-terminus is involved in receptor recognition, and radiolabelled with In-111.[31,32] This was then used with whole body single photon emission computed tomography (SPECT) imaging for diagnostic identification of carcinoids, pancreatic neuroendocrine tumours, and to localise sarcoidosis.[31,32] SPECT imaging using In-111 labelled octreotide was later known as OctreoScan, and the success of OctreoScan in the clinic spurred extensive interest in developing superior analogues. Notably, replacement of the C-terminal alcohol in octreotide with the natural amino acid threonine led to octreotate, an analogue with very high affinity for SST-2R and increased tumour uptake.[33] Furthermore, replacement of Phe3 with the more electron rich Tyr3 led to [Tyr3]octreotate, which demonstrated enhanced reactivity for electrophilic radioiodine (I-125 or I-123) for SPECT imaging.[34] Subsequently, [Tyr3]octreotate was modified with a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelator at the N-terminus and radiolabelled with Ga-68 and Lu-177 to afford optimal theranostics targeting SST-2R (Fig. 2).[35–37]

|
PET imaging using the Ga-68 labelled DOTA-[Tyr3]octreotate and PRRT using the Lu-177 labelled variant have since become the standard of care diagnostic and therapeutic procedures respectively for management of neuroendocrine tumours (NETs) (Fig. 3).[38] The recent randomised phase III clinical study NETTER-1 showed that PRRT using four cycles of [177Lu]Lu-DOTA-[Tyr3]octreotate improved objective response, quality of life, progression free survival, and had an overall survival benefit over long-acting-release double-dose octreotide alone.[39]
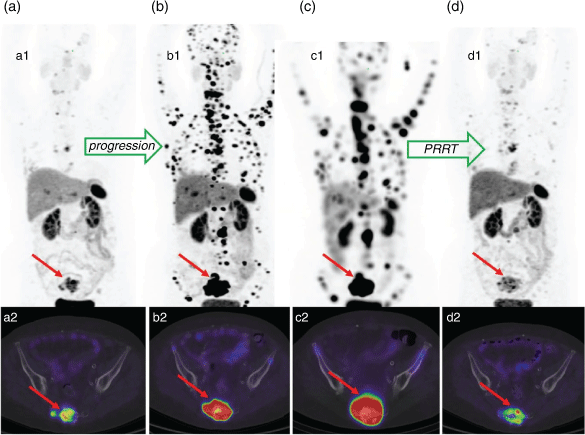
|
Prostate Specific Membrane Antigen in Advanced Prostate Cancer
The prostate specific membrane antigen (PSMA) is a type II transmembrane protein. PSMA acts as a glutamate carboxypeptidase responsible for the sequential hydrolysis of the C-terminal glutamate residues from folate polyglutamate, as well as the hydrolysis of the abundant brain peptide N-acetyl-l-aspartyl-l-glutamate.[40,41] While PSMA is expressed throughout prostate tissue, its expression increases by 100–1000 fold in prostate cancer, making it an excellent target for theranostic application.[42]
In 1996, Jackson et al. developed various high affinity agents targeting PSMA for the treatment of neurological conditions.[43] This work was followed by Kozikowski and co-workers that developed the first potent urea-based inhibitors of PSMA.[44] This urea based scaffold formed the basic structure of all subsequent PSMA avid theranostics in clinical use.[45] Fig. 4 presents some of the most common PSMA targeting theranostics currently in clinical use that employ a myriad of chemical strategies to link the urea based scaffold to the radionuclide binding component. These linkers may serve to improve the pharmacokinetics,[46] biodistribution,[47] cellular internalisation or receptor affinity of the molecule,[48] or provide optimal spacing between the radionuclide and the binding component.[49] Notably, the Ga-68 and F-18 labelled PSMA inhibitors [68Ga]PSMA-11 and [18F]DCFpyL (marketed as PYLARIFY) respectively have both been approved by the Food and Drug Administration (FDA) for use in the diagnosis of prostate cancer.[50]

|
PSMA theranostics are rapidly evolving as a new standard of care for prostate cancer management. Hofman and co-workers from the Peter MacCallum Cancer Centre in Australia and others generated an outstanding body of practice-changing evidence demonstrating the transformative role of PSMA theranostics in the clinic.[51,52] Diagnostic PSMA PET/CT proved to be superior to conventional imaging techniques with higher accuracy that ultimately led to improved clinical management of patients.[51] PSMA therapy using theranostics demonstrated striking responses (Fig. 5) in men with metastatic castrate resistant prostate cancer who had progressed after treatment with conventional therapies.[52,53]

|
Cholecystokinin-2 Receptors in Neuroendocrine Tumours
Cholecystokinin (CCK) receptors are a group of G-protein coupled receptors with high binding affinity for the hormone gastrin.[54] CCK receptors regulate the release of gastric acid in the stomach and play a vital role in the development of the gastrointestinal tract.[54] Importantly, activation of a subset of CCK receptors known as CCK-2R, has been shown to contribute significantly in the development of many malignancies.[55,56] 54.5% of all NETs patients with negative SST-2R expression were found to overexpress CCK-2R.[57] In particular, CCK-2R are overexpressed on medullary thyroid carcinoma, and a neuroendocrine type of lung cancer known as lung carcinoids.[54]
[111In]In-DOTA-DGlu-MG0 is a 13-residue peptide-based on the physiologically active form of the gastrin hormone (G17), it exhibits high binding affinity to CCK-2R, comparable to the parent gastrin hormone.[58,59] However, when therapeutic doses of DGlu-MG0 based theranostics were administered, extensive proteolytic degradation, as well as high kidney uptake and retention was observed, leading to nephrotoxicity (toxicity in the kidney).[60,61] The high, non-specific (i.e. not CCK-2R mediated) kidney uptake was attributed to the N-terminal polyglutamate (Glu2–6), as a result of its high anionic charge.[58] This issue was addressed in the next generation CCK-2R binding peptide, known as CP04 (also known as PP-F11), wherein these Glu2–6 residues were substituted with unnatural d-Glu amino acids (Fig. 6).[59] Subsequent pharmacokinetic studies demonstrated superior stability, decreased kidney uptake, and improved tumour-to-kidney (T/K) ratios, which prompted a European conglomerate to select CP04 as the ‘gold standard’ for targeting CCK-2R in humans.[59,62,63] However, the methionine-11 residue of CP04 was found to be particularly susceptible to radiolytic (catalysed by radiation induced radicals) oxidation affording peptides of lower radiochemical purity. Substitution of Met11 with norleucine afforded PP-F11N (Fig. 6) with improved radiochemical stability and similar biological properties to CP04.[62,63]

|
Our clinical experience with [68Ga]Ga-DOTA-CP04[57] confirms its excellent diagnostic properties (Fig. 7) but disputes its potential for therapeutic application, as its Lu-177 labelled counterpart has been observed to afford suboptimal radiation doses to CCK-2R positive tumours. Von Guggenberg recently reported other truncated analogues of CP04 affording MGS5 with further improvements in metabolic stability and tumour targeting (Fig. 6).[64] [68Ga]Ga-DOTA-MGS5 is currently undergoing clinical translation, demonstrating the concept of CCK-2R PET imaging in patients with MTC.[65]
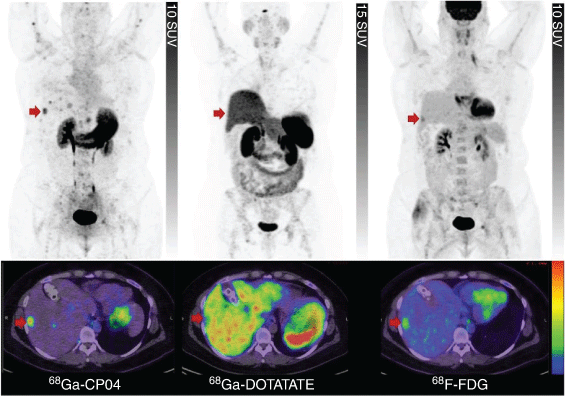
|
There are many other peptide-based diagnostics and/or theranostics currently being investigated in clinical works (Table 2). Detailed description of all theranostics in clinical use is beyond the scope of this review. Our aim is only to show a few well-established examples for peptide-based theranostics impacting clinical management of cancer patients.

|
Constrained Peptides and Foldamers: Optimal Constructs for Theranostics Development
Small peptides are flexible in nature and often do not form highly ordered structures akin to those found in the larger proteins and biomolecules from which they are isolated. The development of effective targeting peptides requires detailed mimicking of the PPI contact points, which often necessitates the recapitulation of not only the primary sequence but also the secondary structure.[71] Small peptides can be evolved through the design of constrained peptides and foldamers to form complex structures that have high binding affinities and prolonged target engagement akin to large biologics, yet they exhibit favourable physiochemical characteristics akin to small molecules, affording ideal theranostics.[72,73]
Samuel Gellman coined the term ‘foldamer’ to describe ‘any polymer with a strong tendency to adopt a specific compact conformation’ without the need for additional structural constraints such as cyclisation or stapling.[74,75] While constrained peptides can include any peptide with conformational constraints (including foldamers) it typically refers to monocyclic, bicyclic, or stapled peptides.
Constrained peptides and foldamers provide several advantages including the ability to design secondary structures, control over the orientation of side-chain functional groups and resistance towards proteolytic degradation.[76] Moreover, constrained peptides and foldamers address the thermodynamic basis of PPIs by minimising conformational degrees of freedom, thereby minimising the entropic penalty paid upon binding, resulting in higher binding affinities (Fig. 8).[71]

|
Secondary structures emerge as a result of the φ and ψ torsion angles between the N-Cα and Cα-C positions within the polypeptide backbone respectively.[77] The range of these angles within a peptide determines its flexibility and ability to adopt certain folds. The two major secondary structures that influence PPI are α-helices and β-turns. These conformations can be stabilised within constrained peptides and foldamers by careful restriction of the φ and ψ torsion angles of the peptidic backbone.[72]
Cyclisation is one useful method commonly employed to generate constrained peptides which mimic β-turn conformations, generally affording theranostics with higher binding affinity and metabolic stability.[78] Head-to-tail amide bond formation is one of the most common methods utilised to generate monocyclic peptides.[79] Furthermore, hetero-chiral amino acids are often used within the turn regions of cyclic peptides in order to mimic the type I’ and type II’ reverse β-turns which are particularly abundant in β-hairpin peptides.[80] Cyclic RGDfK and RGDyK based radiopharmaceuticals are examples of peptides utilising head-to-tail cyclisation through amide bond formation, which also include a hetero-chiral d-phenylanaline/d-tyrosine carefully incorporated in their sequence to mimic a type II’ reverse β-turn (Fig. 9).[81,82]

|
The CXCR4 antagonist, PentixaFor, is another example of a cyclic peptide with enticing potential for theranostic application.[16,83] PentixaFor makes use of head-to-tail amide-bond cyclisation and inclusion of the hetero-chiral d-ornithine.[16] The backbone of PentixaFor is further constrained by N-methylation of d-ornithine to more closely mimic a β-hairpin conformation (Fig. 9).[16,83]
Another common cyclisation strategy is to form disulfide bridges between two cysteine residues, octreotide/octreotate (Fig. 2) are examples of successful theranostics cyclised via disulfide bridges.[31] Moreover, inclusion of the hetero-chiral d-tryptophan in the β-turn region of octreotide/octreotate is able to closely mimic the well defined β-hairpin fold within the Somatostin-14 hormone, leading to their excellent pharmacological properties.[84]
Bicyclic peptides have even greater conformational rigidity and metabolic stability compared with monocyclic peptides.[85] They are typically generated by reacting unprotected cysteine residues with activated electrophiles of a planar aromatic scaffold (Fig. 10).[86–88] Bicyclic peptides are expected to realise the next generation of therapeutics, capable of binding to some of the most challenging targets, but are currently underexplored as moieties for radiopharmaceutical development as methods to rationally develop these structures for cancer specific targets are limited and largely restricted to specific forms of phage display libraries.[85]

|
Maecke and co-workers developed a Ga-68 labelled bicyclic peptide dubbed AM3 for imaging of SST-2R receptors (Fig. 11).[89–91] AM3 demonstrated excellent imaging properties, with high receptor affinity and agonist potency.[89] The design of AM3 retains the octreotide ring, but also includes head-to-tail cyclisation of the terminal residues: arginine and 2,4-diaminobutyric acid.[89] More recently, Teufel and co-workers developed Ga-68 labelled bicyclic peptides targeting the tumour-overexpressed matrix metalloproteinase (MT1-MMP) (Fig. 11) using phage display.[92] Their agents exhibited good tumour penetration, high potency and excellent retention in tumour tissue.[92] These few but compelling examples demonstrate that bicyclic peptides can afford high-contrast theranostic agents. Further research and development is warranted in this sphere.

|
However, cyclisation is not always effective in affording the desired conformation. This is particularly true when a linear peptide is required for functionality, as is the case for many theranostic targets including those peptides which target CCK-2R,[60] Bombesin (BB) receptors[93] and others. Similarly, cyclisation impedes the formation of other important conformations necessary for receptor recognition such as helices in peptides targeting the Glucagon-like peptide 1 (GLP-1), another important target for PET imaging of insulinomas.[94] As such, alternative chemical strategies are needed to accommodate the numerous conformational possibilities and requirements.
Peptide stapling is one approach employed to stabilise α-helical structures which are essential for many PPIs that are otherwise deemed ‘undruggable’ with conventional small molecules.[95] Crosslinking of residues opposite to the binding face in an (i, i + 3), (i, i + 4), or (i, i + 7) arrangement propagates α-helix formation by accommodating the required spatial configuration.[72] This crosslinking can be achieved through a variety of techniques, some typical examples include: the selective inclusion of reactive side-chain functional groups,[96,97] lactamisation,[98] or positioning cysteines in an (i, i + 4) arrangement which can subsequently be cross-linked using a variety of linking groups such as a perfluoroaromatic scaffold (Fig. 12).[99–101] Stitching is able to further stabilise the helical conformation by stapling three residues in an (i, i + 4, i + 7), or (i, i + 4, i + 11) arrangement via ring-closing metathesis to the relevant side chains.[102] Unfortunately, stapling remains very much underexplored for theranostics development, as most peptide-based theranostics currently available are derived from native-hormones or enzymatic substrates that do not adopt α-helical structures.[72,103] However, α-helical structures are one of the most abundant structural motifs responsible for PPIs and as such it is likely many emerging novel peptides targeting new proteins and enzymes will need to emulate these helical conformations for effective binding.[72,103] This leads to an enormous untapped potential for the development of next generation theranostics for oncology relevant biological targets.
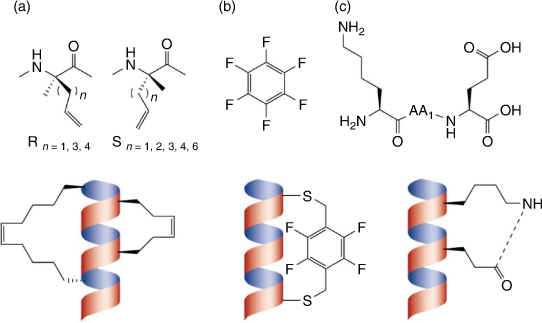
|
Foldamer design is another enticing approach to afford conformational constraints and topologies beyond cyclisation or stapling. There are many different strategies available to achieve desired conformational outcomes when designing foldamers. One common strategy to generate stabilised helical conformations involves α/β hybrid foldamers that contain a combination of natural α-amino acids and unnatural β-amino acids.[74,104] The substitution pattern of β-amino acids has been found to have a profound effect on folding.[74,104] This strategy has been used successfully to synthesise high potency GLP-1 analogues, wherein β-residues where substituted in various positions to enhance α-helix formation.[94] Introduction of the ring-constrained β-residues trans-2-aminocyclopentane-1-carboxylic acid at positions 26 and 30 and trans-4-aminopyrrolidine-3-carboxylic acid at position 34 of GLP-1 yielded an analogue with enhanced helical conformation (as determined by circular dichroism spectrometry) resulting in improved affinity (0.7 ± 0.1 nM) over that of the native GLP-1 peptide (1.6 ± 0.2 nM).[94]
β-Turns and β-hairpin motifs can also be mimicked closely by foldamers without the need for cyclisation.[72] There are a variety of β-turns (including reverse β-turns) formed by different i + 1, i + 2 turn inducing residue combinations, distinguished by their torsional angles.[72] As previously mentioned, β-hairpin structures are typically constructed of type I’ and type II’ reverse turns which necessitate the inclusion of a heterochiral amino acid.[80] Thus, foldamers that mimic a hairpin conformation include a heterochiral residue within their i +1, i + 2 turn region. The following combinations of turn inducing amino acids have been found to be particularly effective in inducing β-hairpin structures: DPro-Gly, DPro-LPro, and any combination of N-methylated Dresidue followed by an N-methylated Lresidue.[105,106] It is also common to incorporate these turn inducing modifications followed by head-to-tail cyclisation to further stabilise a β-hairpin conformation leading to cyclic foldamers with extensive non-covalent intramolecular interactions.[107]
Another useful strategy to generate β-hairpin foldamers is to use turn mimetics or ‘tryptophan zippers’.[108] Tryptophan substituted on opposite sides of a β-turn form cross-strand interaction through their indole rings which stabilised the β-turn and ultimately the β-hairpin structure.[108] Wang et al. demonstrated improved affinity of a programmed death-ligand 1 (PD-L1) inhibitor when two residues were substituted for tryptophan in order to stabilise a β-hairpin central to the peptide.[109]
We have recently introduced the concept of foldamers for theranostics development by preparing linear peptides targeting CCK-2R with enhanced folding capacity affording possible β-hairpin structures.[110] These foldamers were designed using a combination of backbone constrains including N-methylation of key amino acid residues.[110] The conformation of these foldamers were further stabilised by enhancing their amphiphilic structure via substitution of the C-terminus residue with the highly non-polar 1-napthylalanine residue.[110] Our foldamer based radiotracers afforded pico-molar affinity for CCK-2R, presented improved metabolic stability, enhanced cell internalisation, and up to 6-fold increase in tumour uptake compared with the clinically utilised CP04 (Fig. 13).[110] We anticipate that the design of foldamer based theranostics can establish a new paradigm in radiopharmaceutical sciences.

|
Selection of Radioisotope and Radiolabelling Strategy: Considerations when Preparing Theranostics
Research on developing methods for radiolabelling peptides with a variety of radionuclides has intensified in recent years, affording an impressive arsenal of strategies to prepare theranostics from new peptide scaffolds (Table 3).[111] In general, radiolabelling strategies can be parsed into two broad chemistry disciplines, ionic (inorganic) and covalent (organic) radiochemistry.[111] Ionic radiolabelling methods comprise chelation chemistry of radiometals such as Ga-68, Lu-177, and others.[111] Covalent radiolabelling methods include C-11 radiochemistry, and direct radiohalogenation reactions such as those that use F-18 and radioisotopes of iodine.[111]

|
Diagnostic agents utilise radionuclides with relatively short half-lives and a high proportion of detectable or very low linear energy transfer (LET) emissions. Ideally this comprises low momentum positrons for PET or photons of around 140 keV for SPECT, in order to produce the highest quality images while minimising damaging emissions. F-18 and Tc-99m are the prevalent radionuclides utilised in PET and SPECT respectively, due to their favourable emissions, chemistry, and availability.[112,113]
Therapeutic agents utilise radionuclides with moderate half-lives whose decay produce almost entirely therapeutically relevant emissions with suitable characteristics, including α-particles, high LET β-particles, and Auger and Coster-Kronig electrons, whose short path lengths restrict damage within close proximity of the target site. Lu-177 is one popular example, as its decay to Hf-177 always emits a β-particle of therapeutically relevant energy: Eβ(max) = 498.3 keV (79.3 % of decays), 385.3 keV (9.1 % of decays), and 177.0 keV (11.6 % of decays), these LET particles only travel a short distance (1 mm), resulting in minimal damage to surrounding healthy tissue.[114] The excited Hf-177 nucleus resulting from the two lower-energy transitions subsequently results in some useful decay photons. Of particular value is the 177.0 keV 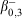 transition that yields an excited Hf-177 nucleus capable of a
transition that yields an excited Hf-177 nucleus capable of a 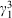 transition that produces 208.4 keV photons, enabling crude SPECT imaging 24 h after administration (Fig. 3).[114] Lu-177’s half-life is also well suited for therapeutic purposes, providing a consistent dose to the target tissue for an extended period.
transition that produces 208.4 keV photons, enabling crude SPECT imaging 24 h after administration (Fig. 3).[114] Lu-177’s half-life is also well suited for therapeutic purposes, providing a consistent dose to the target tissue for an extended period.
Ideal theranostics would make use of different radioisotopes of the same element such as I-124/I-131, Cu-64/Cu-67, and Tb-152/Tb-161, as they would afford agents with identical chemical properties. However, due to numerous issues including limited availability, challenging chemistry, dosimetry problems, or unfavourable biodistribution such combinations are uncommon. Cu-67, for example, suffers from a lack of consistent high activity production,[115] similarly Tb-152/Tb-161 are currently only produced in very few laboratories. While I-124/I-131 agents are often vulnerable to deiodination leading to unwanted accumulation of the free radionuclide in the thyroid,[116] and I-131 itself has some unwanted high energy γ-emissions[117] resulting in patient radiation shielding issues. Hence, most modern theranostics utilise radionuclides of different elements that have comparable behaviour in vivo, a key example is the use of Ga-68 in PET and its therapeutic pair Lu-177 which share similar chelating methods.[118] Such radionuclide pairs are key to theranostic agents and allow for a diagnostic to predict how suitable the equivalent therapeutic will be for treating a given clinical indication and enable guiding dosimetry.[118]
Furthermore, there are successful clinical examples of theranostics for which the diagnostic form is profoundly different in chemical structure and labelling strategy to that of its therapeutic counterpart, yet retains high efficacy in predicting therapeutic response. Both forms of these theranostics target the same receptors, and typically utilise the same targeting scaffold. A key example includes the use of [68Ga]Ga-PSMA-11, [18F]DCFPyL, or [18F]PSMA-1007 as diagnostic equivalents to direct [177Lu]Lu-PSMA-617 or [177Lu]Lu-PSMA-I&T therapy, all of which use the same PMSA targeting moiety but utilise various linkers, chelators, and radionuclides.[52,119–121] This highlights that identical radionuclides and chelating strategies are not always essential when informing clinical decisions, and opens up opportunities for more versatile theranostic radionuclide pairs, such as F-18 and Lu-177.
Although not all targets are as accommodating, hence the choice of radionuclide and radiolabelling strategy need to be fastidiously selected and implemented as they can have as large an influence on the binding affinity, structural conformation, and bio-distribution as the targeting moiety itself. Bifunctional chelating agents (BFCAs) employed for radiometalation of molecules form a significant proportion of their final complexes, e.g. the routinely employed chelator, DOTA, introduces ~400 Da to peptides that are typically smaller than 2000 Da. As such, BCFAs can impact a variety of biological and pharmacological factors of theranostics including the binding affinity and receptor subtype specificity.[16] Hence the use of BCFAs often necessitates inclusion of a linker to optimally space the targeting molecule from the chelator.[122–125]
PentixaFor is an example of a peptide that is severely impacted by the nature of BCFAs and the radiometal used.[16,83] PentixaFor loses its tumour targeting and presents reduced CXCR4 binding affinity when the chelator DOTA is substituted with other chelators such as 1,4,7-triazacyclononane-N,N,N-triacetic acid (NOTA), diethylenetriamine-N,N,N′,N′′,N′′-pentaacetic acid (DTPA), and 2-(4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecan-1-yl)pentanedioic acid (DOTAGA).[16] Furthermore, PentixaFor also demonstrates a loss in tumour targeting with a DOTA chelator when the Ga-68 radionuclide is exchanged with other radiometals including Lu-177, Y-90, and In-111, or when the chelator is left free of metal.[16,83] This is a rather severe circumstance that highlights the detrimental influence of even minor modifications associated with radiolabelling strategies of theranostics. Consequently, the change from one radiometal to another should not be assumed to afford equivalent radiopharmaceuticals, even if the chelation chemistry of the same peptide remains consistent. Never the less, Ga-68 and F-18 labelled radiopharmaceuticals targeting SST-2R and PSMA have been used successfully to stage and select patients, as well as determine the required dosimetry, to treat with their Lu-177 labelled counterparts.[52,118–121,126] This indicates that the use of Ga-68 and F-18 labelled agents to predict biodistribution and therapeutic response to Lu-177 therapy is generally a reliable method but should be assessed carefully.[118,120,126]
There are several clear advantages associated with the use of chelation chemistry for the radiolabelling of theranostics, including:[127–129]
-
High labelling efficiency: Effective complexation of radiometals leading to high radiochemical yields (> 90 %) achieved using relatively small amounts of peptide precursor (typically below 50 µg).[127] The high radiochemical yields, small amount of peptide precursor required, and simplicity associated with BFCA chelation chemistry often means kit-based production of radiopharmaceuticals can be established, eliminating the need for troublesome semi-purification processes.
-
BFCAs increase hydrophilicity and introduce positive charge upon chelation of a radiometal, which jointly improve biodistribution with desirable renal excretion.[127,128]
-
Some BFCAs are amenable to radiolabelling with a variety of radiometals including both diagnostic and therapeutic radiometals, in which case the same precursor can potentially be used for PET, SPECT, and RNT.[127–129] DOTA is one particularly useful and widely employed chelator that has the ability to complex several metal radionuclides (including Ga-68, In-111, Lu-177, Y-90, Ac-225, and others) with high stability.[128,129]
While many radionuclides are available for the preparation of theranostics, the limited availability and half-life characteristics of many impede their wide utility. As such, theranostics that become high-demand radiopharmaceuticals often necessitate the move to specific radionuclides. Ga-68 is a diagnostic radiometal widely used to prepare theranostics as Ga-68 generators are readily available. Ga-68 also has a favourable emission profile that primarily consists of a moderate maximum energy β+ (1.90 MeV, 88 %) and only one high energy γ (1.08 MeV, 3.2 %),[130] and has a reasonable stability constant for a range of common chelators.[131] One key advantage of Ga-68 generators is that they can be housed in hospital environments with little infrastructure enabling wide PET imaging capability. However, Ga-68 generators typically afford less than 3.7 GBq (and commonly under 2 GBq) of activity and this yield reduces over time as the parent Ge-68 decays and the generator degrades.[132] As such, production from one elution of a Ga-68 generator is typically sufficient to afford only 3–4 patient doses at peak performance. Recent developments have led to cyclotron production of Ga-68 using either liquid targets (comprising a [68Zn]Zn(NO3)2 solution)[133] or solid targets (highly enriched metallic zinc-68, usually electroplated onto discs made of pure copper or silver).[134] Irradiation of a liquid target can afford between 2–10 GBq of activity after 1 h of bombardment,[133,135–137] while solid targets can generate multi-curies of activity (up to ~370 GBq) in a period of 1–2 h.[138,139] While these methods don’t require build-up time between runs, both necessitate purification of the resulting Ga-68 before clinical use.[135,139] Moreover, the 68 min half-life[130] of Ga-68 inhibits transportation to distant sites, thereby limiting the availability of Ga-68 labelled theranostics to local hospitals or those with in-house production capacity. As such, theranostics that achieve demand by a large population of patients necessitate radiolabelling with more flexible and widely available diagnostic radionuclides, such as fluorine-18, in order to scale up production and distribution effectively. F-18 is currently the workhorse radionuclide for PET imaging as it can be produced in significant quantities from low energy cyclotrons, with up to ~1000 GBq able to be produced over a 5 h bombardment.[140] F-18 possesses a half-life (110 min)[114] long enough to make its radiolabelled products suitable for transportation, but also short enough that it undergoes more than 10 half-lives to reach background levels within 24 h, thereby limiting a patients radiation exposure.
We will now discuss in some detail the methods for radiometalation (relevant to Ga-68 and Lu-177) and radiofluorination of peptides to generate theranostics.
Ga-68 and Lu-177 Radiolabelling Methods for the Preparation of Theranostics
There are two general classes of BFCAs for the coordination of the radionuclides Ga-68 and Lu-177: macrocyclic and acyclic chelators (MCC and ACC respectively).[128] While ACCs generally have faster metal-binding kinetics, MCCs produce complexes that are more kinetically inert due to their pre-organised conformations.[141] Kinetic inertness of the radiometal complex is often more relevant than thermodynamic stability for successful in vivo application of theranostics.[128,129] MCCs generally require heating and a higher precursor concentration to coordinate radionuclides efficiently.[128,129] Heating a reaction mixture in the presence of ionising radiation often catalyses radiolysis (i.e. decomposition of molecules by ionizing radiation).[142] However, there are well established procedures available to combat radiolytic breakdown of peptides.[143–145] All things considered MCCs are the better suited choice for most Ga-68 and Lu-177 labelling.[129]
The development of BFCAs for the coordination of Ga-68 and Lu-177 are governed by the properties of the corresponding metal. Gallium is a metal that belongs to group-13 of the periodic table with an ionic radius of 47–62 pm.[146,147] Gallium exists as free hydrated Ga3+ ion in acidic aqueous solution but can form insoluble gallium hydroxide (Ga(OH)3) within neutral or mildly acidic solutions.[128,147] At pH > 7 gallium hydroxide redissolves by forming (Ga(OH)4)−.[147] Gallium is classified as a hard Lewis acid thus favouring the formation of pseudo-octahedral complexes with Lewis bases, such as nitrogen and oxygen.[128,129,147] In general, carboxylic and amino groups form strong six-coordinate complexes with gallium that are thermodynamically stable and kinetically inert at physiological conditions.[128,129,147] Many acyclic and cyclic polyaminopolycarboxylic ligands have been investigated as scaffolds for Ga-68 coordination, including those presented in Fig. 14.[148–154] Of particular interest are the cyclic chelators NOTA and DOTA for Ga-68 labelling (Fig. 14).[149,155] The Ga3+ ionic radius is uniquely suited for use with NOTA, affording stable gallium complexes rapidly at room temperature.[149,156,157] However, DOTA remains the chelator of choice for Ga-68 as it also lends itself efficiently for use with Lu-177 and other radiometals, enabling PET imaging and RNT using the same chelation chemistry.[128,148]

|
Lutetium on the other hand is the last member of the lanthanide series with 71 electrons configured as 4f145d16s2. Similar to gallium, lutetium generates +3 metal cationic species through the loss of the two outermost electrons in the 6s orbital as well as its single 5d electron.[158] Extraordinarily, Lu3+ exhibits the smallest ionic radius (86–103 pm)[146] of any lanthanide ion (Ln3+) due to the lanthanide contraction phenomenon.[158] Like Ga, Lu exhibits hard Lewis acid properties favouring complexation with hard donor atoms like oxygen and nitrogen, but forms thermodynamically stable complexes with coordination numbers 8 or 9.[158] In particular, the DOTA chelator binds Lu3+ efficiently to form a nine-coordinated complex (including the complexation of a water molecule)[158] with high thermodynamic stability and kinetic inertness, making it the BFCA of choice for Lu-177 radiometalation.[148,152,154] Other commonly used acyclic and cyclic BFCAs for radiometallation have also been investigated for Lu-177 chelation with less success, affording lower stability constants (log KML) and lower kinetic inertness than DOTA (Fig. 14).[148,152,154]
Finally, it is important to refer briefly to the conjugation chemistries commonly employed to link BFCAs to peptides. BFCAs are typically attached to peptides through the reaction of one of their carboxylates with a free amine available on the corresponding peptide to form a stable amide bond. Phenyl isothiocyanate functionalised BCFAs are another common intermediate attached to peptides via reaction with the ϵ-amine of a lysine residue on a peptide to form chemically stable thioureas.[159] One major shortcoming associated with this conjugation strategy is that thiols are commonly prone to radiolytic oxidation, affording radiopharmaceuticals of compromised purity and radiolytic stability.[110,160,161] As such when possible the use of isothiocyanate containing chelators should be avoided.
Fluorine-18 Radiolabelling of Peptides
F-18 was first produced by Snell in 1936[162] and it found rapid utility in health-related studies only 4 years after its discovery.[163] The ideal chemical and nuclear characteristics of F-18 resulted in a surge in PET diagnosis and pharmacological research. Moreover, it exhibits ideal physical properties for PET imaging with a high positron decay ratio (97 %) and a low maximum positron energy (634 keV) that leads to a short diffusion range (< 2.4 mm), enabling the highest-resolution images.[114] These favourable characteristics have led to increased developments in radiofluorination methods for peptides.[164,165] However, labelling peptides with F-18 is generally challenging and can result in a more lipophilic product with undesirable biodistribution.[166] This increased lipophilicity necessitates the use of further chemical modifications that can compensate and improve the overall performance of the resulting diagnostics.[123,167] Some commonly employed strategies to increase the hydrophilicity and improve the biodistribution of overly lipophilic theranostic peptides include PEGylation[124] and glycosylation.[123,167] We have previously established a novel means of increasing the hydrophilic nature of radiofluorinated small cyclic peptides through the site specific sulfonation of tyrosine residues.[168] Furthermore, we demonstrated the superiority of sulfonation in comparison to glycosylation or PEGylation when applied to cyclic RGD peptides (Fig. 15).[168]

|
The most common method for producing F-18 involves the bombardment of oxygen-18 enriched water with protons, expelling a neutron to generate F-18. This nuclear reaction is depicted as 18O(p,n)18F and it generates F-18 in high molar activity (Am – the ratio between radioactivity and amount of substance in moles).[169] A high Am is of paramount importance to avoid toxicity and saturation of receptor binding sites by the competing non-radioactive F-19 containing counterpart. The 18O(p,n)18F nuclear reaction affords [18F]fluoride ions tightly hydrated (ΔHhydr = 506 kJ mol−1) in the oxygen-18 enriched water, rendering it unreactive towards nucleophilic reactions.[170] Therefore, F-18 is typically processed in many ways after isolation from the cyclotron to afford an anhydrous form of ‘naked’ fluoride-18 ions that are highly reactive and readily available for nucleophilic reactions.[170,171]
Another common method used to generate F-18 involves the bombardment of neon gas containing 0.1–2 % of F2 gas with deuterium ions in order to expel an α-particle, leading to [18F]F2. This nuclear reaction is represented as 20Ne(d, α)18F. [18F]F2 is a highly reactive form of electrophilic F-18 that is obtained in low molar activity due to the carrier F2 gas used to recover it from the target.[172] This and the noxious nature of F2 gas used in the process makes the use of electrophilic F-18 an undesirable approach for the radiofluorination of peptides.
Therefore, nucleophilic radiofluorination is the most significant route for obtaining F-18 labelled compounds and peptides. The use of non-carrier added (NCA; affording high Am) fluoride-18 ions for nucleophilic substitution reactions of aliphatic systems progresses through an SN2-mechanism, involving the substitution of an appropriate leaving group such as a bromine or a tosylate, resulting in stereochemical inversion.[170] Nucleophilic aromatic radiofluorination is another important class of reactions that is widely employed for the preparation of radiopharmaceuticals, resulting in excellent chemical and metabolic stability at the [18F]F-aromatic bond. Nucleophilic aromatic radiofluorination is, however, a more challenging task, requiring the use of aromatic molecules activated by strong electron withdrawing groups with high Hammett constants[173] (such as CN, NO2, esters, and aldehydes) in the ortho- or para- positions relative to the leaving group. Common leaving groups in this reaction system include the trimethylammonium and nitro groups.[170]
A major breakthrough was described by Pike and Aigbirhio in 1995 while describing the use of diaryl-λ3-iodanes (diaryliodinium salts) as ‘hyper’ leaving groups to facilitate radiofluorination of some of the most inert aromatic systems.[174] The radiofluorination yields were generally higher when an electron withdrawing group was positioned ortho- or para- to the iodinium salt.[175] Pike and co-workers pioneered this approach for the preparation of F-18 labelled radiopharmaceuticals with wide substrate scope.[175] This led to increased interest in the use of hypervalent iodane complexes as precursors for radiofluorination. We previously explored the use of diacetoxyiodo arenes as substrates for radiofluorination with limited success.[176] Liang et al. introduced spirocyclic iodonium ylides as precursors that effectively yield F-18 labelled non-activated and hindered aromatics.[177] A common complexity associated with these approaches is the non-trivial preparation of the λ3-iodane precursor, especially when associated with a complex chemical substrate.
Key developments in recent years relate to improvements in copper catalysed late-stage radiofluorination of aryl boronates and stannanes.[178–181] Aryl boronate and stannane precursors are common intermediates in organic synthesis and can be prepared readily using a range of chemical transformations.[182] More importantly radiofluorination of the precursor proceeds efficiently regardless of the presence or absence of electron withdrawing group on the aromatic ring.[178–181] These methodologies facilitate radiofluorination of a wide range of chemical substrates, enabling the preparation of highly complex and inert F-18 labelled compounds (Fig. 16).

|
However, the methods described thus far relate to the direct radiofluorination of compounds. Peptides were long considered unamenable to direct radiofluorination due to their chemical sensitivity and intolerance to harsh radiofluorination conditions involving elevated temperatures and basic conditions.[183] This necessitated the use of indirect radiofluorination methodologies utilising a prosthetic group that is radiofluorinated directly as discussed above and later conjugated to a peptide under mild conditions.[164,184] An extensive number of methods have been developed for indirect labelling of peptides, the discussion of which is beyond the scope of this review.[183,185] However, it would be prudent to briefly discuss one of the most commonly used indirect labelling methods: [18F]fluoroacylation, as it affords stable amides upon conjugation to peptides. [18F]fluoroacylation involves the use of a F-18 labelled activated ester to acylate a nucleophilic group on the peptide such as the ϵ-amine of a lysine side chain.[183] Common [18F]fluoroacylation prosthetic groups include 4-nitrophenyl 2-[18F]fluoropropionate,[186] N-succinimidyl 4-[18F]fluorobenzoate[187] and 2,3,5,6-tetrafluorophenyl 6-[18F]fluoronicotinate[188] (Fig. 17). [18F]fluoroacylation reactions are effective but they involve multistep, laborious, and complex processes that often include initial radiofluorination of a prosthetic group, followed by activation of the fluorinated prosthetic group to effect conjugation, before coupling to a peptide.[183] 2,3,5,6-Tetrafluorophenyl 6-[18F]fluoronicotinate was the first example describing the simplified radiofluorination of peptides in two chemical steps, involving only F-18 labelling of a pre-activated prosthetic group, followed by peptide acylation.[188] This development was facilitated by the facile nucleophilic radiofluorination of the pyridine scaffold under mild conditions (40°C) preserving the pre-activated ester (as the 2,3,5,6-tetrafluorophenyl ester) from hydrolysis.[188] The increased reactivity of the nicotinic scaffold can be explained by the pyridine nitrogen’s electronegativity that ‘swallows’ the electron density building up as a consequence of a nucleophile’s attack at the ipso-carbon atom.[189] The mild radiofluorination conditions required to retain the integrity of the pre-activated 2,3,5,6-tetrafluorophenyl ester limits the wide utility of this simplified methodology. Unlike heteroarenes (such as pyridines), the radiofluorination of arenes require elevated temperatures around ~100°C or more. We have recently developed pre-activated 4-nitrophenyl esters as superior intermediates that can facilitate radiofluorination at elevated temperatures (100°C), while still activated enough to acylate peptides quantitatively at room temperature.[190] This methodology was adopted to prepare 4-nitrophenyl 2-[18F]fluoropropionate[190,191] and analogues of the most useful prosthetic groups, N-succinimidyl 4-[18F]fluorobenzoate and 2,3,5,6-tetrafluorophenyl 6-[18F]fluoronicotinate, in one step[191] (Fig. 17). These developments constitute a major breakthrough for the indirect radiofluorination of peptides.[191,192]

|
More recently, the notion that peptides are unamenable to direct radiofluorination, necessitating the use of F-18 labelled prosthetic groups, has been challenged. Both F-18 labelled PSMA analogues PSMA-1007 and DCFPyl (discussed above) are now being routinely prepared by direct radiofluorination of the fully unprotected peptide with a pyridine quaternary ammonium salt precursor (Fig. 18).[193–195]

|
A myriad of other methods have been described to facilitate direct radiofluorination of unprotected peptides via the use of pyridines or highly activated aromatic systems with strong electron withdrawing groups (such as CN or CF3) in the ortho-position to the leaving group.[185,196] However, the general applicability of these methods to a range of peptides with diverse functional groups, conformations, and molecular sizes remains unknown. As such there is a need for intensive research to understand and optimise the factors that govern direct radiofluorination of peptides and to evaluate its adaptability to diverse peptide scaffolds.
Conclusion
Peptides exhibit ideal properties for theranostics application. In this review, we have discussed existing and evolving methods for the generation of effective peptides as radiopharmaceuticals for imaging and therapy. Furthermore, we describe common radiolabelling methodologies to yield peptide-based radiopharmaceuticals useful within oncology. Peptide-based theranostics is a rapidly growing field impacting clinical management of complex diseases and leading to ever-increasing research in radiopharmaceutical sciences. We are increasingly witnessing the integration of more complex and advanced peptide chemistries in radiopharmaceutical science leading to more effective and innovative theranostics. As more tumour-targeting peptides are developed, there will be increasing demand to optimise their biological properties and improve radiolabelling methodologies to produce diverse theranostics effectively.
Data Availability Statement
Data sharing is not applicable for this as no new data were generated or analysed throughout this review. All data used in this review are accessible through their respective references.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
Some parts of our work described in this review were funded by the Peter MacCallum Cancer Centre through Peter MacCallum Cancer Foundation Grant #1728 and the National Health and Medical Research Council (NHMRC) for New Investigator grant funding (APP1158863).
References
[1] S. S. Kelkar, T. M. Reineke, Bioconjug. Chem. 2011, 22, 1879.| Crossref | GoogleScholarGoogle Scholar | 21830812PubMed |
[2] S. S. Gambhir, Nat. Rev. Cancer 2002, 2, 683.
| Crossref | GoogleScholarGoogle Scholar | 12209157PubMed |
[3] C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai, S. S. Jurisson, Chem. Rev. 2013, 113, 858.
| Crossref | GoogleScholarGoogle Scholar | 23198879PubMed |
[4] L. G. Milroy, T. N. Grossmann, S. Hennig, L. Brunsveld, C. Ottmann, Chem. Rev. 2014, 114, 4695.
| Crossref | GoogleScholarGoogle Scholar | 24735440PubMed |
[5] A. C.-L. Lee, J. L. Harris, K. K. Khanna, J.-H. Hong, Int. J. Mol. Sci. 2019, 20, 2383.
| Crossref | GoogleScholarGoogle Scholar |
[6] S. Asati, V. Pandey, V. Soni, Int. J. Pept. Res. Ther. 2019, 25, 49.
| Crossref | GoogleScholarGoogle Scholar |
[7] V. Ambrosini, M. Fani, S. Fanti, F. Forrer, H. R. Maecke, J. Nucl. Med. 2011, 52, 42S.
| Crossref | GoogleScholarGoogle Scholar | 22144555PubMed |
[8] C. A. Lipinski, F. Lombardo, B. W. Dominy, P. J. Feeney, Adv. Drug Deliv. Rev. 1997, 23, 3.
| Crossref | GoogleScholarGoogle Scholar |
[9] A. K. Ghose, V. N. Viswanadhan, J. J. Wendoloski, J. Comb. Chem. 1999, 1, 55.
| Crossref | GoogleScholarGoogle Scholar | 10746014PubMed |
[10] R. N. Waterhouse, Mol. Imaging Biol. 2003, 5, 376.
| Crossref | GoogleScholarGoogle Scholar | 14667492PubMed |
[11] V. W. Pike, Curr. Med. Chem. 2016, 23, 1818.
| Crossref | GoogleScholarGoogle Scholar | 27087244PubMed |
[12] M. Schottelius, J. Šimeček, F. Hoffmann, M. Willibald, M. Schwaiger, H.-J. Wester, EJNMMI Res. 2015, 5, 22.
| Crossref | GoogleScholarGoogle Scholar | 25918675PubMed |
[13] A.-C. Baranski, M. Schäfer, U. Bauder-Wüst, M. Roscher, J. Schmidt, E. Stenau, T. Simpfendörfer, D. Teber, L. Maier-Hein, B. Hadaschik, U. Haberkorn, M. Eder, K. Kopka, J. Nucl. Med. 2018, 59, 639.
| Crossref | GoogleScholarGoogle Scholar | 29191856PubMed |
[14] S. Robu, A. Schmidt, M. Eiber, M. Schottelius, T. Günther, B. Hooshyar Yousefi, M. Schwaiger, H.-J. Wester, EJNMMI Res. 2018, 8, 30.
| Crossref | GoogleScholarGoogle Scholar | 29651565PubMed |
[15] M. Benešová, C. A. Umbricht, R. Schibli, C. Müller, Mol. Pharm. 2018, 15, 934.
| Crossref | GoogleScholarGoogle Scholar | 29400475PubMed |
[16] A. Poschenrieder, M. Schottelius, M. Schwaiger, H. Kessler, H. J. Wester, EJNMMI Res. 2016, 6, 36.
| 27112767PubMed |
[17] M. Schottelius, T. Osl, A. Poschenrieder, F. Hoffmann, S. Beykan, H. Hänscheid, A. Schirbel, A. K. Buck, S. Kropf, M. Schwaiger, U. Keller, M. Lassmann, H.-J. Wester, Theranostics 2017, 7, 2350.
| Crossref | GoogleScholarGoogle Scholar | 28744319PubMed |
[18] K. Pohle, J. Notni, J. Bussemer, H. Kessler, M. Schwaiger, A. J. Beer, Nucl. Med. Biol. 2012, 39, 777.
| Crossref | GoogleScholarGoogle Scholar | 22444238PubMed |
[19] Z. Wu, Z. B. Li, K. Chen, W. Cai, L. He, F. T. Chin, F. Li, X. Chen, J. Nucl. Med. 2007, 48, 1536.
| Crossref | GoogleScholarGoogle Scholar | 17704249PubMed |
[20] M. Maurin, P. Garnuszek, P. Baran, D. Pawlak, R. Mikołajczak, Nucl. Med. Rev. 2015, 18, 51.
| Crossref | GoogleScholarGoogle Scholar |
[21] J. Toms, J. Kogler, S. Maschauer, C. Daniel, C. Schmidkonz, T. Kuwert, O. Prante, J. Nucl. Med. 2020, 61, 1806.
| Crossref | GoogleScholarGoogle Scholar | 32332144PubMed |
[22] M. Benesová, M. Schäfer, U. Bauder-Wüst, A. Afshar-Oromieh, C. Kratochwil, W. Mier, U. Haberkorn, K. Kopka, M. Eder, J. Nucl. Med. 2015, 56, 914.
| Crossref | GoogleScholarGoogle Scholar | 25883127PubMed |
[23] E. Vegt, M. de Jong, J. F. Wetzels, R. Masereeuw, M. Melis, W. J. Oyen, M. Gotthardt, O. C. Boerman, J. Nucl. Med. 2010, 51, 1049.
| Crossref | GoogleScholarGoogle Scholar | 20554737PubMed |
[24] N. Sarvilinna, H. Eronen, S. Miettinen, A. Vienonen, T. Ylikomi, Int. J. Cancer 2006, 118, 832.
| Crossref | GoogleScholarGoogle Scholar | 16152593PubMed |
[25] G. P. V. Reddy, E. R. Barrack, Q. P. Dou, M. Menon, R. Pelley, F. H. Sarkar, S. Sheng, J. Cell. Biochem. 2006, 98, 1408.
| Crossref | GoogleScholarGoogle Scholar |
[26] D. Kwekkeboom, E. P. Krenning, M. De Jong, J. Nucl. Med. 2000, 41, 1704.
| 11038002PubMed |
[27] J. C. Reubi, J. C. Schär, B. Waser, S. Wenger, A. Heppeler, J. S. Schmitt, H. R. Mäcke, Eur. J. Nucl. Med. 2000, 27, 273.
| Crossref | GoogleScholarGoogle Scholar | 10774879PubMed |
[28] J. C. Reubi, Endocr. Rev. 2003, 24, 389.
| Crossref | GoogleScholarGoogle Scholar | 12920149PubMed |
[29] Y. C. Patel, Front. Neuroendocrinol. 1999, 20, 157.
| Crossref | GoogleScholarGoogle Scholar | 10433861PubMed |
[30] A. G. Harris, Gut 1994, 35, S1.
| Crossref | GoogleScholarGoogle Scholar | 7911441PubMed |
[31] W. Bauer, U. Briner, W. Doepfner, R. Haller, R. Huguenin, P. Marbach, T. J. Petcher, J. Pless, Life Sci. 1982, 31, 1133.
| Crossref | GoogleScholarGoogle Scholar | 6128648PubMed |
[32] W. H. Bakker, R. Albert, C. Bruns, W. A. P. Breeman, L. J. Hofland, P. Marbach, J. Pless, D. Pralet, B. Stolz, J. W. Koper, S. W. J. Lamberts, T. J. Visser, E. P. Krenning, Life Sci. 1991, 49, 1583.
| Crossref | GoogleScholarGoogle Scholar | 1658515PubMed |
[33] M. De Jong, W. A. P. Breeman, W. H. Bakker, P. P. M. Kooij, B. F. Bernard, L. J. Hofland, T. J. Visser, A. Srinivasan, M. A. Schmidt, J. L. Erion, J. E. Bugaj, H. R. Mäcke, E. P. Krenning, Cancer Res. 1998, 58, 437.
| 9458086PubMed |
[34] W. H. Bakker, E. P. Krenning, W. A. Breeman, J. W. Koper, P. P. Kooij, J.-C. Reubi, J. G. Klijn, T. J. Visser, R. Docter, S. W. Lamberts, J. Nucl. Med. 1990, 31, 1501.
| 1975618PubMed |
[35] D. J. Kwekkeboom, W. H. Bakker, P. P. Kooij, M. W. Konijnenberg, A. Srinivasan, J. L. Erion, M. A. Schmidt, J. L. Bugaj, M. De Jong, E. P. Krenning, Eur. J. Nucl. Med. 2001, 28, 1319.
| Crossref | GoogleScholarGoogle Scholar | 11585290PubMed |
[36] J. S. Lewis, M. Wang, R. Laforest, F. Wang, J. L. Erion, J. E. Bugaj, A. Srinivasan, C. J. Anderson, Int. J. Cancer 2001, 94, 873.
| Crossref | GoogleScholarGoogle Scholar | 11745491PubMed |
[37] M. De Jong, W. A. P. Breeman, B. F. Bernard, W. H. Bakker, M. Schaar, A. van Gameren, J. E. Bugaj, J. Erion, M. Schmidt, A. Srinivasan, E. P. Krenning, Int. J. Cancer 2001, 92, 628.
| Crossref | GoogleScholarGoogle Scholar | 11340564PubMed |
[38] R. J. Hicks, Cancer Imaging 2010, 10, S83.
| Crossref | GoogleScholarGoogle Scholar | 20880795PubMed |
[39] J. Strosberg, G. El-Haddad, E. Wolin, A. Hendifar, J. Yao, B. Chasen, E. Mittra, P. L. Kunz, M. H. Kulke, H. Jacene, D. Bushnell, T. M. O’Dorisio, R. P. Baum, H. R. Kulkarni, M. Caplin, R. Lebtahi, T. Hobday, E. Delpassand, E. Van Cutsem, A. Benson, R. Srirajaskanthan, M. Pavel, J. Mora, J. Berlin, E. Grande, N. Reed, E. Seregni, K. Öberg, M. L. Sierra, P. Santoro, T. Thevenet, J. L. Erion, P. Ruszniewski, D. Kwekkeboom, E. Krenning, N. Engl. J. Med. 2017, 376, 125.
| Crossref | GoogleScholarGoogle Scholar | 28076709PubMed |
[40] A. Ghosh, W. D. W. Heston, J. Cell. Biochem. 2004, 91, 528.
| Crossref | GoogleScholarGoogle Scholar | 14755683PubMed |
[41] A. Ghosh, W. D. W. Heston, in The Oncogenomics Handbook (Eds W. J. LaRochelle, R. A. Shimkets) 2005, pp. 597–615 (Humana Press: Totowa, NJ).
[42] M. Perera, N. Papa, D. Christidis, D. Wetherell, M. S. Hofman, D. G. Murphy, D. Bolton, N. Lawrentschuk, Eur. Urol. 2016, 70, 926.
| Crossref | GoogleScholarGoogle Scholar | 27363387PubMed |
[43] P. F. Jackson, D. C. Cole, B. S. Slusher, S. L. Stetz, L. E. Ross, B. A. Donzanti, D. A. Trainor, J. Med. Chem. 1996, 39, 619.
| Crossref | GoogleScholarGoogle Scholar | 8558536PubMed |
[44] A. P. Kozikowski, F. Nan, P. Conti, J. Zhang, E. Ramadan, T. Bzdega, B. Wroblewska, J. H. Neale, S. Pshenichkin, J. T. Wroblewski, J. Med. Chem. 2001, 44, 298.
| Crossref | GoogleScholarGoogle Scholar | 11462970PubMed |
[45] E. Eppard, A. Fuente, M. Benešová, A. Khawar, R. Bundschuh, F. Gaertner, B. Kreppel, K. Kopka, M. Essler, F. Roesch, Theranostics 2017, 7, 4359.
| Crossref | GoogleScholarGoogle Scholar | 29158832PubMed |
[46] A.-C. Baranski, M. Schäfer, U. Bauder-Wüst, A. Wacker, J. Schmidt, C. Liolios, W. Mier, U. Haberkorn, M. Eisenhut, K. Kopka, M. Eder, Bioconjug. Chem. 2017, 28, 2485.
| Crossref | GoogleScholarGoogle Scholar | 28787147PubMed |
[47] H.-T. Kuo, J. Pan, Z. Zhang, J. Lau, H. Merkens, C. Zhang, N. Colpo, K.-S. Lin, F. Bénard, Mol. Pharm. 2018, 15, 3502.
| Crossref | GoogleScholarGoogle Scholar | 29920108PubMed |
[48] M. Benešová, U. Bauder-Wüst, M. Schäfer, K. D. Klika, W. Mier, U. Haberkorn, K. Kopka, M. Eder, J. Med. Chem. 2016, 59, 1761.
| Crossref | GoogleScholarGoogle Scholar | 26878194PubMed |
[49] T. Liu, J. R. Nedrow-Byers, M. R. Hopkins, C. E. Berkman, Bioorg. Med. Chem. Lett. 2011, 21, 7013.
| Crossref | GoogleScholarGoogle Scholar | 22018464PubMed |
[50] United States Food and Drug Administration, FDA approves second PSMA-targeted PET imaging drug for men with prostate cancer. Drug Safety and Availability 2021. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-second-psma-targeted-pet-imaging-drug-men-prostate-cancer
[51] M. S. Hofman, N. Lawrentschuk, R. J. Francis, C. Tang, I. Vela, P. Thomas, N. Rutherford, J. M. Martin, M. Frydenberg, R. Shakher, L.-M. Wong, K. Taubman, S. T. Lee, E. Hsiao, P. Roach, M. Nottage, I. Kirkwood, D. Hayne, E. Link, P. Marusic, A. Matera, A. Herschtal, A. Iravani, R. J. Hicks, S. Williams, D. G. Murphy, Lancet 2020, 395, 1208.
| Crossref | GoogleScholarGoogle Scholar | 32209449PubMed |
[52] M. S. Hofman, J. Violet, R. J. Hicks, J. Ferdinandus, S. Ping Thang, T. Akhurst, A. Iravani, G. Kong, A. Ravi Kumar, D. G. Murphy, P. Eu, P. Jackson, M. Scalzo, S. G. Williams, S. Sandhu, Lancet Oncol. 2018, 19, 825.
| Crossref | GoogleScholarGoogle Scholar | 29752180PubMed |
[53] M. S. Hofman, L. Emmett, S. Sandhu, A. Iravani, A. M. Joshua, J. C. Goh, D. A. Pattison, T. H. Tan, I. D. Kirkwood, S. Ng, R. J. Francis, C. Gedye, N. K. Rutherford, A. Weickhardt, A. M. Scott, S.-T. Lee, E. M. Kwan, A. A. Azad, S. Ramdave, A. D. Redfern, W. Macdonald, A. Guminski, E. Hsiao, W. Chua, P. Lin, A. Y. Zhang, M. M. McJannett, M. R. Stockler, J. A. Violet, S. G. Williams, A. J. Martin, I. D. Davis, A. A. Azad, W. Chua, I. D. Davis, N. Dhiantravan, L. Emmett, K. Ford, N. Rana, S. Yip, Lancet 2021, 397, 797.
| Crossref | GoogleScholarGoogle Scholar | 33581798PubMed |
[54] J. C. Reubi, J.-C. Schaer, B. Waser, Cancer Res. 1997, 57, 1377.
| 9102227PubMed |
[55] J. C. Reubi, B. Waser, Int. J. Cancer 1996, 67, 644.
| Crossref | GoogleScholarGoogle Scholar | 8782652PubMed |
[56] C. Chao, X. Han, K. Ives, J. Park, A. A. Kolokoltsov, R. A. Davey, M. P. Moyer, M. R. Hellmich, Int. J. Cancer 2010, 126, 864.
| 19697327PubMed |
[57] M. B. Haskali, P. D. Roselt, D. Binns, A. Hetsron, S. Poniger, C. A. Hutton, R. J. Hicks, EJNMMI Radiopharm. Chem. 2019, 4, 23.
| Crossref | GoogleScholarGoogle Scholar | 31659509PubMed |
[58] M. Béhé, G. Kluge, W. Becker, M. Gotthardt, T. M. Behr, J. Nucl. Med. 2005, 46, 1012.
| 15937313PubMed |
[59] P. Laverman, L. Joosten, A. Eek, S. Roosenburg, P. K. Peitl, T. Maina, H. Mäcke, L. Aloj, E. von Guggenberg, J. K. Sosabowski, M. de Jong, J.-C. Reubi, W. J. G. Oyen, O. C. Boerman, Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1410.
| Crossref | GoogleScholarGoogle Scholar | 21461732PubMed |
[60] S. Roosenburg, P. Laverman, F. L. Van Delft, O. C. Boerman, Amino Acids 2011, 41, 1049.
| Crossref | GoogleScholarGoogle Scholar | 20198494PubMed |
[61] T. M. Behr, N. Jenner, M. Béhé, C. Angerstein, S. Gratz, F. Raue, W. Becker, J. Nucl. Med. 1999, 40, 1029.
| 10452322PubMed |
[62] L. Aloj, M. Aurilio, V. Rinaldi, L. D’ambrosio, D. Tesauro, P. K. Peitl, T. Maina, R. Mansi, E. von Guggenberg, L. Joosten, J. K. Sosabowski, W. A. P. Breeman, E. De Blois, S. Koelewijn, M. Melis, B. Waser, K. Beetschen, J. C. Reubi, M. de Jong, Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1417.
| Crossref | GoogleScholarGoogle Scholar | 21523391PubMed |
[63] M. Ocak, A. Helbok, C. Rangger, P. K. Peitl, B. A. Nock, G. Morelli, A. Eek, J. K. Sosabowski, W. A. P. Breeman, J. C. Reubi, C. Decristoforo, Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1426.
| Crossref | GoogleScholarGoogle Scholar | 21528387PubMed |
[64] M. Klingler, D. Summer, C. Rangger, R. Haubner, J. Foster, J. Sosabowski, C. Decristoforo, I. Virgolini, E. Von Guggenberg, J. Nucl. Med. 2019, 60, 1010.
| Crossref | GoogleScholarGoogle Scholar | 30530828PubMed |
[65] C. Uprimny, E. von Guggenberg, A. Svirydenka, R. Mikołajczak, A. Hubalewska-Dydejczyk, I. J. Virgolini, Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 935.
| Crossref | GoogleScholarGoogle Scholar | 32748048PubMed |
[66] X. Li, H. Cai, X. Wu, L. Li, H. Wu, R. Tian, Front Chem. 2020, 8, 583309.
| 33335885PubMed |
[67] H. Chen, G. Niu, H. Wu, X. Chen, Theranostics 2016, 6, 78.
| Crossref | GoogleScholarGoogle Scholar | 26722375PubMed |
[68] M. Kircher, P. Herhaus, M. Schottelius, A. K. Buck, R. A. Werner, H.-J. Wester, U. Keller, C. Lapa, Ann. Nucl. Med. 2018, 32, 503.
| Crossref | GoogleScholarGoogle Scholar | 30105558PubMed |
[69] P. Windisch, D. R. Zwahlen, S. A. Koerber, F. L. Giesel, J. Debus, U. Haberkorn, S. Adeberg, Cancers (Basel) 2020, 12, 2629.
[70] T. J. P. Jansen, S. A. M. van Lith, M. Boss, M. Brom, L. Joosten, M. Béhé, M. Buitinga, M. Gotthardt, J. Labelled Comp. Radiopharm. 2019, 62, 656.
| Crossref | GoogleScholarGoogle Scholar |
[71] C. E. A. Chang, W. Chen, M. K. Gilson, Proc. Natl. Acad. Sci. USA 2007, 104, 1534.
| Crossref | GoogleScholarGoogle Scholar |
[72] K. Bozovičar, T. Bratkovič, Int. J. Mol. Sci. 2021, 22, 1611.
| Crossref | GoogleScholarGoogle Scholar | 33562633PubMed |
[73] H. Yin, ISRN Biochem. 2012, 2012, 692190.
| Crossref | GoogleScholarGoogle Scholar | 25969758PubMed |
[74] D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell, S. H. Gellman, J. Am. Chem. Soc. 1996, 118, 13071.
| Crossref | GoogleScholarGoogle Scholar |
[75] S. H. Gellman, Acc. Chem. Res. 1998, 31, 173.
| Crossref | GoogleScholarGoogle Scholar |
[76] R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury, E. Valeur, J. Med. Chem. 2016, 59, 9599.
| Crossref | GoogleScholarGoogle Scholar | 27362955PubMed |
[77] G. N. Ramachandran, C. Ramakrishnan, V. Sasisekharan, J. Mol. Biol. 1963, 7, 95.
| Crossref | GoogleScholarGoogle Scholar | 13990617PubMed |
[78] O. Khakshoor, B. Demeler, J. S. Nowick, J. Am. Chem. Soc. 2007, 129, 5558.
| Crossref | GoogleScholarGoogle Scholar | 17419629PubMed |
[79] J. S. Davies, J. Pept. Sci. 2003, 9, 471.
| Crossref | GoogleScholarGoogle Scholar | 12952390PubMed |
[80] V. J. Hruby, Nat. Rev. Drug Discov. 2002, 1, 847.
| Crossref | GoogleScholarGoogle Scholar | 12415245PubMed |
[81] M. A. Dechantsreiter, E. Planker, B. Mathä, E. Lohof, G. Hölzemann, A. Jonczyk, S. L. Goodman, H. Kessler, J. Med. Chem. 1999, 42, 3033.
| Crossref | GoogleScholarGoogle Scholar | 10447947PubMed |
[82] K. Temming, R. M. Schiffelers, G. Molema, R. J. Kok, Drug Resist. Updates 2005, 8, 381.
| Crossref | GoogleScholarGoogle Scholar |
[83] O. Demmer, E. Gourni, U. Schumacher, H. Kessler, H. J. Wester, ChemMedChem 2011, 6, 1789.
| Crossref | GoogleScholarGoogle Scholar | 21780290PubMed |
[84] J. Rivier, M. Brown, W. Vale, Biochem. Biophys. Res. Commun. 1975, 65, 746.
| Crossref | GoogleScholarGoogle Scholar | 1096897PubMed |
[85] C. A. Rhodes, D. Pei, Chemistry (Easton) 2017, 23, 12690.
[86] P. Timmerman, J. Beld, W. C. Puijk, R. H. Meloen, ChemBioChem 2005, 6, 821.
| Crossref | GoogleScholarGoogle Scholar | 15812852PubMed |
[87] S. Chen, J. Morales-Sanfrutos, A. Angelini, B. Cutting, C. Heinis, ChemBioChem 2012, 13, 1032.
| Crossref | GoogleScholarGoogle Scholar | 22492661PubMed |
[88] C. Ernst, J. Sindlinger, D. Schwarzer, P. Koch, F. M. Boeckler, ACS Omega 2018, 3, 12361.
| Crossref | GoogleScholarGoogle Scholar | 30411004PubMed |
[89] M. Fani, A. Mueller, M. L. Tamma, G. Nicolas, H. R. Rink, R. Cescato, J. C. Reubi, H. R. Maecke, J. Nucl. Med. 2010, 51, 1771.
| Crossref | GoogleScholarGoogle Scholar | 20956465PubMed |
[90] D. F. Veber, F. W. Holly, W. J. Paleveda, R. F. Nutt, S. J. Bergstrand, M. Torchiana, M. S. Glitzer, R. Saperstein, R. Hirschmann, Proc. Natl. Acad. Sci. USA 1978, 75, 2636.
| Crossref | GoogleScholarGoogle Scholar | 208068PubMed |
[91] E. Falb, Y. Salitra, T. Yechezkel, M. Bracha, P. Litman, R. Olender, R. Rosenfeld, H. Senderowitz, S. Jiang, M. Goodman, Bioorg. Med. Chem. 2001, 9, 3255.
| Crossref | GoogleScholarGoogle Scholar | 11711301PubMed |
[92] M. Eder, S. Pavan, U. Bauder-Wüst, K. van Rietschoten, A. C. Baranski, H. Harrison, S. Campbell, C. L. Stace, E. H. Walker, L. Chen, G. Bennett, G. Mudd, U. Schierbaum, K. Leotta, U. Haberkorn, K. Kopka, D. P. Teufel, Cancer Res. 2019, 79, 841.
| Crossref | GoogleScholarGoogle Scholar | 30606721PubMed |
[93] D. H. Coy, N. Y. Jiang, S. H. Kim, J. P. Moreau, J. T. Lin, H. Frucht, J. M. Qian, L. W. Wang, R. T. Jensen, J. Biol. Chem. 1991, 266, 16441.
| Crossref | GoogleScholarGoogle Scholar | 1715866PubMed |
[94] L. M. Johnson, S. Barrick, M. V. Hager, A. McFedries, E. A. Homan, M. E. Rabaglia, M. P. Keller, A. D. Attie, A. Saghatelian, A. Bisello, S. H. Gellman, J. Am. Chem. Soc. 2014, 136, 12848.
| Crossref | GoogleScholarGoogle Scholar | 25191938PubMed |
[95] A. M. Ali, J. Atmaj, N. Van Oosterwijk, M. R. Groves, A. Dömling, Comput. Struct. Biotechnol. J. 2019, 17, 263.
| Crossref | GoogleScholarGoogle Scholar | 30867891PubMed |
[96] C. E. Schafmeister, J. Po, G. L. Verdine, J. Am. Chem. Soc. 2000, 122, 5891.
| Crossref | GoogleScholarGoogle Scholar |
[97] H. E. Blackwell, R. H. Grubbs, Angew. Chem. Int. Ed. 1998, 37, 3281.
| Crossref | GoogleScholarGoogle Scholar |
[98] N. E. Shepherd, H. N. Hoang, G. Abbenante, D. P. Fairlie, J. Am. Chem. Soc. 2005, 127, 2974.
| Crossref | GoogleScholarGoogle Scholar | 15740134PubMed |
[99] A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. S. Lin, B. L. Pentelute, J. Am. Chem. Soc. 2013, 135, 5946.
| Crossref | GoogleScholarGoogle Scholar | 23560559PubMed |
[100] N. Assem, D. J. Ferreira, D. W. Wolan, P. E. Dawson, Angew. Chem. Int. Ed. 2015, 54, 8665.
| Crossref | GoogleScholarGoogle Scholar |
[101] F. M. Brunel, P. E. Dawson, Chem. Commun. 2005, 2552.
| Crossref | GoogleScholarGoogle Scholar |
[102] G. J. Hilinski, Y. W. Kim, J. Hong, P. S. Kutchukian, C. M. Crenshaw, S. S. Berkovitch, A. Chang, S. Ham, G. L. Verdine, J. Am. Chem. Soc. 2014, 136, 12314.
| Crossref | GoogleScholarGoogle Scholar | 25105213PubMed |
[103] M. Pelay-Gimeno, A. Glas, O. Koch, T. N. Grossmann, Angew. Chem. Int. Ed. 2015, 54, 8896.
| Crossref | GoogleScholarGoogle Scholar |
[104] D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi, S. H. Gellman, Nature 1997, 387, 381.
| Crossref | GoogleScholarGoogle Scholar | 9163422PubMed |
[105] D. Ghosh, P. Lahiri, H. Verma, S. Mukherjee, J. Chatterjee, Chem. Sci. 2016, 7, 5212.
| Crossref | GoogleScholarGoogle Scholar | 29449932PubMed |
[106] W. G. Lesniak, E. Sikorska, H. Shallal, B. Behnam Azad, A. Lisok, M. Pullambhatla, M. G. Pomper, S. Nimmagadda, Mol. Pharm. 2015, 12, 941.
| Crossref | GoogleScholarGoogle Scholar | 25590535PubMed |
[107] A. C. Gibbs, T. C. Bjorndahl, R. S. Hodges, D. S. Wishart, J. Am. Chem. Soc. 2002, 124, 1203.
| Crossref | GoogleScholarGoogle Scholar | 11841288PubMed |
[108] A. G. Cochran, N. J. Skelton, M. A. Starovasnik, Proc. Natl. Acad. Sci. USA 2001, 98, 5578.
| Crossref | GoogleScholarGoogle Scholar | 11331745PubMed |
[109] K. Wang, Y. Song, Y. Su, Y. Liang, L. Wang, Biochem. Biophys. Res. Commun. 2020, 527, 453.
| Crossref | GoogleScholarGoogle Scholar | 32336542PubMed |
[110] A. Corlett, M.-A. Sani, J. Van Zuylekom, C.-S. Ang, E. von Guggenberg, C. Cullinane, B. Blyth, R. J. Hicks, P. D. Roselt, P. E. Thompson, C. A. Hutton, M. B. Haskali, J. Med. Chem. 2021, 64, 4841.
| 33826325PubMed |
[111] S. Boschi, F. Lodi, in Basic Science of PET Imaging (Ed. M. Khalil) 2017, pp. 79–103 (Springer: Cham).
[112] P. W. Miller, N. J. Long, R. Vilar, A. D. Gee, Angew. Chem. Int. Ed. 2008, 47, 8998.
| Crossref | GoogleScholarGoogle Scholar |
[113] S. Liu, D. S. Edwards, Chem. Rev. 1999, 99, 2235.
| Crossref | GoogleScholarGoogle Scholar | 11749481PubMed |
[114] M. M. Be, V. Chiste, C. Dulieu, E. Browne, V. Chechev, N. Kuzmenko, R. Helmer, A. Nichols, E. Schonfeld, R. Dersch, Table of Radionuclides 2004 (Bureau International des Poids et Mesures: France).
[115] A. Gopalakrishna, S. V. Suryanarayana, H. Naik, T. S. Dixit, B. K. Nayak, A. Kumar, P. Maletha, K. Thakur, A. Deshpande, R. Krishnan, K. Kamaldeep, S. Banerjee, A. Saxena, Radiochim. Acta 2018, 106, 549.
| Crossref | GoogleScholarGoogle Scholar |
[116] F. F. Knapp, M. M. Goodman, A. P. Callahan, L. A. Ferren, G. W. Kabalka, K. A. R. Sastry, J. Med. Chem. 1983, 26, 1293.
| Crossref | GoogleScholarGoogle Scholar | 6887204PubMed |
[117] M. M. Be, V. Chiste, C. Dulieu, M. A. Kellett, X. Mougeot, A. Arinc, V. P. Chechev, N. K. Kuzmenko, T. Kibedi, A. Luca, A. L. Nichols, Table of Radionuclides 2016 (Bureau International des Poids et Mesures: France).
[118] R. A. Werner, C. Bluemel, M. S. Allen-Auerbach, T. Higuchi, K. Herrmann, Ann. Nucl. Med. 2015, 29, 1.
| Crossref | GoogleScholarGoogle Scholar | 25139472PubMed |
[119] W. Jones, K. Griffiths, P. C. Barata, C. J. Paller, Cancers (Basel) 2020, 12, 1367.
| Crossref | GoogleScholarGoogle Scholar |
[120] R. A. Werner, T. Derlin, C. Lapa, S. Sheikbahaei, T. Higuchi, F. L. Giesel, S. Behr, A. Drzezga, H. Kimura, A. K. Buck, F. M. Bengel, M. G. Pomper, M. A. Gorin, S. P. Rowe, Theranostics 2020, 10, 1.
| Crossref | GoogleScholarGoogle Scholar | 31903102PubMed |
[121] K. Rahbar, A. Afshar-Oromieh, H. Jadvar, H. Ahmadzadehfar, Mol. Imaging 2018, 17, 1.
| Crossref | GoogleScholarGoogle Scholar |
[122] P. M. Smith-Jones, B. Stolz, R. Albert, H. Knecht, C. Bruns, Nucl. Med. Biol. 1997, 24, 761.
| Crossref | GoogleScholarGoogle Scholar | 9428603PubMed |
[123] R. Haubner, H. J. Wester, F. Burkhart, R. Senekowitsch-Schmidtke, W. Weber, S. L. Goodman, H. Kessler, M. Schwaiger, J. Nucl. Med. 2001, 42, 326.
| 11216533PubMed |
[124] X. Chen, R. Park, A. H. Shahinian, J. R. Bading, P. S. Conti, Nucl. Med. Biol. 2004, 31, 11.
| Crossref | GoogleScholarGoogle Scholar | 14741566PubMed |
[125] I. Dijkgraaf, S. Liu, J. A. W. Kruijtzer, A. C. Soede, W. J. G. Oyen, R. M. J. Liskamp, F. H. M. Corstens, O. C. Boerman, Nucl. Med. Biol. 2007, 34, 29.
| Crossref | GoogleScholarGoogle Scholar | 17210459PubMed |
[126] P. Laverman, W. J. McBride, R. M. Sharkey, A. Eek, L. Joosten, W. J. G. Oyen, D. M. Goldenberg, O. C. Boerman, J. Nucl. Med. 2010, 51, 454.
| Crossref | GoogleScholarGoogle Scholar | 20150268PubMed |
[127] M. W. Brechbiel, Q. J. Nucl. Med. Mol. Imaging 2008, 52, 166.
| 18043537PubMed |
[128] E. W. Price, C. Orvig, Chem. Soc. Rev. 2014, 43, 260.
| Crossref | GoogleScholarGoogle Scholar | 24173525PubMed |
[129] S. Liu, Adv. Drug Deliv. Rev. 2008, 60, 1347.
| Crossref | GoogleScholarGoogle Scholar | 18538888PubMed |
[130] M. M. Be, V. Chiste, C. Dulieu, X. Mougeot, V. P. Chechev, F. G. Kondev, A. L. Nichols, X. Huang, B. Wang, Table of Radionuclides 2013 (Bureau International des Poids et Mesures: France).
[131] J. D. G. Correia, A. Paulo, P. D. Raposinho, I. Santos, Dalton Trans. 2011, 40, 6144.
| Crossref | GoogleScholarGoogle Scholar |
[132] K. P. Zhernosekov, D. V. Filosofov, R. P. Baum, P. Aschoff, H. Bihl, A. A. Razbash, M. Jahn, M. Jennewein, F. Rösch, J. Nucl. Med. 2007, 48, 1741.
| Crossref | GoogleScholarGoogle Scholar | 17873136PubMed |
[133] M. K. Pandey, J. F. Byrne, H. Jiang, A. B. Packard, T. R. DeGrado, Am. J. Nucl. Med. Mol. Imaging 2014, 4, 303.
| 24982816PubMed |
[134] M. Sadeghi, T. Kakavand, S. Rajabifar, L. Mokhtari, A. Rahimi-Nezhad, Nukleonika 2009, 54, 25.
[135] M. K. Pandey, J. F. Byrne, K. N. Schlasner, N. R. Schmit, T. R. DeGrado, Nucl. Med. Biol. 2019, 74–75, 49.
| Crossref | GoogleScholarGoogle Scholar | 31085059PubMed |
[136] M. Nair, S. Happel, T. Eriksson, M. K. Pandey, T. R. DeGrado, K. Gagnon, Eur. J. Nucl. Med. Mol. Imaging 2017, 44, S275.
[137] S. Riga, G. Cicoria, D. Pancaldi, F. Zagni, S. Vichi, M. Dassenno, L. Mora, F. Lodi, M. P. Morigi, M. Marengo, Phys. Med. 2018, 55, 116.
| Crossref | GoogleScholarGoogle Scholar | 30473059PubMed |
[138] M. Lin, G. J. Waligorski, C. G. Lepera, Appl. Radiat. Isot. 2018, 133, 1.
| Crossref | GoogleScholarGoogle Scholar | 29272820PubMed |
[139] H. Thisgaard, J. Kumlin, N. Langkjær, J. Chua, B. Hook, M. Jensen, A. Kassaian, S. Zeisler, S. Borjian, M. Cross, P. Schaffer, J. H. Dam, EJNMMI Radiopharm. Chem. 2021, 6, 1.
| Crossref | GoogleScholarGoogle Scholar | 33411034PubMed |
[140] S. Eberl, T. Eriksson, O. Svedberg, J. Norling, D. Henderson, P. Lam, M. Fulham, Appl. Radiat. Isot. 2012, 70, 922.
| Crossref | GoogleScholarGoogle Scholar | 22476015PubMed |
[141] R. Delgado, V. Félix, L. M. P. Lima, D. W. Price, Dalton Trans. 2007, 2734.
| Crossref | GoogleScholarGoogle Scholar | 17592589PubMed |
[142] A. Meents, S. Gutmann, A. Wagner, C. Schulze-Briese, Proc. Natl. Acad. Sci. USA 2010, 107, 1094.
| Crossref | GoogleScholarGoogle Scholar | 20080548PubMed |
[143] W. Brand-Williams, M. E. Cuvelier, C. Berset, Lebensm. Wiss. Technol. 1995, 28, 25.
| Crossref | GoogleScholarGoogle Scholar |
[144] S. Liu, D. S. Edwards, Bioconjug. Chem. 2001, 12, 554.
| Crossref | GoogleScholarGoogle Scholar | 11459460PubMed |
[145] S. Liu, C. E. Ellars, D. S. Edwards, Bioconjug. Chem. 2003, 14, 1052.
| Crossref | GoogleScholarGoogle Scholar | 13129412PubMed |
[146] R. Shannon, Acta Crystallogr. A 1976, 32, 751.
| Crossref | GoogleScholarGoogle Scholar |
[147] T. J. Wadas, E. H. Wong, G. R. Weisman, C. J. Anderson, Chem. Rev. 2010, 110, 2858.
| Crossref | GoogleScholarGoogle Scholar | 20415480PubMed |
[148] W. P. Cacheris, S. K. Nickle, A. D. Sherry, Inorg. Chem. 1987, 26, 958.
| Crossref | GoogleScholarGoogle Scholar |
[149] E. T. Clarke, A. E. Martell, Inorg. Chim. Acta 1991, 181, 273.
| Crossref | GoogleScholarGoogle Scholar |
[150] E. T. Clarke, A. E. Martell, Inorg. Chim. Acta 1991, 190, 27.
| Crossref | GoogleScholarGoogle Scholar |
[151] E. T. Clarke, A. E. Martell, Inorg. Chim. Acta 1991, 190, 37.
| Crossref | GoogleScholarGoogle Scholar |
[152] M. Kodama, T. Koike, A. B. Mahatma, E. Kimura, Inorg. Chem. 1991, 30, 1270.
| Crossref | GoogleScholarGoogle Scholar |
[153] K. Kumar, C. A. Chang, L. C. Francesconi, D. D. Dischino, M. F. Malley, J. Z. Gougoutas, M. F. Tweedle, Inorg. Chem. 1994, 33, 3567.
| Crossref | GoogleScholarGoogle Scholar |
[154] K. Kumar, C. A. Chang, M. F. Tweedle, Inorg. Chem. 1993, 32, 587.
| Crossref | GoogleScholarGoogle Scholar |
[155] A. E. Martell, R. J. Motekaitis, E. T. Clarke, R. Delgado, Y. Sun, R. Ma, Supramol. Chem. 1996, 6, 353.
| Crossref | GoogleScholarGoogle Scholar |
[156] R. Delgado, Y. Sun, R. J. Motekaitis, A. E. Martell, Inorg. Chem. 1993, 32, 3320.
| Crossref | GoogleScholarGoogle Scholar |
[157] I. Velikyan, H. Maecke, B. Langstrom, Bioconjug. Chem. 2008, 19, 569.
| Crossref | GoogleScholarGoogle Scholar | 18205327PubMed |
[158] S. Banerjee, M. R. A. Pillai, F. F. Knapp, Chem. Rev. 2015, 115, 2934.
| Crossref | GoogleScholarGoogle Scholar | 25865818PubMed |
[159] J. Fichna, A. Janecka, Bioconjug. Chem. 2003, 14, 3.
| Crossref | GoogleScholarGoogle Scholar | 12526687PubMed |
[160] T. C. Owen, M. T. Brown, J. Org. Chem. 1969, 34, 1161.
| Crossref | GoogleScholarGoogle Scholar |
[161] G. Xu, M. R. Chance, Anal. Chem. 2004, 76, 1213.
| Crossref | GoogleScholarGoogle Scholar | 14987073PubMed |
[162] A. H. Snell, Proceedings of the American Physical Society: Physical Review 1937, 51, 142.
[163] J. F. Volker, H. C. Hodge, H. J. Wilson, S. N. Van Voorhis, J. Biol. Chem. 1940, 134, 543.
| Crossref | GoogleScholarGoogle Scholar |
[164] O. Jacobson, D. O. Kiesewetter, X. Chen, Bioconjug. Chem. 2015, 26, 1.
| Crossref | GoogleScholarGoogle Scholar | 25473848PubMed |
[165] N. A. Meanwell, J. Med. Chem. 2018, 61, 5822.
| Crossref | GoogleScholarGoogle Scholar | 29400967PubMed |
[166] H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander, M. Stahl, ChemBioChem 2004, 5, 637.
| Crossref | GoogleScholarGoogle Scholar | 15122635PubMed |
[167] R. Haubner, B. Kuhnast, C. Mang, W. A. Weber, H. Kessler, H. J. Wester, M. Schwaiger, Bioconjug. Chem. 2004, 15, 61.
| Crossref | GoogleScholarGoogle Scholar | 14733584PubMed |
[168] M. B. Haskali, D. Denoyer, W. Noonan, C. Culinane, C. Rangger, N. Pouliot, R. Haubner, P. D. Roselt, R. J. Hicks, C. A. Hutton, Mol. Pharm. 2017, 14, 1169.
| Crossref | GoogleScholarGoogle Scholar | 28191977PubMed |
[169] E. Hess, S. Takács, B. Scholten, F. Tárkányi, H. H. Coenen, S. M. Qaim, Radiochim. Acta 2001, 89, 357.
| Crossref | GoogleScholarGoogle Scholar |
[170] H. H. Coenen, in PET Chemistry (Eds P. A. Schubiger, L. Lehmann, M. Friebe) 2007, pp. 15–50 (Springer: Berlin).
[171] O. Jacobson, X. Chen, Curr. Top. Med. Chem. 2010, 10, 1048.
| Crossref | GoogleScholarGoogle Scholar | 20388116PubMed |
[172] E. Hess, G. Blessing, H. H. Coenen, S. M. Qaim, Appl. Radiat. Isot. 2000, 52, 1431.
| Crossref | GoogleScholarGoogle Scholar | 10855672PubMed |
[173] C. Hansch, A. Leo, R. W. Taft, Chem. Rev. 1991, 91, 165.
| Crossref | GoogleScholarGoogle Scholar |
[174] V. W. Pike, F. I. Aigbirhio, J. Labelled Comp. Radiopharm. 1995, 37, 120.
[175] W. Qu, X. Li, in Fluorination (Eds J. Hu, T. Umemoto) 2018, pp. 1–12 (Springer: Singapore).
[176] M. B. Haskali, S. Telu, Y.-S. Lee, C. L. Morse, S. Lu, V. W. Pike, J. Org. Chem. 2016, 81, 297.
| Crossref | GoogleScholarGoogle Scholar | 26641128PubMed |
[177] B. H. Rotstein, N. A. Stephenson, N. Vasdev, S. H. Liang, Nat. Commun. 2014, 5, 4365.
| Crossref | GoogleScholarGoogle Scholar | 25007318PubMed |
[178] A. V. Mossine, A. F. Brooks, K. J. Makaravage, J. M. Miller, N. Ichiishi, M. S. Sanford, P. J. H. Scott, Org. Lett. 2015, 17, 5780.
| Crossref | GoogleScholarGoogle Scholar | 26568457PubMed |
[179] J. S. Wright, T. Kaur, S. Preshlock, S. S. Tanzey, W. P. Winton, L. S. Sharninghausen, N. Wiesner, A. F. Brooks, M. S. Sanford, P. J. H. Scott, Clin. Transl. Imaging 2020, 8, 167.
| Crossref | GoogleScholarGoogle Scholar | 33748018PubMed |
[180] K. J. Makaravage, A. F. Brooks, A. V. Mossine, M. S. Sanford, P. J. H. Scott, Org. Lett. 2016, 18, 5440.
| Crossref | GoogleScholarGoogle Scholar | 27718581PubMed |
[181] S. Preshlock, S. Calderwood, S. Verhoog, M. Tredwell, M. Huiban, A. Hienzsch, S. Gruber, T. C. Wilson, N. J. Taylor, T. Cailly, M. Schedler, T. L. Collier, J. Passchier, R. Smits, J. Mollitor, A. Hoepping, M. Mueller, C. Genicot, J. Mercier, V. Gouverneur, Chem. Commun. 2016, 52, 8361.
| Crossref | GoogleScholarGoogle Scholar |
[182] D. G. Hall, in Boronic Acids (Eds D. G. Hall) 2011, pp. 1–133 (Wiley-VCH: Weinheim, Germany).
[183] H. J. Wester, M. Schottelius, in PET Chemistry (Eds P. A. Schubiger, L. Lehmann, M. Friebe) 2007, pp. 79–111 (Springer: Berlin).
[184] K. A. Jacobson, D. C. Furlano, K. L. Kirk, J. Fluor. Chem. 1988, 39, 339.
| Crossref | GoogleScholarGoogle Scholar | 26523070PubMed |
[185] H. S. Krishnan, L. Ma, N. Vasdev, S. H. Liang, Chem. – Eur. J. 2017, 23, 15553.
| Crossref | GoogleScholarGoogle Scholar | 28704575PubMed |
[186] S. Guhlke, H. H. Coenen, G. Stöcklin, Appl. Radiat. Isot. 1994, 45, 715.
| Crossref | GoogleScholarGoogle Scholar |
[187] H. J. Wester, K. Hamacher, G. Stöcklin, Nucl. Med. Biol. 1996, 23, 365.
| Crossref | GoogleScholarGoogle Scholar | 8782249PubMed |
[188] D. E. Olberg, J. M. Arukwe, D. Grace, O. K. Hjelstuen, M. Solbakken, G. M. Kindberg, A. Cuthbertson, J. Med. Chem. 2010, 53, 1732.
| Crossref | GoogleScholarGoogle Scholar | 20088512PubMed |
[189] M. Schlosser, R. Ruzziconi, Synthesis 2010, 2010, 2111.
| Crossref | GoogleScholarGoogle Scholar |
[190] M. B. Haskali, P. D. Roselt, J. A. Karas, W. Noonan, C. W. Wichmann, A. Katsifis, R. J. Hicks, C. A. Hutton, J. Labelled Comp. Radiopharm. 2013, 56, 726.
| Crossref | GoogleScholarGoogle Scholar | 24339012PubMed |
[191] M. B. Haskali, A. L. Farnsworth, P. D. Roselt, C. A. Hutton, RSC Med. Chem. 2020, 11, 919.
| Crossref | GoogleScholarGoogle Scholar | 33479687PubMed |
[192] M. B. Haskali, P. D. Roselt, R. J. Hicks, C. A. Hutton, J. Labelled Comp. Radiopharm. 2018, 61, 61.
| Crossref | GoogleScholarGoogle Scholar | 29143364PubMed |
[193] J. Cardinale, R. Martin, Y. Remde, M. Schäfer, A. Hienzsch, S. Hübner, A. M. Zerges, H. Marx, R. Hesse, K. Weber, R. Smits, A. Hoepping, M. Müller, O. C. Neels, K. Kopka, Pharmaceuticals (Basel) 2017, 10, 77.
| Crossref | GoogleScholarGoogle Scholar |
[194] S. Naka, T. Watabe, K. Kurimoto, M. Uemura, F. Soeda, O. C. Neels, K. Kopka, M. Tatsumi, H. Kato, N. Nonomura, E. Shimosegawa, J. Cardinale, F. L. Giesel, J. Hatazawa, EJNMMI Radiopharm. Chem. 2020, 5, 18.
| Crossref | GoogleScholarGoogle Scholar | 32728815PubMed |
[195] M. H. Dornan, J.-M. Simard, A. Leblond, D. Juneau, G. Delouya, F. Saad, C. Ménard, J. N. DaSilva, J. Labelled Comp. Radiopharm. 2018, 61, 757.
| Crossref | GoogleScholarGoogle Scholar |
[196] M. Richard, S. Specklin, M. Roche, F. Hinnen, B. Kuhnast, Chem. Commun. 2020, 56, 2507.
| Crossref | GoogleScholarGoogle Scholar |