

Emerging Ionic Polymers for CO2 Conversion to Cyclic Carbonates: An Overview of Recent Developments*
Rabia Jamil A , Liliana C. Tomé B , David Mecerreyes C D and Debbie S. Silvester A E
A E
A School of Molecular and Life Sciences, Curtin University, GPO Box U1987, Perth, WA 6845, Australia.
B LAQV-REQUIMTE, Chemistry Department, NOVA School of Science and Technology, Universidade Nova de Lisboa, 2829-516 Caparica, Portugal.
C POLYMAT University of the Basque Country UPV/EHU, 20018 Donostia-San Sebastian, Spain.
D IKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain.
E Corresponding author. Email: d.silvester-dean@curtin.edu.au

Rabia Jamil completed her M.Sc. at Quaid-i-Azam University, Islamabad, Pakistan. Her M.Phil. research was carried out in the same university under the supervision of Dr Muhammad Shahid Ansari, focussed on the synthesis of nanomaterials with various surface modifications for electrocatalysis and materials chemistry for renewable energy. She will soon begin her Ph.D. studies under the supervision of A/Prof. Debbie Silvester (her start has been delayed due to travel restrictions following COVID-19), focussing on the use of ionic liquids and poly(ionic liquids) for gas sensing and electrochemical applications. |

Liliana C. Tomé received a M.Sc. in Materials Derived from Renewable Resources (2008) from the University of Aveiro and a Ph.D. in Engineering and Technology Sciences – Chemical Engineering (2014) from Instituto de Tecnologia Química e Biológica António Xavier (ITQB NOVA), Portugal. After her post-doc (2015–2018) at the same institute, she moved to POLYMAT – University of the Basque Country (Spain) with a Marie Skłodowska-Curie Individual Fellowship. In 2020, she joined LAQV-REQUIMTE, NOVA School of Science and Technology | FCT NOVA (Portugal), where she is an Assistant Researcher and recently received the L’Oréal Portugal Medal of Honor for Women in Science 2020. Her areas of research are centered on iongels and poly(ionic liquid)-based materials for gas separation and bioelectronics. |

David Mecerreyes is an Ikerbasque Research Professor at POLYMAT-University of the Basque Country (Spain). He received his Ph.D. from University of Liege in 1998. After a post-doc (1998–2000) at IBM Almaden Research Center (California), he moved to Spain to work at CIDETEC. In 2010, he moved to POLYMAT where he became the leader of the Innovative Polymers Group. His area of expertise is polymer synthesis of ionic and electroactive polymers, such as poly(ionic liquid)s, polymer electrolytes, redox polymers, and PEDOT type conductive polymers. His current research interests involve the application of innovative polymers in emerging technologies such as organic batteries and bioelectronic devices. |

Debbie Silvester is an Associate Professor and Australian Research Council (ARC) Future Fellow at the School of Molecular and Life Sciences at Curtin University in Perth, Australia. She completed her M.Sci. in Chemistry at the University of Bristol and her Ph.D. at the University of Oxford, UK. Her research interests include electrochemistry in ionic liquids, electrochemical reaction mechanisms, gas sensing, explosives detection, electrodeposition, and formation of modified electrodes for sensing applications. She has been awarded several national and international awards for research, including the 2021 Le Fèvre medal (Australian Academy of Science) and the 2021 Analyst Emerging Investigator Lectureship (Royal Society of Chemistry). |
Australian Journal of Chemistry 74(11) 767-777 https://doi.org/10.1071/CH21182
Submitted: 31 July 2021 Accepted: 24 September 2021 Published: 25 October 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
In this mini review, we highlight some key work from the last 2 years where ionic polymers have been used as a catalyst to convert CO2 into cyclic carbonates. Emerging ionic polymers reported for this catalytic application include materials such as poly(ionic liquid)s (PILs), ionic porous organic polymers (iPOPs) or ionic covalent organic frameworks (iCOFs) among others. All these organic materials share in common the ionic moiety cations such as imidazolium, pyridinium, viologen, ammonium, phosphonium, and guanidinium, and anions such as halides, [BF4]–, [PF6]–, and [Tf2N]–. The mechanistic aspects and efficiency of the CO2 conversion reaction and the polymer design including functional groups and porosity are discussed in detail. This review should provide valuable information for researchers to design new polymers for important catalysis applications.
Keywords: ionic liquids, polymers, CO2 fixation, cycloaddition reaction, heterogeneous catalysis, poly(ionic liquids), CO2 conversion, catalysts.
Introduction
The growing atmospheric concentration of greenhouse gases (in particular carbon dioxide, CO2) from industrial processes is a matter of widespread public concern due to the resulting environmental problems associated with global warming and unpredictable climate change.[1–3] In order to meet CO2 emission reduction targets, several strategies have been sought to capture CO2 directly from different sources, in particular carbon capture and storage (CCS).[4] Nevertheless, the processes associated to this approach are highly energy intensive and less economically viable compared with CO2 capture and utilization (CCU),[5] which focusses on the chemical transformation of CO2 and its utilization as a carbon source for the production of fuels and commodity chemicals.[6–8] Therefore, a viable strategy to remove CO2 from the atmosphere is to convert the gas via chemical processes to useful chemicals[9,10] or polymers.[11]
Using CO2 as an environmentally friendly and economically viable source of carbon can only be achieved by addressing the challenges in the underlying principles of CO2 activation, designing appropriate reactors and integration of processes. One of the most effective routes for CO2 fixation is cycloaddition of CO2 with epoxides to produce cyclic carbonates without by-products (see Fig. 1), in a complete atom economy reaction that meets the criteria of green chemistry.[12–14] Cyclic carbonates are of particular interest in applications such as aprotic polar solvents and electrolytes in secondary batteries, precursors for polycarbonate and polymeric materials synthesis, and intermediates in the production of value-added chemicals such as dialkylcarbonates, glycols, carbamates, pyrimidines, and purines.[13,15,16]
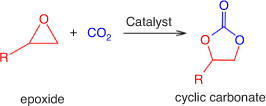
|
The use of a catalyst in this reaction is essential for reducing the temperature and the pressure to make it efficient and economically viable. The development of new catalysts has been extensively investigated over the last few years, in particular homogeneous catalysts such as organometallic complexes and metal oxides, onium salts, ionic liquids, Schiff bases, and mono or binary metal-free catalysts.[17] To bridge the gap between homogeneous and heterogeneous catalysts in the CO2–epoxide coupling reaction, organic polymers have emerged over the past several years as efficient catalysts with unprecedented inherent structural features.[16,18–20] The main feature of these polymers is that they are ionic in nature and include many ionic groups such as cations and anions similar to the ones used in ionic liquid chemistry (shown in Fig. 2). Owing to their design versatility and polymeric materials, several families of polymers including materials with high surface area such as porous organic polymers have been designed including ionic groups. The goal of this mini review is to show some examples of the different types of ionic polymers that have been reported in the scientific literature in the last 2 years for CO2 conversion.

|
Recent reviews have focussed on the design of and progress around ionic polymers for the CO2 cycloaddition reaction,[14,16,18,21,22] with minor mentions of poly(ionic liquid)s or ionic polymers. To our knowledge, only one review focussed only on imidazolium-functionalized ionic polymers as heterogeneous catalysts, only partially addressing the subject reviewed here.[18] To inspire the design of new ionic polymers, it is timely to summarize and discuss recent strategies adopted for the CO2 conversion reaction that could be applied to other catalytic reactions. In this mini review, we briefly overview the numerous categories of ionic polymers, with a discussion on the careful selection of cation and anion pairs, polymer backbone, and functional groups of newly emerging ionic polymers. Only ionic functionalized organic materials are considered for this review while ionic liquid-supported inorganic materials (graphene oxide (GO), SiO2, CNTs, TiO2, etc.) and inorganic–organic hybrid materials (polyhedral oligomeric silsesquioxane (POSS), metal organic frameworks (MOFs), zeolitic imidazolate frameworks (ZIFs), etc.) are intentionally excluded. First, an introduction to the mechanism of CO2 conversion and some crucial insights into CO2 activation are provided, followed by a brief overview of the various sub-classes of ionic polymeric materials used for CO2 conversion. Special emphasis is given to the strategies for improving catalytic performance of the charged polymers. The aim of the review is to provide some insights into the possibilities of using emerging ionic polymeric materials for the CO2 cycloaddition reaction.
Mechanism of CO2 Cycloaddition Reaction
CO2 is thermodynamically and kinetically stable, which limits its utilization as a feedstock for organic synthesis. One way to tackle this challenge is to choose products with a fairly high free energy, thus making the reaction with CO2 thermodynamically feasible. An example is the synthesis of ethylene carbonate from ethylene oxide and CO2, which is a highly exothermic reaction with ΔHrxn = –144 kJ mol−1 and has an almost 100 % atomic efficiency. The ring-opening of the three-membered epoxide releases ring strain energy, which is sufficient to drive the reaction.[23]
In principle, a cycloaddition reaction of CO2 with an epoxide to generate a cyclic carbonate often follows two pathways: (1) epoxide activation; or (2) CO2 activation; both pathways are depicted in Fig. 3.[19,23] In the epoxide activation route, a nucleophile attacks the less hindered carbon atom on the epoxide, while in the CO2 activation route, the nucleophile attacks the electrophilic carbon on CO2. For epoxide activation, the generally accepted mechanism using polymeric ionic liquids or ionic polymer catalysts involves the following steps, as shown schematically in Fig. 4. Step (1): epoxide activation by the interaction of Lewis or Brønsted acid sites on the poly(ionic liquid) (PIL) cations with the weakly basic oxygen atom on the epoxide. In metal-containing catalysts (Lewis acid site), the interaction is via a metal–oxygen coordinative bond, whereas hydoxyl-, amine- or carboxyl-containing ionic polymers (Brønsted acid site) interact with epoxide through the hydrogen bond. Step (2): ring-opening of the epoxide initiated by nucleophilic attack of the halide anion (X–) on the least hindered carbon atom of the epoxide, leading to the formation of an alkoxide intermediate. The ring-opened epoxide is stabilized by the counter cation present in the polymeric framework. Step (3): CO2 insertion requires activation of CO2 by nucleophilic or electrophilic attack. CO2 is a linear, apolar molecule with two polar (C=O) bonds, with electropositive and electronegative charges on the carbon and oxygen atoms, respectively. The electronegative oxygen acts as a nucleophile and activates CO2 by attacking the electrophilic carbon atom to form a carbonate intermediate. Step (4): the open-chained carbonate undergoes intramolecular cyclic elimination. Step (5): finally, displacement of X– leads to the formation of a five-membered cyclic carbonate, together with regeneration of the catalyst. The choice of the PIL or ionic polymer cation, and the use of a halide anion (as the nucleophile) are crucial for the generation of five-membered cyclic carbonates.[21] The nucleophile should have a good balance of nucleophilicity – to facilitate the ring-opening process – as well as the ability to act as a leaving group so that it can be easily displaced to ensure cyclic carbonate formation. Similarly, to insert CO2 into the molecule, the PIL or ionic polymer cation needs to stabilize the alkoxide intermediate through favourable chemical interactions.[23]

|
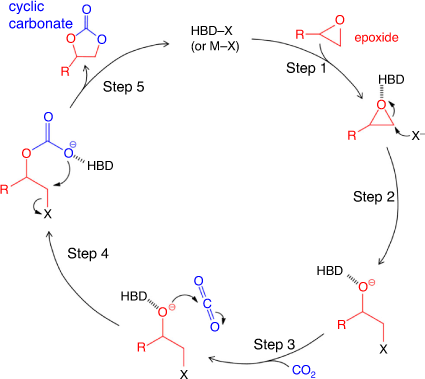
|
The CO2 activation pathway proceeds when a nucleophile such as an N-heterocyclic carbene (NHC), e.g. 1,5,7-triazabicyclo[4.4.0]dec-5-ene or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), attacks the electrophilic carbon of CO2, as depicted in Fig. 5. Recent studies show that PILs/ionic polymers can act as pre-catalysts for polyNHCs.[21] A possible CO2 activation pathway using NHCs is as follows: (i) nucleophilic attack of CO2 by an NHC catalyst to form a zwitterionic NHC carboxylate adduct; (ii) the zwitterionic adduct attacks the epoxide to produce a new zwitterion; (iii) then, the alkoxy anion nucleophile formed attacks the electropositive carbonyl carbon atom to produce a cyclic carbonate by intramolecular cyclic elimination.[21]
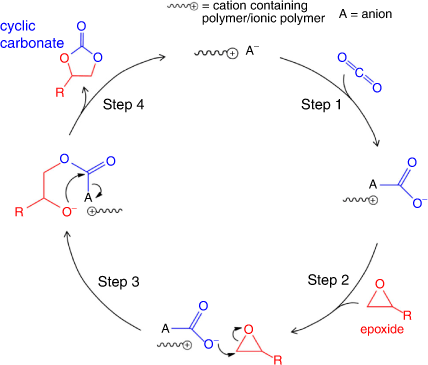
|
In the next sections, different emerging ionic polymers are discussed from the standpoint of ease of introduction of the desired functional group, and the atomic-level control over their structures and compositions. It is worth noting that the classification of these emerging ionic polymers is not easy, and we have kept the nomenclature and names proposed by the authors. In general, most of the emerging ionic polymers have in common the ionic functionalities (anions and cations), the inclusion of other functionalities, and their use as heterogeneous catalysts that can be recovered and reused several times, as well as the different types of porosity.
Functionalized Ionic Polymers for CO2 Conversion
Ionic polymers contain abundant ionic sites that retain some of the molecular ionic compounds, such as ionic liquids, making them attractive as heterogeneous catalysts for CO2 conversion. Different approaches have been adopted to modulate the type and density of ionic sites, as well as the porosity of ionic polymers, to upgrade their catalytic performances.[18,26–28] One efficient way is to utilize the unique advantages of ionic hyper-crosslinked polymers due to their large surface area, versatile architectures, and diverse ionic building blocks.[29,30] Wang et al. synthesized imidazolium-functionalized hyper-crosslinked polymers (HCPs) using a series of benzylimidazole salts as the (charged) ionic monomers and α,α′-dibromo-p-xylene as a (neutral) cross-linking agent.[31] The connection between the phenyl ring and the imidazolium cation facilitated a hyper-crosslinking reaction through FeCl3-catalyzed Friedel–Crafts alkylation. The fascinating structural design with large surface area (up to 1017 m2 g−1) showed a remarkable CO2 uptake (up to 3.05 mmol g−1 at 273 K), as well as better CO2 adsorptive selectivity over N2. In addition, ionic HCPs catalyzed the epoxide reaction using simulated flue gas (15 % CO2 in N2 v/v) with high yield in the absence of solvents and additives.[31]
Several strategies have been developed for tethering hydrogen bond donor (HBD) groups onto ionic moieties of ionic polymers.[18,32,33] HBD groups strengthen the H-bonding interaction between the catalyst and the substrate and stabilize the reaction intermediates. The impact of surface groups on the catalytic activity of imidazolium-based ionic polymers follows the order hydroxyl > alkyl > amino > carboxyl > sulfite group.[29] The better performance of the hydroxyl-tethered ionic polymer is attributed to the formation of medium-strength hydrogen bonds, which is an important pre-requisite for epoxide activation and ring opening.[29] By increasing the number of adjacent hydrogen-bond donors, the catalytic activity of the ionic polymer can be increased. For instance, a high density of urea groups in the ionic polymer backbone serves as dual hydrogen bond donors for the activation of epoxide C−O bonds. Urea-functionalized imidazolium-based ionic polymers exhibited a 2-fold higher catalytic activity compared with their counterparts without urea groups.[34]
Poly(Ionic Liquid)s (PILs)
The reactivity of PILs for CO2 fixation or conversion can be altered by manipulating the composition of the PIL or varying the chemical structures of the ions. A new family of PILs based on aromatic crosslinked poly(azomethinepyridinium) salts containing Cl–, [TFSI]– (TFSI = bis(trifluoromethyl)sulfonylimide), and [PF6]– anions were prepared in a one-step synthesis from diamine methylpyridinium salts and isophthaldehyde.[35] The different anions were found to influence the thermal stability, morphology, and catalytic efficiency of the resulting poly(azomethinepyridinium) PIL. PILs containing the chloride anion were the most promising catalysts, with 98 % substrate conversion from 0.5 mol-% of catalyst at 100°C and 3 bar (300 kPa) of CO2.[35]
Amino acid ionic liquids are acid–base multifunctional materials that possess a carboxylic acid functionality, an amino group, and nucleophile in a single molecule. Zhou et al. prepared amino acid paired PILs (AA-MPILs) as highly active metal- and halogen-free heterogeneous organocatalysts (see Fig. 6).[36] It was found that manipulating the amino groups on the cation was crucial to increase catalytic efficiency. The amino acid groups exhibit an ‘enhancement effect’ even stronger than that of hydroxyl or halogen anions, due to the better leaving ability of the carboxyl group. Among the various amino acid groups examined, those with dual carboxyl groups exhibited better efficiencies.[36] Apart from this study, investigations on PILs containing amino acid ionic liquids as ionic moieties for CO2 conversion are extremely scarce.

|
Functionalized PILs
Introducing Brønsted acid sites and Lewis basic sites in catalysts is helpful for promoting smooth cycloaddition reactions by activating and accelerating the ring-opening process of epoxides.[37] Therefore, designing PILs with high-density nucleophilic and electrophilic sites is a promising approach to attain high catalytic activity.[25,37] For this reason, functionalization of PILs by introducing HBD groups (e.g. hydroxyl, amine, carboxyl), or using a halogen as the anion are useful strategies. The synergistic effect of an HBD and a nucleophile can lower the activation energy for the rate-determining step for CO2 fixation into cyclic carbonates.[9,38]
In order to investigate the effects of different groups on the catalytic activity of PILs, Ying et al. used 2-(dimethylamino)ethyl methacrylate (DMAEMA) as a precursor to tailor different ionic liquids (ILs) and their corresponding PILs by self-polymerization.[39] The activity of the PILs decreased in the order: P(DMAEMA-EtOH)Br > P(DMAEMA-PrOH)Br > P(DMAEMA-Pr)Br > P(DMAEMA-Bu)Br > P(DMAEMA-Et)Br > P(DMAEMA-Pr)SO3. The basicity of the Br– anion significantly boosted the activity compared with the SO3– anion. Hydroxyl-rich PILs showed equivalent activity to their bulk ILs, but superior activity to other PILs due to the synergistic effect of the hydroxyl anion and the halogen nucleophile.[39] Another study showed that the yield of propylene carbonate (PC) can be increased from 72 % to 94 % (at 105°C, 2 MPa CO2, 3 h) by functionalizing the PIL with an HBD, giving a yield comparable with that using bulk ILs (95 %).[37] Both hydroxyl groups of the ILs and HBDs can form strong H-bonds with propylene oxide (PO) and help to initiate the ring-opening process. The functionalized PILs demonstrated good conversion and selectivity (>98 %) for various epoxides to form their corresponding cyclic carbonates.[37]
Li et al. synthesized heterogeneous PILs with thiourea (as an electron-deficient centre) and ammonium bromide (as a nucleophilic site).[40] The two hydrogen atoms on thiourea form strong H-bonds with epoxide; thus they can activate and initiate the reaction in synergy with the bromide ion.[40] Zhang and co-workers covalently integrated porous poly(ionic liquid)s (PPILs) with covalent organic frameworks (COFs) to form a core–shell hybrid (denoted PPIL@COF) for the first time.[41] The hydroxyl groups in the interfacial layer of the PPIL@COF act as Brønsted acid sites that could cooperate with bromide for the activation of epoxides.[41] Zhang et al. combined both homogeneous and heterogeneous catalysts, which resulted in high catalytic activity and easy separation of catalysts from the reaction products.[42] A proof-of-concept study used an amine-functionalized imidazolium PIL monolayer attached on mesoporous silica particles to ensure easy separation of catalysts from the reaction system, while swelling of the surface-attached PIL chains allowed a local homogeneous catalytic reaction to occur. Lewis acid–base interactions with the amine, together with the reactive carbon at the C(2)-position of the imidazole ring could activate CO2 for the cycloaddition reaction. The counter ions of the PILs were found to significantly influence the efficiency of the catalyst, in the order: Br– > [PF6]– > CH3COO–, owing to differences in electrophilicity and steric hindrance.[42]
Apart from HBDs, metal sites can also act as electrophiles, which together with the halogen anion greatly enhance reaction kinetics owing to the dual activation mode.[43] As a proof of concept, Xie et al. synthesized a PIL containing an electrophile and a nucleophile by free-radical polymerization of imidazolium-based monomers and bipyridine (Py)-based monomers, followed by efficient post-synthetic metallization. Cyclic carbonate yields of >92 % (1 atm (100 kPa) and 30°C for 18–48 h) were obtained for a broad range of epoxides owing to the very high density of bromide anions and zinc sites (six bromide anions in a monomer, 5.15 wt-% loading of Zn).[44] In another study, PIL-supported NHC silver complexes (poly(NHC-Ag/IL)) containing active sites of ILs and NHC-Ag complexes in the network were able to catalyze the cycloaddition reaction under mild conditions without co-catalyst.[45] A proposed mechanism for the reaction of epoxide with CO2, catalyzed by poly(NHC-Ag/IL) is a dual activation of the epoxide ring with a Lewis acid site (NHC-Ag) together with a nucleophilic reagent (Br–), resulting in ring-opening of the epoxide, followed by the addition of CO2. The poly(NHC-Ag/IL) exhibited a higher yield (83 %) compared with the corresponding PIL (74 %).[45]
Hypercrosslinked PILs
Until now, great efforts have been dedicated to modulate the chemical composition and porosity of PILs for CO2 absorption and conversion.[21] Hierachical meso and macroporous PILs have drawn tremendous attention because of their high surface area. The density of nano pores as well as the pore diameters are essential to promote rapid mass transfer during CO2 capture and conversion.[43] The synthetic strategy should be carefully tuned to take full advantage of so-called ionic HCPs. The predominant microporosity and high surface area of HCPs is advantageous for CO2 capture, and ILs or functionalized-IL monomers provide the catalytic sites for CO2 conversion.[46] A Friedel–Crafts alkylation reaction involving an electrophilic substitution step is commonly used to prepare HCPs. The choice of the cation in IL monomers can affect the linking between the cross-linkers and IL monomers.[29,46] In order to regulate the pore structures, surface functional groups, and density of ionic moieties, Sang and Huang prepared benzimidazole-based hyper-crosslinked PILs (denoted HPILs). In the FeCl3-catalyzed Friedel–Crafts alkylation reaction, the quaternization reaction between benzimidazole and benzyl halides produced imidazolium salts for HPILs. The amount of benzimidazole was used to control the N content and the porosity of the resultant materials (see Fig. 7).[46]
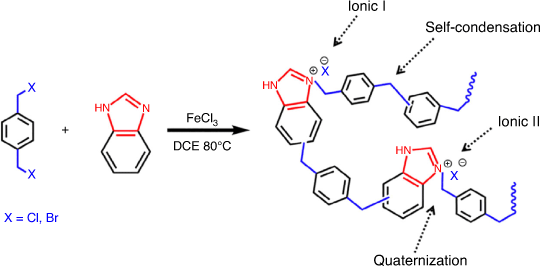
|
In another study, Gai and co-workers demonstrated the importance of using an appropriate number of benzene rings in IL monomers to promote the Friedel−Crafts alkylation to form HPILs. It was shown that using a small number of benzene rings impedes Friedel−Crafts alkylation owing to the limited number of active sites, while a large number of benzene rings in the structure creates steric hindrance, preventing the formation of an HCP.[47] The resultant porous benzene-rich PIL had a good CO2 absorption strength, high selectivity for CO2 over N2, and good subsequent CO2 conversion to cyclic compounds.
The majority of porous PILs have amorphous structures, meaning they do not contain ordered porosity spacing. Wang et al. proposed the concept of an ordered porous crystalline PIL as a way to improve both the adsorption capacity of CO2 as well as its conversion.[48] Structure-controlled, ordered crystalline porous PILs were synthesized via a thermodynamically controlled Schiff base condensation from IL-derived ionic salts and neutral monomers. In the case of an ordered porous material, the chemical composition and pore structure play a vital role in determining the surface affinity for CO2, while the ionic moieties act as active catalytic sites for CO2 conversion.
Ionic Porous Organic Polymers (iPOPs) or Porous Ionic Polymers (PIPs)
Porous organic polymers (POPs) are new and highly versatile organic polymers with high surface areas, tuneable pore sizes, and tuneable catalytic active sites.[18,49] POPs include HCPs, porous aromatic frameworks (PAFs), porous polymer networks (PPNs), conjugated microporous polymers (CMPs), COFs, and covalent triazine frameworks (CTFs).[14] Furthermore, the functionalization of POPs with nitrogen-containing groups (e.g. amines, imines, triazines, carbazoles, imidazoles, and azo linkages) has been shown to increase CO2 capture and conversion abilities.[14] Porous ionic polymers (PIPs) are a class of POPs that have controllable porous structures, abundant ionic sites, good ion exchange properties, and tuneable chemical functionalities.[50] In particular, halogen-based PIPs have better CO2 capture ability due to the strong dipole–quadrupole interactions of their ionic moieties with CO2. As discussed previously, the halogen anion plays the role of an active nucleophile for the CO2 cycloaddition reaction.[50] Moreover, IL moieties in the polymer backbone have an intrinsic hydrophilicity that can chemically absorb nearby water molecules via H-bonding. A sufficient amount of water can enhance the coupling of CO2 with epoxide by forming H-bonds with the oxygen atom on the epoxide.[37,50]
Viologen-Based iPOPs
Dicationic quaternary salts of 4,4′-bipyridine are referred to as the viologen family. Viologen-based ionic polymers exhibit unique properties because of their active ionic sites and adjustable redox states, rendering them useful for diverse applications.[50] Different synthetic methods have been adopted, including ionothermal trimerization, C–C coupling reactions (e.g. Sonogashira coupling reaction and Heck reaction), the Zincke reaction, imine condensation, and the Menshutkin reaction. Among them, the Menshutkin reaction is a convenient, highly efficient method that represents a catalyst-free route to achieving viologen-based iPOPs.[50] Zhang et al. synthesized a task-specific crystalline polymer enriched with Br– anions paired with viologen ionic sites to catalyze CO2 into cyclic carbonates and highlighted the role of chemically absorbed water molecules, which are often observed in ILs and IL-based solid materials.[50] Chemically absorbed water molecules can be regarded as HBD co-catalysts that work together with Br– anions in the activation and ring opening of epoxide (see Fig. 8).[50] A comparison between Br– and Cl– anions as nucleophiles revealed that viologen paired with a Br– anion showed better activity owing to its higher nucleophilicity and better leaving ability than the Cl– anion.[50] In another study, viologen-based iPOPs containing dual active sites i.e. Si−OH groups (HBD) and the Br– anion, demonstrated CO2 fixation under ambient pressure and low temperature without using co-catalysts or solvents.[51]

|
Metallosalen-Based iPOPs
Metallosalen-based PIPs combine the merits of both homogeneous organometallics and heterogeneous porous ionic catalysts for the CO2 cycloaddition reaction.[30] In contrast to traditional metallosalen catalysts, incorporating metallosalen complexes into a porous framework containing free mobile anions can provide a co-operative environment to promote catalytic CO2 conversion.[52] The nature of the metal, the spatial distance between the metal active site and the halogen anion, and the ratio of ionic:metal units in metallosalen-based iPOPs play a crucial role in the catalytic activity enhancement towards CO2 cycloaddition.[30] In order to study the cooperative activation from the metal (Lewis acid) and halogen anions (nucleophile), Li et al. prepared a series of porous metallosalen (M = Co, Al, Zn) hyper-crosslinked ionic polymers by Friedel−Crafts alkylation and quaternization.[30] Among them, the cobalt salen-based catalyst showed the highest activity. Theoretical investigations revealed that the reduced energy barrier of the ring-opening process associated with Co is due to its efficient coordination with the oxygen atoms of epoxide compared with Al and Zn.[30] After the polarization of the C–O bond by cobalt ions, bromide anions attack the less-hindered side of the coordinated C on the epoxide to form a Co-coordinated bromide alkoxide intermediate, which then attacks CO2 to form alkylcarbonate anions. Lastly, an intramolecular ring-closing reaction leads to the formation of cyclic carbonates (see Fig. 9).[30] Valence state and corresponding coordination groups can influence the rate-determining step of the cycloaddition reaction and nucleophilicity of the halogen anions. Density functional theory results showed that Co-salen with CoII exhibited the lowest energy barrier of the reactions, as well as an enhanced nucleophilicity of the Br– anion.[52]
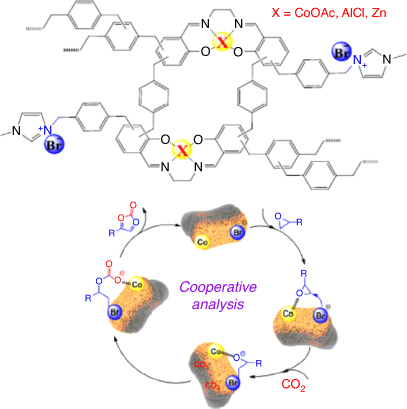
|
Metalloporphyrin-Based iPOPs
Porphyrins and their derivatives are π-conjugated macrocycles rich in nitrogen atoms that facilitate the coordination of metal ions.[17] When integrated into functional polymeric materials, metalloporphyrins provide electron-deficient Lewis acid centres for epoxide activation and ring opening.[17] Using an ‘all-in-one’ strategy, Luo et al. integrated imidazole, a halogen anion, and a metal centre into a single catalyst and prepared a heterogeneous zinc porphyrin-based cationic polymer (ZnPor-CP) for CO2 fixation.[17] The abundant imidazolium functionalities in the covalently linked skeletons and the spatially separated active Zn-porphyrin sites co-operatively enhanced the reaction under mild conditions with good recyclability.[17] In another study, the same authors demonstrated the use of a click-based reaction for the synthesis of a Co porphyrin-based ionic polymer with abundant acidic metalloporphyrin and imidazolium IL sites. The activity of the Co porphyrin ionic polymer decreased with the introduction of halogen anions in the order Br– > Cl– > I–. It was suggested that the Br– anion provides the optimum balance of nucleophilicity and leaving ability compared with the Cl– and I– anions.[53]
Porous Ionic Copolymers
Typical flue gases contain ~15 % CO2 along with large amounts of N2. The direct conversion of diluted CO2 is challenging, because the catalyst needs to be highly active and selective towards relatively small concentrations of CO2 in the presence of N2. Hui et al. synthesized an ionic co-polymer by combining protic ILs with porous polymers for efficient conversion of diluted ‘waste’ CO2 (1.0 bar (100 kPa), 15 % CO2 and 85 % N2) into cyclic carbonates under ambient conditions.[54] In another study, Sun et al. co-polymerized bi-functionalized ILs containing ether and amine groups with divinylbenzene (DVB) to produce halogen-free ionic porous copolymers.[24] The maximum Brunauer–Emmett–Teller (BET) specific surface area of the copolymer was 508 m2 g−1 with a pore volume of 0.90 cm3 g−1. The construction of porous materials and the addition of functional groups amplified both CO2 capture capacity and catalytic performance of the IL polymer for CO2 conversion.[24]
Ionic Porous Organic Frameworks
Porous organic frameworks are a subclass of POPs with a certain degree of ordered networks. Among them, N-rich ionic porous organic frameworks (POFs) contain exchangeable counter ions that act as nucleophiles for the epoxide ring-opening process. Recently, Hussain and co-workers employed metal-loaded triaminoguanidinium-based cationic frameworks for the catalytic conversion of CO2.[55] The guanidinium core not only provides suitable anchoring sites for Zn ions, but also compensates for the poor physiochemical instability of iPOFs. ZnII (47.2 %) acts as a Lewis acid to augment the catalytic activity related with such frameworks.[55]
Ionic Covalent Organic Frameworks (iCOFs)
COFs are crystalline, porous materials with two-dimensional (2D) or three-dimensional (3D) structures. They are synthesized from organic building blocks using reversible covalent bonds. Significant features include high stability, accessible open channels or nanopores with uniform sizes (ranging from angstroms to nanometres), and extremely low density.[56,57] Although COFs have been used as heterogeneous catalysts for the CO2 cycloaddition reaction, the low catalytic efficiency and/or use of co-catalysts limits their application. One way to overcome this is to functionalize a COF with an imidazolium salt, which provides cationic and anionic sites in the nanoscopic channels as active centres towards CO2 conversion. Qiu et al. demonstrated that an imidazolium salt-functionalized COF exhibited ~50 times better turnover efficiency compared with the transition metal-free COF catalyst.[56] In another study, ILs grafted onto the channel walls of COFs exhibited good catalytic activity for the synthesis of cyclic carbonates from epoxides and CO2 in the absence of solvents and co-catalysts.[57]
Zhong et al. upgraded the catalytic ability of COFs by devising a ‘host–guest’ catalytic system via integrating a nitrogen-rich COF and an imidazolium-based ionic polymer. The unification of two catalytically active components not only facilitated the epoxide and CO2 cycloaddition reaction, but also gave high absorption selectivity of CO2 over N2.[58] Hence, the entrapment ability of COFs makes them a good host to integrate with ILs or ionic polymers.
Ionic Covalent Organic Polymers (iCOPs)
Covalent organic polymers (COPs) are composed of well-defined periodic frameworks of covalently bonded organic groups. Functional building units control the catalytic properties of COPs, as well as the environment around catalytically active sites.[59] A wide flexibility in monomer selection and tailored surface manipulation with polar groups allow COPs to be used for the cycloaddition reaction of CO2. For example, Kim et al. reported a nanoporous hydrocarbon framework with grafted quaternary ammonium salts for the CO2 cycloaddition reaction.[60] The steric hinderance of the alkyl units decreased the catalytic activity in the order CH3NMe3+ > CH3NEt3+ > CH3NiPr2Et+. Thus, a higher loading of less sterically hindered ammonium salt led to higher CO2 absorption and subsequent faster cycloaddition reaction kinetics.[60] A cationic COP based on an aluminium salen (Al salen) ligand (Lewis acid site) synthesized via a post-synthetic metalation strategy exhibited good catalytic efficiency. Furthermore, COP-Al retained its efficiency for up to five reaction cycles.[61] Recently, Ayhan et al. reported efficient catalytic activity of viologen-porphyrin-based iCOPs due to the presence of multiple CO2-philic sites and efficient non-covalent interactions with CO2.[62]
Charged Covalent Triazine Frameworks (cCTFs)
CTFs are nitrogen-rich porous structures with ultra-strong aromatic C=N linkages. CTFs are versatile materials for heterogeneous catalysis owing to their high specific surface area with adjustable pore size, designable surface-active sites, and exceptional physicochemical stability.[19] In comparison with typical CTFs, charged CTFs are co-integrated with ionic moieties containing both electrophilic and nucleophilic centres within the framework, eliminating the need for a co-catalyst. Coskun and co-workers first prepared a charged CTF via a conventional ZnCl2-catalyzed trimerization reaction using 1,1′-bis(4-cyanophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride as a feedstock under ionothermal reaction conditions.[63] The microporous ionic CTF exhibited high specific surface area (1247 m2 g−1), high CO2 uptake capacity (3.0 mmol g−1 at 273 K), and high yield (up to 99 %) for propylene oxide under 1.0 MPa of CO2 at 90°C for 12 h. Cao and co-workers demonstrated the effects of polymerization temperature and the monomer/ZnCl2 ratio on porosity and ionic surface properties of CTFs.[64] Although higher polymerization temperatures increased the mesoporosity, the decomposition of imidazolium moieties and loss of spatially confined nucleophilic chloride anions dramatically reduced the catalytic performance of imidazolium-functionalized cationic CTFs synthesized at elevated temperatures.[64] The presence of abundant imidazolium and triazine units is beneficial for excellent CO2 uptake capacity, but the synergistic effect between ionic moieties is crucial for efficient catalytic activity. Considering the fact that both the metal active sites and halogen anions can significantly enhance the CO2 cycloaddition reaction under ambient conditions, the same group integrated cobalt porphyrin into imidazolium-functionalized cationic CTFs.[65] These mixed monomers led to stable CTFs with multiple catalytically active sites that not only increased CO2 absorption capacity, but also subsequent CO2 conversion.[65] It is noteworthy that the choice of an appropriate polymerization temperature is important to obtain abundant hierarchical porous structures without losing the high density of active sites.
Recyclability and Stability of Ionic Polymers
In addition to high efficiency, recyclability and stability are also important factors for catalysts before they can be used in practical applications. IL-supported inorganic–organic hybrid materials offer multiple conversions of CO2; however, the main drawback of this type of hybrid material is its coordination bond because leaching of IL from the catalyst during the reaction can occur, decreasing both the reactivity and stability.[18] Ionic polymers with both a high activity and easy separation from the product have therefore been the focus of recent research. Owing to the rich covalent chemistry and cross-linked network, ionic polymers offer sufficient stability for CO2-based liquid-phase reactions. Table 1 demonstrates the excellent recyclability and stability of ionic polymers; the majority can be reused at least five times without obvious loss of catalytic activity and selectivity. The choice of an efficient synthesis protocol and the introduction of targeted active sites (such as HBD groups, metal sites, nucleophiles) with high density and good dispersion can further enhance the stability of ionic polymers.[44] For example, a Co porphyrin-based ionic polymer synthesized via a click-based reaction was recovered and reused eight times without change in its composition.[53] The internal structure of the catalyst was unchanged and its activity remained above 98 % after eight runs. Therefore, abundant metal and IL sites and the presence of stable covalent bonds in the structure can not only improve the catalytic activity but also its reusability.

|
Conclusions and Future Prospects
Table 1 summarizes the reactions discussed in this mini review, including different epoxide substrates, emerging ionic polymers, and reaction conditions. The variety of substrates and reaction conditions make it difficult to establish a comparison and to benchmark among the different types of emerging ionic polymers. In any case, integrating ionic moieties in organic functional polymers provides tremendous opportunities to efficiently catalyze the cycloaddition of CO2 under ambient conditions owing to the strong van der Waals forces and electrostatic interactions between ILs and CO2. Cations such as imidazolium, pyridinium, viologen, ammonium, phosphonium, and guanidinium, and anions such as halides, [BF4]–, [PF6]–, and [Tf2N]– have been thoroughly explored for the CO2 cycloaddition reaction. Moreover, the task-specific design of ionic polymers and including porosity in their structure allow easy control over the catalytically active sites and cooperative interactions of the functional groups, giving rise to highly selective CO2 adsorption and simultaneous conversion. It is likely that the perfect combination of a suitable design strategy and synthetic protocol will open the doors for ionic polymers to be used in industrial applications. But there are still several future challenges and areas for research that we suggest to further develop these materials, including:
-
developing innovative ionic polymers with a high surface area (SBET), tuneable porosity, and high density of ionic moieties for improved CO2 capture and simultaneous conversion
-
upgrading synthetic strategies to reduce the cost of PILs and ILs
-
extending the range of substrates (to more than epoxide) to realize the practical application of CO2 conversion into other fine chemicals
-
co-operating with computational and theoretical chemists to understand the CO2 capture and catalytic mechanism, especially with emerging task-specific ionic polymers containing different functional groups
-
building an encouraging environment between synthetic chemists and chemical engineers to pave the way for the introduction of ionic polymer-based materials into the industrial sector. This will shift the current proof-of-concept applications to more commercial applications.
To sum up, this review shows that emerging ionic polymers are being designed owing to the rich and available ionic chemistry coming from ILs and emerging approaches to produce porous polymers. Although several works have been already reported as shown here, we believe that the area is still young and there is plenty of room for the design of more efficient catalysts that allow CO2 conversion reactions to be carried out more efficiently and at lower temperatures and pressures. It is worth remarking that although most of these polymers are being investigated in CO2 conversion reactions, they also have many possibilities as heterogeneous catalysts in other types of chemical transformations.
Data Availability Statement
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
D.S.S. thanks the Australian Research Council for funding through a Future Fellowship (FT170100315). DM thanks the Agencia Española de Investigación (AEI) for funding through project PID2020–119026GB-I00. LCT is grateful to FCT (Fundação para a Ciência e a Tecnologia) in Portugal for her research contract under Scientific Employment Stimulus (2020.01555.CEECIND). Associate Laboratory for Green Chemistry – LAQV also acknowledge the financial support from FCT/MCTES (UIDB/50006/2020 and UIDP/50006/2020).
References
[1] L. Al-Ghussain, Environ. Prog. Sustain. Energy 2019, 38, 13.| Crossref | GoogleScholarGoogle Scholar |
[2] M. Li, J. Wang, P. Li, K. Chang, C. Li, T. Wang, B. Jiang, H. Zhang, H. Liu, Y. Yamauchi, N. Umezawa, J. Ye, J. Mater. Chem. A Mater. Energy Sustain. 2016, 4, 4776.
| Crossref | GoogleScholarGoogle Scholar |
[3] Z.-L. Wang, J. Choi, M. Xu, X. Hao, H. Zhang, Z. Jiang, M. Zuo, J. Kim, W. Zhou, X. Meng, Q. Yu, Z. Sun, S. Wei, J. Ye, G. G. Wallace, D. L. Officer, Y. Yamauchi, ChemSusChem 2020, 13, 929.
| Crossref | GoogleScholarGoogle Scholar | 31880398PubMed |
[4] M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox, N. Mac Dowell, Energy Environ. Sci. 2018, 11, 1062.
| Crossref | GoogleScholarGoogle Scholar |
[5] I. Ghiat, T. Al-Ansari, J. CO Util. 2021, 45, 101432.
| Crossref | GoogleScholarGoogle Scholar |
[6] I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen, A. C. Alba-Rubio, Ind. Eng. Chem. Res. 2020, 59, 17612.
| Crossref | GoogleScholarGoogle Scholar |
[7] P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber, T. E. Müller, Energy Environ. Sci. 2012, 5, 7281.
| Crossref | GoogleScholarGoogle Scholar |
[8] J. Choi, J. Kim, P. Wagner, J. Na, G. G. Wallace, D. L. Officer, Y. Yamauchi, J. Mater. Chem. A Mater. Energy Sustain. 2020, 8, 14966.
| Crossref | GoogleScholarGoogle Scholar |
[9] C. Yang, Y. Chen, Y. Qu, J. Zhang, J. Sun, Sustain. Energy Fuels 2021, 5, 1026.
| Crossref | GoogleScholarGoogle Scholar |
[10] Y. Zhao, L. Zheng, D. Jiang, W. Xia, X. Xu, Y. Yamauchi, J. Ge, J. Tang, Small 2021, 17, 2006590.
| Crossref | GoogleScholarGoogle Scholar |
[11] B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij, C. Detrembleur, Chem. Soc. Rev. 2019, 48, 4466.
| Crossref | GoogleScholarGoogle Scholar | 31276137PubMed |
[12] C. Martín, G. Fiorani, A. W. Kleij, ACS Catal. 2015, 5, 1353.
| Crossref | GoogleScholarGoogle Scholar |
[13] B.-H. Xu, J.-Q. Wang, J. Sun, Y. Huang, J.-P. Zhang, X.-P. Zhang, S.-J. Zhang, Green Chem. 2015, 17, 108.
| Crossref | GoogleScholarGoogle Scholar |
[14] Z.-J. Li, J.-F. Sun, Q.-Q. Xu, J.-Z. Yin, ChemCatChem 2021, 13, 1848.
| Crossref | GoogleScholarGoogle Scholar |
[15] S. Ghosh, A. Modak, A. Samanta, K. Kole, S. Jana, Mater. Adv. 2021, 2, 3161.
| Crossref | GoogleScholarGoogle Scholar |
[16] P. P. Pescarmona, Curr. Opin. Green Sustain. Chem. 2021, 29, 100457.
| Crossref | GoogleScholarGoogle Scholar |
[17] X. Liu, F. Zhou, M. Chen, W. Xu, H. Liu, J. Zhong, R. Luo, ChemistrySelect 2021, 6, 583.
| Crossref | GoogleScholarGoogle Scholar |
[18] R. Luo, X. Liu, M. Chen, B. Liu, Y. Fang, ChemSusChem 2020, 13, 3945.
| Crossref | GoogleScholarGoogle Scholar | 32478431PubMed |
[19] R. Luo, W. Xu, M. Chen, X. Liu, Y. Fang, H. Ji, ChemSusChem 2020, 13, 6509.
| Crossref | GoogleScholarGoogle Scholar | 33118279PubMed |
[20] P. Bhanja, A. Modak, A. Bhaumik, ChemCatChem 2019, 11, 244.
| Crossref | GoogleScholarGoogle Scholar |
[21] X. Zhou, J. Weber, J. Yuan, Curr. Opin. Green Sustain. Chem. 2019, 16, 39.
| Crossref | GoogleScholarGoogle Scholar |
[22] Z.-W. Liu, B.-H. Han, Curr. Opin. Green Sustain. Chem. 2019, 16, 20.
| Crossref | GoogleScholarGoogle Scholar |
[23] A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad, A. Harvey, J. Environ. Chem. Eng. 2021, 9, 105113.
| Crossref | GoogleScholarGoogle Scholar |
[24] L. Sun, J. Luo, M. Gao, S. Tang, React. Funct. Polym. 2020, 154, 104636.
| Crossref | GoogleScholarGoogle Scholar |
[25] Y. He, D. Jiang, X. Li, J. Ding, H. Li, H. Wan, G. Guan, J. CO Util. 2021, 44, 101427.
| Crossref | GoogleScholarGoogle Scholar |
[26] Y. Zhang, E.-S. M. El-Sayed, K. Su, D. Yuan, Z. Han, J. CO Util. 2020, 42, 101301.
| Crossref | GoogleScholarGoogle Scholar |
[27] W. Zhang, Y. Mei, P. Wu, H.-H. Wu, M.-Y. He, Catal. Sci. Technol. 2019, 9, 1030.
| Crossref | GoogleScholarGoogle Scholar |
[28] C. Cui, R. Sa, Z. Hong, H. Zhong, R. Wang, ChemSusChem 2020, 13, 180.
| Crossref | GoogleScholarGoogle Scholar | 31710182PubMed |
[29] D. Jia, L. Ma, Y. Wang, W. Zhang, J. Li, Y. Zhou, J. Wang, Chem. Eng. J. 2020, 390, 124652.
| Crossref | GoogleScholarGoogle Scholar |
[30] J. Li, Y. Han, T. Ji, N. Wu, H. Lin, J. Jiang, J. Zhu, Ind. Eng. Chem. Res. 2020, 59, 676.
| Crossref | GoogleScholarGoogle Scholar |
[31] W. Zhang, F. Ma, L. Ma, Y. Zhou, J. Wang, ChemSusChem 2020, 13, 341.
| Crossref | GoogleScholarGoogle Scholar | 31709710PubMed |
[32] Y. Zhang, G. Chen, L. Wu, K. Liu, H. Zhong, Z. Long, M. Tong, Z. Yang, S. Dai, Chem. Commun. 2020, 3309.
| Crossref | GoogleScholarGoogle Scholar |
[33] Y. Lei, Y. Wan, W. Zhong, D. Liu, Z. Yang, Polymers 2020, 12, 596.
| Crossref | GoogleScholarGoogle Scholar |
[34] M. A. Ziaee, Y. Tang, H. Zhong, D. Tian, R. Wang, ACS Sustain. Chem. & Eng. 2019, 7, 2380.
| Crossref | GoogleScholarGoogle Scholar |
[35] E. M. Maya, E. Verde-Sesto, D. Mantione, M. Iglesias, D. Mecerreyes, Eur. Polym. J. 2019, 110, 107.
| Crossref | GoogleScholarGoogle Scholar |
[36] Y. Zhou, W. Zhang, L. Ma, Y. Zhou, J. Wang, ACS Sustain. Chem. & Eng. 2019, 7, 9387.
| Crossref | GoogleScholarGoogle Scholar |
[37] H. Gou, X. Ma, Q. Su, L. Liu, T. Ying, W. Qian, L. Dong, W. Cheng, Phys. Chem. Chem. Phys. 2021, 23, 2005.
| Crossref | GoogleScholarGoogle Scholar | 33443524PubMed |
[38] J. Zhang, X. Li, Z. Zhu, T. Chang, X. Fu, Y. Hao, X. Meng, B. Panchal, S. Qin, Adv. Sustainable Syst. 2021, 5, 2000133.
| Crossref | GoogleScholarGoogle Scholar |
[39] T. Ying, X. Tan, Q. Su, W. Cheng, L. Dong, S. Zhang, Green Chem. 2019, 21, 2352.
| Crossref | GoogleScholarGoogle Scholar |
[40] T. Dong, Y.-J. Zheng, G.-W. Yang, Y.-Y. Zhang, B. Li, G.-P. Wu, ChemSusChem 2020, 13, 4121.
| Crossref | GoogleScholarGoogle Scholar | 32662576PubMed |
[41] Y.-R. Du, G.-R. Ding, Y.-F. Wang, B.-H. Xu, S.-J. Zhang, Green Chem. 2021, 23, 2411.
| Crossref | GoogleScholarGoogle Scholar |
[42] R. Qu, Z. Ren, N. Li, F. Zhang, Z. J. Zhang, H. Zhang, J. CO Util. 2020, 38, 168.
| Crossref | GoogleScholarGoogle Scholar |
[43] Y. Fu, Y. Xu, Z. Zeng, A.-R. Ibrahim, J. Yang, S. Yang, Y. Xie, Y. Hong, Y. Su, H. Wang, Y. Wang, L. Peng, J. Li, W. L. Queen, Green Energy Environ. 2021,
| Crossref | GoogleScholarGoogle Scholar |
[44] Y. Xie, J. Liang, Y. Fu, J. Lin, H. Wang, S. Tu, J. Li, J. CO Util. 2019, 32, 281.
| Crossref | GoogleScholarGoogle Scholar |
[45] L. Qin, Y. Ji, T. Ding, B. Liu, R. Wang, L. Ji, G. Gao, Catal. Lett. 2020, 150, 1196.
| Crossref | GoogleScholarGoogle Scholar |
[46] Y. Sang, J. Huang, Chem. Eng. J. 2020, 385, 123973.
| Crossref | GoogleScholarGoogle Scholar |
[47] H. Song, Y. Wang, Y. Liu, L. Chen, B. Feng, X. Jin, Y. Zhou, T. Huang, M. Xiao, F. Huang, H. Gai, ACS Sustain. Chem. & Eng. 2021, 9, 2115.
| Crossref | GoogleScholarGoogle Scholar |
[48] J. Cao, W. Shan, Q. Wang, X. Ling, G. Li, Y. Lyu, Y. Zhou, J. Wang, ACS Appl. Mater. Interfaces 2019, 11, 6031.
| Crossref | GoogleScholarGoogle Scholar | 30648855PubMed |
[49] R. Luo, M. Chen, X. Liu, W. Xu, J. Li, B. Liu, Y. Fang, J. Mater. Chem. A Mater. Energy Sustain. 2020, 8, 18408.
| Crossref | GoogleScholarGoogle Scholar |
[50] Y. Zhang, K. Zhang, L. Wu, K. Liu, R. Huang, Z. Long, M. Tong, G. Chen, RSC Adv. 2020, 10, 3606.
| Crossref | GoogleScholarGoogle Scholar |
[51] Y. Zhang, K. Liu, L. Wu, H. Zhong, N. Luo, Y. Zhu, M. Tong, Z. Long, G. Chen, ACS Sustain. Chem. & Eng. 2019, 7, 16907.
| Crossref | GoogleScholarGoogle Scholar |
[52] J. Li, Y. Han, H. Lin, N. Wu, Q. Li, J. Jiang, J. Zhu, ACS Appl. Mater. Interfaces 2020, 12, 609.
| Crossref | GoogleScholarGoogle Scholar | 31799826PubMed |
[53] Y. Chen, R. Luo, Q. Ren, X. Zhou, H. Ji, Ind. Eng. Chem. Res. 2020, 59, 20269.
| Crossref | GoogleScholarGoogle Scholar |
[54] W. Hui, X.-M. He, X.-Y. Xu, Y.-M. Chen, Y. Zhou, Z.-M. Li, L. Zhang, D.-J. Tao, J. CO Util. 2020, 36, 169.
| Crossref | GoogleScholarGoogle Scholar |
[55] M. D. W. Hussain, V. Bhardwaj, A. Giri, A. Chande, A. Patra, Chem. Sci. 2020, 11, 7910.
| Crossref | GoogleScholarGoogle Scholar |
[56] J. Qiu, Y. Zhao, Z. Li, H. Wang, Y. Shi, J. Wang, ChemSusChem 2019, 12, 2421.
| Crossref | GoogleScholarGoogle Scholar | 30895744PubMed |
[57] Y. Zhang, H. Hu, J. Ju, Q. Yan, V. Arumugam, X. Jing, H. Cai, Y. Gao, Chin. J. Catal. 2020, 41, 485.
| Crossref | GoogleScholarGoogle Scholar |
[58] H. Zhong, J. Gao, R. Sa, S. Yang, Z. Wu, R. Wang, ChemSusChem 2020, 13, 6323.
| Crossref | GoogleScholarGoogle Scholar | 32710471PubMed |
[59] Y. Hao, X. Yan, Z. Zhu, T. Chang, X. Meng, X. Fu, B. Panchal, L. Kang, S. Qin, Sustain. Energy Fuels 2021, 5, 2943.
| Crossref | GoogleScholarGoogle Scholar |
[60] D. Kim, S. Subramanian, D. Thirion, Y. Song, A. Jamal, M. S. Otaibi, C. T. Yavuz, Catal. Today 2020, 356, 527.
| Crossref | GoogleScholarGoogle Scholar |
[61] R.-Y. Zhang, Y. Zhang, J. Tong, L. Liu, Z.-B. Han, Catal. Lett. 2021, 151, 2833.
| Crossref | GoogleScholarGoogle Scholar |
[62] S. T. Kostakoğlu, Y. Chumakov, Y. Zorlu, A. E. Sadak, S. Denizaltı, A. G. Gürek, M. M. Ayhan, Mater. Adv. 2021, 2, 3685.
| Crossref | GoogleScholarGoogle Scholar |
[63] O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim, A. Coskun, ACS Appl. Mater. Interfaces 2017, 9, 7209.
| Crossref | GoogleScholarGoogle Scholar | 28177215PubMed |
[64] T.-T. Liu, R. Xu, J.-D. Yi, J. Liang, X.-S. Wang, P.-C. Shi, Y.-B. Huang, R. Cao, ChemCatChem 2018, 10, 2036.
| Crossref | GoogleScholarGoogle Scholar |
[65] Q.-J. Wu, M.-J. Mao, J.-X. Chen, Y.-B. Huang, R. Cao, Catal. Sci. Technol. 2020, 10, 8026.
| Crossref | GoogleScholarGoogle Scholar |
* Debbie S. Silvester was awarded the 2021 Le Fèvre Medal from the Australian Academy of Science.