

My 37 years of working with nitrogen heterocycles and alkaloids
Stephen G. Pyne A *
A *
A School of Chemistry and Molecular Bioscience, University of Wollongong, Wollongong, NSW 2522, Australia.
Australian Journal of Chemistry 75(11) 923-944 https://doi.org/10.1071/CH22144
Submitted: 23 June 2022 Accepted: 11 August 2022 Published: 10 November 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
This account highlights work from my laboratory at the University of Wollongong (UOW), concerning nitrogen heterocycles and alkaloids, from my appointment as lecturer in Chemistry in February 1985 to the present time as an Emeritus Professor since 2022. I am thankful to the Royal Australian Chemical Institute for the recognition of my work through the recent award of a Distinguished Fellow at the national conference in Brisbane in July 2022.
Keywords: 1,2-amino alcohols, alkaloid, borono-Mannich reaction, cycloadditions, metal catalysis, structural corrections, sulfoxide, sulfoximine, vinyl epoxides.
Introduction
My first experience in a research laboratory was at the University of Adelaide in my BSc (Honours) year working with Dr. Ralph Massey-Westropp. My project involved the synthesis of the proline derived alkaloid odorine, isolated from a plant growing naturally in Indonesia.[1] The purpose of this work was to determine the absolute configuration of the two stereogenic centers in this molecule. This project sparked my interest in the asymmetric synthesis of natural products as a means of validating their structures and determining their configurations, a theme that continued through my academic career at Wollongong. Before my academic appointment I completed a PhD at the ANU with Lew Mander (gibberellic acid synthesis),[2] and post-doctoral fellowships with Philip Fuchs (Purdue University, on alkaloid synthesis)[3–5] and E. J. Corey (Harvard, on leukotriene synthesis and methodology).[6–9] A more senior colleague once announced at a conference ‘never work with children, animals or nitrogen compounds’, fortunately I did not heed that advice and spent my career working on the synthesis of such molecules and the isolation and synthesis of alkaloids.
Chiral sulfoxime and sulfoxide chemistry
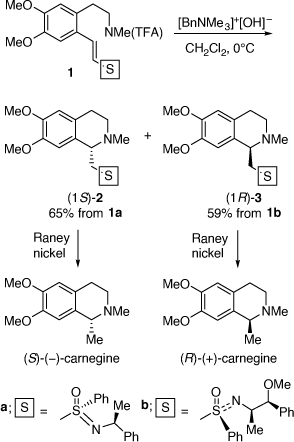
My first ARC grant supported a project on asymmetric synthesis using chiral vinyl sulfoximines and sulfoxides as chiral auxiliaries. This grant proposal was written at La Trobe University during my short time there as an independent research fellow. While my first publication resulting from this grant was on the asymmetric synthesis of chiral molecules from the addition of carbon nucleophiles (organometallic reagents) to chiral vinyl sulfoximes,[10] we soon discovered at UOW that the intramolecular addition of amine nucleophiles to these substrates was useful for the asymmetric synthesis of alkaloids. In 1986 we reported the asymmetric synthesis of (S)-(−)- and (R)-(+)-carnegine from the intramolecular addition of amine nucleophiles to chiral (S)- and (R)-vinyl sulfoximes.[11] These were prepared in situ from base hydrolysis of their N-trifluoroacetamides, 1a and 1b, respectively (Scheme 1). While the diastereoselectivities of these reactions were rather modest (diastereomeric ratios (dr) 74:26–71:29), the pure major diastereomers (1S)-2 and (1R)-3 could be isolated in respectable yields, 65 and 59%, respectively. Treatment of these individual products with Raney nickel gave (S)-(−)-carnegine ([α]D −23.5 (~0.15, EtOH); lit. ([α]D −24.9 (~4.45, EtOH));[11] and (R)-(+)-carnegine ([α]D +23.2 (~0.18, EtOH), respectively. The analogous reactions of the related (E)-vinyl (S)-tolylsulfoxides were less diastereoselective, however, those of the corresponding (Z)-vinyl (S)-tolylsulfoxides proceeded with enhanced diastereoselectivity (dr up to 84:16) allowing the synthesis of (R)-(+)-canadine.[12]
|
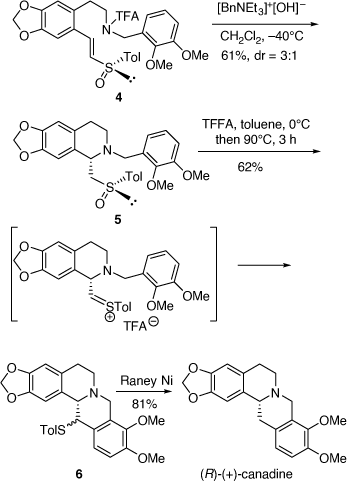
Our asymmetric synthesis of (R)-(+)-canadine involved a similar cyclization step of the chiral (E)-vinyl (R)-tolylsulfoxide 4 to form the 1-tolylsufinylmethyltetrahydroisoquinoline derivative 5 (dr = 3:1) which could be isolated as a pure diastereomer after purification by column chromatography (Scheme 2). In contrast to our earlier work,[12] the (Z)-isomer of 4 cyclized to also give 5 as the major diastereomer and with improved diastereoselectivity (dr = 4:1). The major diastereomer of 5 underwent a Pummerer reaction to give the 13-[(4-methylphenyl)thio]-6H-dibenzo[a,g]quinolizine derivatives 6 (dr = 1:1) which upon desulfurization over Raney nickel gave (R)-(+)-canadine ([α]D +273 (~0.04, CHCl3); lit. ([α]D +299 (~0.04, CHCl3)) (Scheme 2).[13]
|
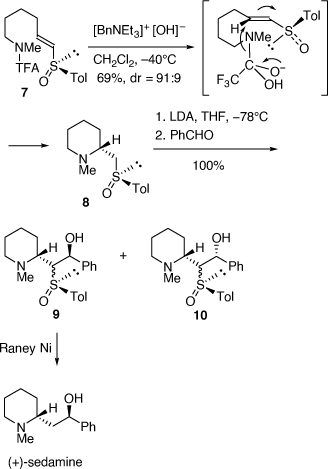
A related strategy was employed to prepare (+)- and (−)-sedamine via base-catalyzed cyclization of the (E)- and (Z)-vinyl (R)-tolylsulfoxides (Scheme 3).[14] Cyclization of the (E)-vinyl (R)-sulfoxide 7 gave the piperidine 8 (dr = 91:9) which was isolated diastereomerically pure after column chromatography. Since these cyclization reactions proceed at low temperature (−40°C) we proposed that they occur via an incipient amide ion, as shown in Scheme 3, rather than the free amine, since we found that the intermolecular reactions of amines with vinyl sulfoxides and sulfoximine require elevated temperatures (50% conversion after 6 days in refluxing EtOH) and an excess amount of the amine.[15,16] Deprotonation of 8 with lithium diisopropylamide (LDA) at −78°C and then quenching the resulting sulfoxide stabilized carbanion with benzaldehyde gave a mixture of diastereomeric aldol-like products 9 and 10, each as a mixture of syn- and anti-isomers. Reductive desulfurization of 9 gave (+)-sedamine. The (Z)-vinyl (R)-sulfoxide analogue of 7 allowed for the synthesis of (−)-sedamine.[14]
|
It is noteworthy that in Schemes 2, 3 we employed the chiral sulfoxide in two diastereoselective reactions, the intramolecular addition of an amine to a vinyl sulfoxide and then in an intramolecular Pummerer cyclization reaction to form a tetracyclic product (Scheme 2) and in an aldol-like reaction to form a new C–C bond (Scheme 3).
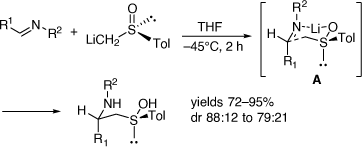
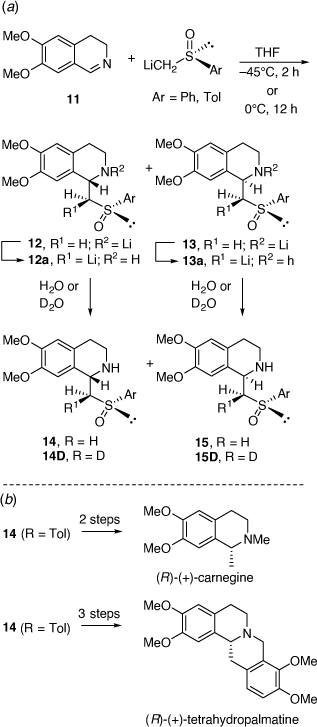
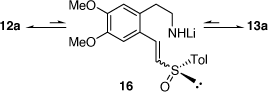
In parallel to these studies, we also studied the synthesis of related compounds via the addition of α-lithiated sulfoxides (Scheme 4) and sulfoximines to imines[17–21] and nitrones.[22] The reaction of lithiated rac-methylphenylsulfoxide (LiCH2S(O)Ar, Ar = Ph) with 3,4-dihydro-6,7-dimethoxyisoquinolne 11 at −45°C for 2 h gave in 64% yield, a 23:77 mixture of the diastereomers 14 (R = Ph) and 15 (R = Ph), respectively (Scheme 5a).[18] However, when this reaction was run at 0°C for 12 h the diastereoselectivity reversed to 89:11 in favor of 14 (R = Ph). Quenching this reaction with D2O, rather than H2O, resulted in isolation of the deuterated derivatives 14D and 15D indicating proton transfer to the nitrogen of the initially formed nitrogen-anion intermediate. The reaction of 11 with lithiated (R)-methyltolylsulfoxide ((R)-LiCH2S(O)Ar, Ar = Tol) at 0°C afforded a 92:8 mixture of diastereomers 14 (R = Tol) and 15 (R = Tol), respectively. These higher temperature results and the deuteration experiment suggested that products were formed under reversible (thermodynamic controlled) conditions. We suggested that the intermediates 12a and 13a are in equilibrium via the intermediate vinyl sulfoximine 16 (Scheme 6), allowing equilibration between the kinetically favoured intermediate 13a and the thermodynamically more favoured intermediate 12a. Compound 14 was converted into (R)-(+)-tetrahydropalmatine and (R)-(+)-carnagine (Scheme 5b).[18]
|
|
|
Other examples that have or could be employed in alkaloid synthesis include the addition of lithiated rac-N-t-butyldiphenylsilyl-S-benzyl-S-methylsulfoxime to the BF3 complex of 3,4-dihydro-6,7-dimethoxyisoquinolne 11 to give a 1-benzyltetrahydroisoquinoline with high diastereoselectivity (dr = 92:8).[19]
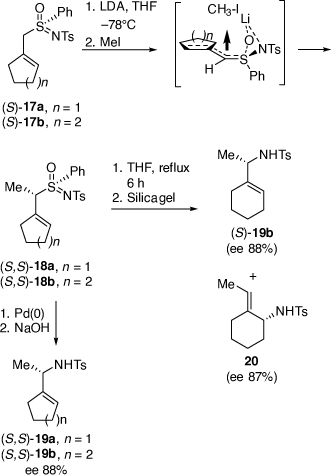
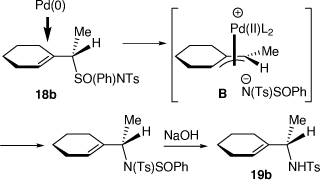
We also developed a palladium-catalyzed rearrangement of chiral allylic sulfoximines for the asymmetric synthesis of chiral N-tosylallylic amines (Scheme 7).[23–25] Treatment of allylic (S)-sulfoximines 17a or 17b with lithium diisopropylamide (LDA) and then methyl iodide gave the corresponding methylated products 18a and 18b, respectively (dr = 96–98%).[25] Heating a solution of 18b in THF for 6 h and then purification over silica gel gave a 45:55 mixture of the N-tosyl allylic sulfonamides 19b (88% ee or 92% ee corrected for the ee of 17b) and 20 (87% ee or 91% ee corrected for the ee of 17b), respectively, which were separated by semi-preparative HPLC in a combined yield of 73%. The configuration of 18b was established by its conversion into (S)-N-tosyl(1-cyclohexylethyl)amine 19b, indicating the configuration of 18b was (S,S), consistent with our earlier investigations.[26] In contrast, treatment of 18a or 18b with 5 mol% [Pd(PPh3)4] in THF at room temperature for 10 min, followed by treatment with 10% aqueous NaOH at room temperature gave only the N-tosyl allylic amines 19a and 19b, respectively, in 36 and 59% yields, both in 88% ee (92% ee corrected for the ee of 17a, b). The intermediate B that we proposed to be involved in these Pd-catalyzed reactions, can account for the observed configurations of the isolated products, is shown in Scheme 8.[25]
|
|
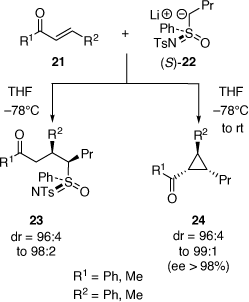
In this period, the diastereoselective reactions of lithiated sulfoximines (e.g. (S)-22 (ee 97%)) at −78°C with enones 21 was developed to give ketones 23,[26,27] with the discovery that when these reactions were warmed to room temperature then chiral cyclopropane products 24 were obtained in high ee (98%) involving cyclization of the initially formed enolate intermediate with the sulfoximine moiety acting as a leaving group (Scheme 9).[27]
|
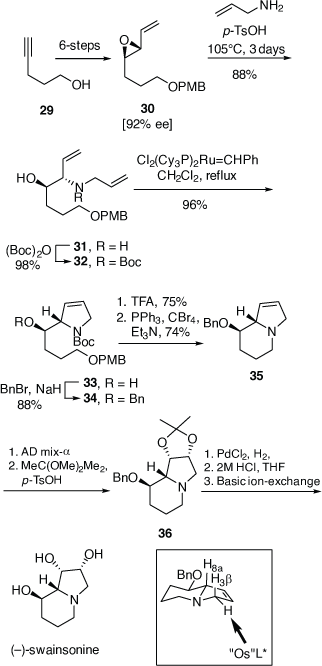
Diastereoselective synthesis of polyfunctional-pyrrolidines via vinyl epoxide aminolysis/ring-closing metathesis
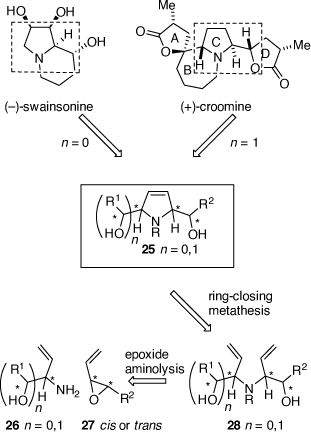
At this point in time, Sharpless had developed the catalytic asymmetric epoxidation and asymmetric dihydroxylation (ADH) reactions and other chiral catalysts for asymmetric synthesis were being developed. Chiral auxiliaries that were not recyclable, like those that we were using, were taking second place. We therefore moved out of chiral sulfur chemistry (a much-appreciated move by my colleagues in the School of Chemistry) and explored aminolysis reactions of chiral vinyl epoxides (VE) 27, now readily available via Sharpless asymmetric epoxidation reactions of their precursor allylic alcohols, as ways to access more complex alkaloids that had, as a common structural feature, a 1,2-amino alcohol (1,2-AA) moiety (e.g. (−)-swainsonine and (+)-croomine, Scheme 10). This method was teamed up with the newly described ruthenium-catalyzed ring-closing metathesis (RCM) reaction developed by Grubbs to provide rapid access to chiral 2,5-dihydropyrroles (e.g. 25) and pyrrolizidines (Scheme 10).[28–30] Using this methodology the total synthesis of (−)-swainsonine, (+)-1,2,8-tri-epi-swainsonine and (+)-1,2-di-epi-swainsonine,[31,32] and (−)-7-epiaustraline, (+)-1-epiaustraline and (+)-1,7-diepiaustraline[33,34] and (+)-(1R,2S,9S,9aR)-octahydro-1H-pyrrolo[1,2-a]azepine-1,2,9-triol, a potential glycosidase inhibitor.[35] Model studies on the synthesis of croomine resulted in the synthesis of the tricyclic B,C,D-ring core structure.[36] Unfortunately, our attempted synthesis of australine was unsuccessful using this methodology,[34] however, a latter and different strategy that we developed was.
|
Total synthesis of (−)-swainsonine
(−)-Swainsonine is a natural product and a powerful inhibitor of α-d-mannosidase and mannosidase II. Its ability to inhibit the processing of glycoproteins is likely linked with its several biological activities, including anticancer activity.[37] For our synthesis of (−)-swainsonine, 4-pentyn-1-ol 29 was converted into the VE 30 (ee 92%) in six synthetic steps. (Scheme 11).[31] Heating a solution 30 in allylamine (10 equiv.) with pTsOH·H2O (0.1 equiv.) at 105°C for 3 days gave the anti-1,2-AA 31 in 88% yield as a single diastereoisomer, the reaction proceeding with inversion of configuration at the allylic carbon. N-Boc protection of 31 gave 32 which upon RCM using Grubbs first generation catalyst (Cl2(Cy3P)2Ru = CHPh, Grubbs I) yielded, in 96% yield, the 2,5-dihydropyrrole 33. The pyrrolizidine 35 was obtained after O-benzylation of 33 and then N-Boc and O-PMB removal with TFA/anisole and finally cyclization of the resulting amino alcohol under Appel conditions (Ph3P, CBr4, Et3N, 0°C). Cis-dihydroxylation (DH) of 35 using OsO4/N-methylmorpholine N-oxide (NMO), gave a 2:1 mixture of diols favouring that required for the synthesis of (−)-swainsonine. However, DH of 35 using ADmix-α was highly diastereoselective (dr = 98:2). When ADmix-β was used there was a slight reduction in diastereoselectivity (95:5), however, the same major diastereomer was formed. The diol from DH of 35 with ADmix-α was transformed to the known acetonide 36. Its specific rotation (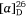 −54 (~0.6, CHCl3), lit.
−54 (~0.6, CHCl3), lit. 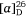 −58.9 (~0.27, CHCl3))[31] compared favourably with the literature value, considering the 92% ee of VE 30. Compound 36 was then converted into (−)-swainsonine, as shown in Scheme 11, in 94% overall yield. The high diastereoselectivity in the DH reaction of 35 with either ADmix-α or β is consistent with addition of the bulky osmium reagent to the less hindered α-face of the molecule, anti to the sterically demanding pseudo-axial protons H8a and H3β (highlighted in Scheme 11).
−58.9 (~0.27, CHCl3))[31] compared favourably with the literature value, considering the 92% ee of VE 30. Compound 36 was then converted into (−)-swainsonine, as shown in Scheme 11, in 94% overall yield. The high diastereoselectivity in the DH reaction of 35 with either ADmix-α or β is consistent with addition of the bulky osmium reagent to the less hindered α-face of the molecule, anti to the sterically demanding pseudo-axial protons H8a and H3β (highlighted in Scheme 11).
|
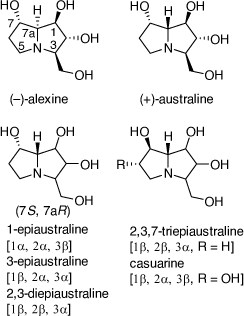
Total synthesis of (+)-1,7-di-epiaustraline and (−)-7-epiaustraline
Alexine was isolated from the seeds of Castanospermum australe and was the first alkaloid to be isolated with the 3-hydroxymethylpyrrolizine structure.[38] In later studies australine, 7a-epi-alexine, was described followed by the epimeric australines shown in Scheme 12.[38] 2,3,7-Triepiaustraline, having the identical configuration at C-7, C-7a as casurine, was the first 7-epiaustraline alkaloid to be isolated. Australine, 6-O-α-d-glucopyranosylcasurine and 2-O-β-d-glucopyranosyl-1-epiaustraline, were the most specific and potent glycosidase inhibitors. Further studies have shown that these and related compounds have anti-retroviral and antiviral activities.[38]
|
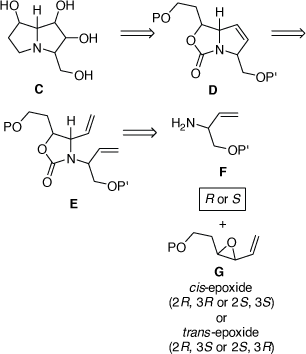
Towards the total synthesis of some of these compounds and their diastereomers we established a modular synthetic approach which could in principle be applied to the synthesis of any natural 3-hydroxymethylpyrrolizidine alkaloid or their diastereomers as outlined in Scheme 13. Aminolysis of an enantiomerically enriched trans- or cis-VE G, with the R or S enantiomer of the allylic amine F, could, in principle, provide the corresponding 1,2-AA with any required configuration. The 2-oxazolidinone E could then be obtained by protection of the 1,2-AA moiety. A RCM reaction of E would then provide the pyrrolo[1,2-c]oxazol-3-one D. The bicyclic nature of D would then allow for the stereoselective DH of the 3,4-double bond of D, a problem that we had experienced in our synthesis of (−)-swainsonine (Scheme 11). To examine the viability of this synthetic tactic we chose (+)-1,7-diepiaustraline and (−)-7-epiaustaline as our initial targets.[33]
|
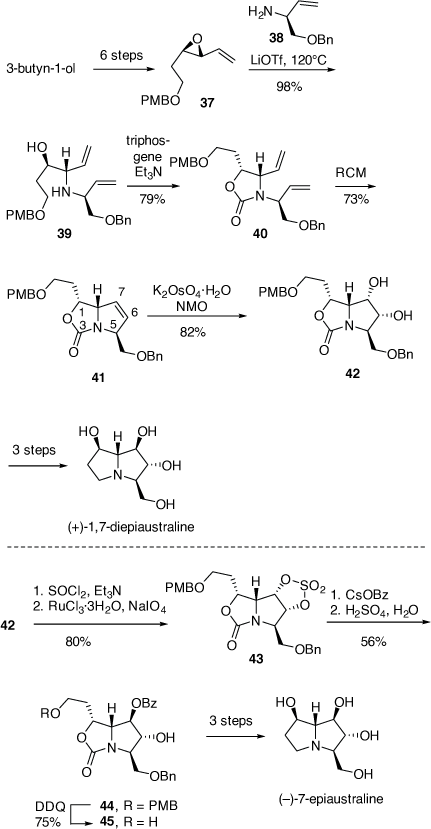
For the synthesis of 1,7-diepiaustraline the VE (+)-(2R, 3R)-37 was prepared in six steps from 3-butyn-1-ol. These steps included the Sharpless epoxidation reaction (94% ee), the Swern oxidation and a Wittig-olefination reaction (Scheme 14).[33] Treatment of 37 with (S)-allylamine 38 (1.4 equiv) and LiOTf (1.5 equiv) at 120°C for 72 h afforded the 1,2-AA 39 in 98% yield, via a regioselective SN2 ring opening reaction. A RCM reaction on the corresponding 2-oxazolidinone derivative 40 provided the pyrrolo[1,2-c]oxazol-3-one 41 in 73% yield. A DH reaction of compound 41 gave the diol 42 (82% yield), with the DH occurring at the least hindered face of the 6,7-alkene in 41 (cf. Scheme 11). The absolute configuration of 41 was confirmed from its conversion into (+)-1,7-di-epiaustraline. Its specific rotation matched closely with the literature value.[33]
|
The synthesis of (−)-7-epiaustraline required an inversion of configuration at C-7 in the pyrrolo[1,2-c]oxazol-3-one 42. Consequently, the diol moiety of 42 was transformed to its corresponding cyclic-sulfate 43, which underwent regioselective ring opening with cesium benzoate, to give the benzoate 44 in 56% yield. Nucleophilic attack on 43 had occurred regioselectively at C-7 since attack at C-6 was hindered by the β-C-5 benzyloxymethyl substituent. Treatment of 44 with DDQ provided the alcohol 45 (75% yield) which was converted into (−)-7-epiaustaline in three synthetic steps. The spectroscopic data and specific rotation of this compound were consistent with those reported.[33]
The Stemona group of alkaloids comprises over one hundred members where the pyrrolo[1,2-a]azepine nucleus is shared by most of these alkaloids (e.g. croomine).[39–41] In 2003, in collaboration with scientists from Chang Mai University, we reported the isolation and structure determination of stemocurtisine from the roots of S. curtisii.[42] This was the first Stemona alkaloid having a pyrido[1,2-a]azepine A,B-ring system. Others having this same structural feature have since been isolated.[43,44] Extracts of the roots of Stemona plants have been used in traditional Chinese medicine as anthelmintic agents and for the treatment of various respiratory diseases. The relative complexity and structural diversity of these alkaloids has resulted in many synthetic studies.[40] More recently we identified some naturally occurring and some semi-synthetic stemofoline derivatives that inhibited P-glycoprotein in multidrug resistant human leukemic cells allowing for their more efficacious chemotherapeutic treatment with standard anticancer drugs.[45–47]
In 2004, we reported our progress toward developing a convergent synthesis of croomine.[36] Our retro-synthetic analysis (Scheme 15), indicated that the A and D rings could be realized from the tetrol 46 by an oxidative lactonization reaction, while an N-alkylation reaction could be used to prepare the B-ring. A RCM reaction of the diene 47 could be employed to prepare the tetrol 46, which could be obtainable by reactions between the VPs 48 or 51 and the allylic amines 49 and 50, respectively.
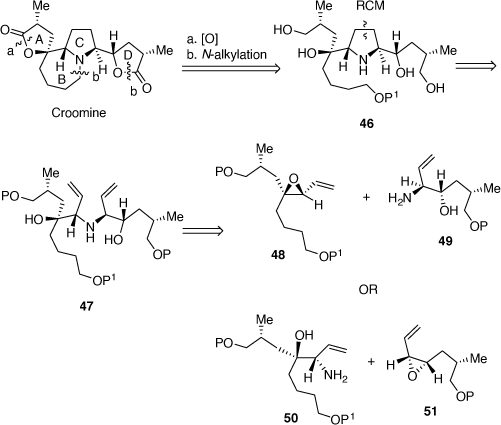
|
To test the feasibility of this approach a model study was performed using the allylic amine 54, and the VP 55, the nor-methyl analogue of 51. Compound 54 was prepared from the chiral cis-VP 53 (ee 92–94%), which was readily available in five steps (38% overall) from O-PMB protected 5-hexyn-1-ol 52 (Scheme 16). Aminolysis of 53 in aqueous ammonia at 110°C for 30 min was highly regioselective and gave the corresponding 1,2-AA in 98% yield which was protected as its O-TBS ether 54 in 85% yield (Scheme 16).
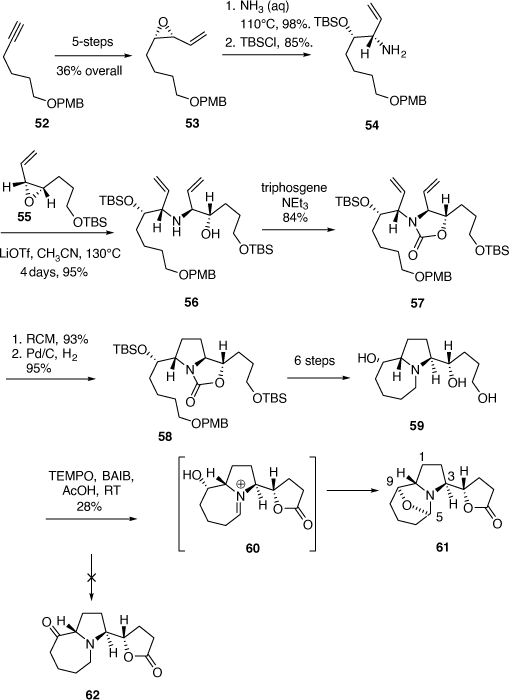
|
Thermolysis of a mixture of 54, 55 and LiOTf (1.5 equiv) in acetonitrile at 130°C for 4 days gave the AA 56 (78%, ee estimated ~95%) and its diastereomer (not shown, 17%) (Scheme 15). Compound 56 was converted into the oxazolidinone 57 which upon RCM gave the expected 2,5-dihydropyrrole product (93%) which upon hydrogenation over Pd/C smoothly gave the pyrrolidine 58 with the PMB group still intact. Compound 58 was then transformed to the triol 59 in six synthetic steps. The realization of the keto-lactone 62 by oxidation of triol 59 was problematic. For example, the use of tetrapropylammoniumperruthenate (TPAP)/NMO gave a mixture of products while the use of 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO, catalytic)/bis-acetoxy iodobenzene (BAIB, stoichiometric) in acetic acid, gave the novel 5,9-epoxy-1H-pyrrolo[1,2-a]azepine 61 in 28% yield. This compound most likely arises from the cyclic iminium ion 60 followed by ring closure by the secondary hydroxy group.
Petasis Borono-Mannich reaction (PBMR)
While our above achievements in alkaloid synthesis using chiral VEs were valuable, one major problem was that their synthesis from commercially available starting materials often required six or more synthetic steps. Thus, an alternative, shorter method for synthesizing chiral 1,2-AAs was warranted. At this point in time Petasis had reported on the PBMR as a highly diastereoselective way of preparing anti-1,2-AAs from a one-pot, three-component reaction between α-hydroxyaldehydes, 1° or 2° amines and vinyl or aryl boronates or boronic acids.[48] We have pioneered the development of the PBMR in the total synthesis of many biologically important indolizidine and pyrrolizidine aza-sugar alkaloids, and the nortropane alkaloid, calystegine B4. The α-hydroxyaldehydes that we have examined include unprotected or partially protected sugars and those prepared in situ from the ADH reactions of vinyl sulfones or the chemoselective oxidation of chiral 1,2-diols. The success of these achievements further demonstrated the use of an oxazolidinone as a protecting group for the anti-1,2-AA products for a subsequent RCM reaction of the diene moiety to build a 1,5-dihydropyrrolidine ring which then allowed diastereoselective DH of the resulting pyrrolo[1,2-c]oxazol-3-one (Scheme 17).
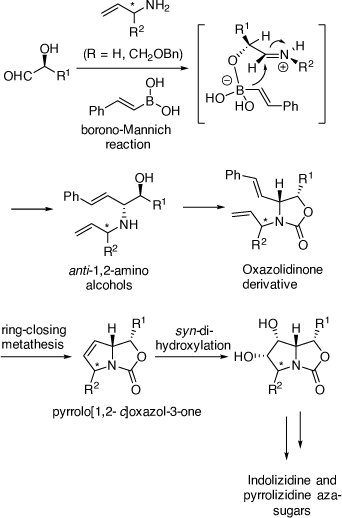
|
Correcting the structure of uniflorine A
In 2003 I had a new PhD student (Andrew Davis) who was given as his synthetic target molecule the alkaloid uniflorine A. This proposed polyhydroxylindolizidine alkaloid was isolated from the aqueous extracts of the leaves the Paraguayan tree Eugenia uniflora L.[49] These extracts were used as an antidiabetic agent in Paraguayan traditional medicine. Uniflorine A was shown to inhibit the α-glucosidases, maltase (IC50 = 12 μM) and sucrase (IC50 = 3.1 μM), consistent with the antidiabetic activity of the leaf extract. Its initial chemical structure was proposed from NMR analysis to be that shown in Scheme 18.[49] This purported indolizidine structure is related to that of castanospermine, except for the configuration at C-1 and the extra hydroxy substituent at C-2.
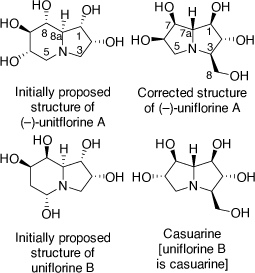
|
To achieve the synthesis of this proposed structure we employed the PBMR using l-xylose as the α-hydroxyaldehyde component, allyl amine and β-(E)-styrenylboronic acid (Scheme 19).[50] The resulting amino-tetrol 63 was obtained in 73% yield. This was transformed to the N-Boc derivative 64 (51% yield) and then the primary alcohol was selectively protected as its O-trityl ether 65 (68% yield). A RCM reaction of 65 readily gave the 2,3-dihydropyrrole 66 which upon DH furnished the pentol 67 in 88% yield as a single diastereomer due to the stereodirecting effect of the C-2 ring substituent in 66. The pentol 67 was readily converted into its penta-O-benzyl derivative 68, then selective deprotection of the secondary amino and primary hydroxy groups of 68 was achieved by treatment of 68 with TFA/anisole. Unexpectedly, this reaction resulted in a mixture of the desired AA 69 (37%) and the indolizidine 70 (54%) (Scheme 19). The AA 69 underwent cyclization under Appel conditions (Ph3P/CBr4/Et3N) to give the same indolizidine 70 (54%). Debenzylation of 70 over PdCl2/H2 gave the proposed structure of uniflorine A in 63% in a total of eight synthetic steps from l-xylose. The structure of this compound was confirmed from the single crystal X-ray analysis of its pentaacetate derivative. The 1H and 13C NMR spectroscopic data of this compound, however, did not match those reported for uniflorine A. We therefore concluded that the structure assigned to uniflorine A was not correct. After a more critical examination of the NMR data in the original isolation paper we proposed that uniflorines A and B were not indolizidines as originally proposed but were the pyrrolizidine alkaloids shown in Scheme 18.[51]
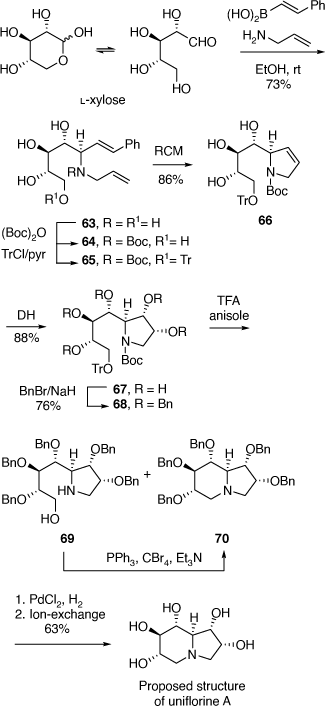
|
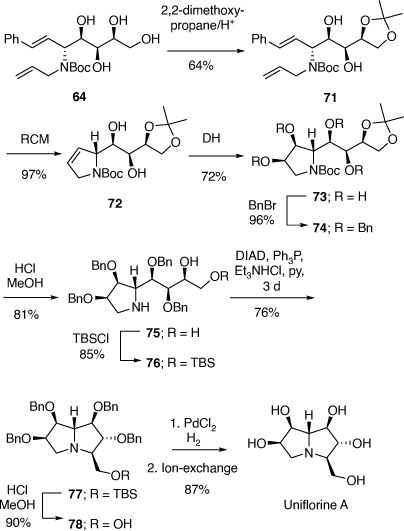
To prove our hypothesis we first prepared (+)-uniflorine A from the less expensive l-xylose[52] and then later natural (−)-uniflorine A from the more costly d-xylose (Scheme 20).[53] The terminal diol functionality of the previously prepared N-Boc-tetrol 64 (Scheme 19) was converted into its corresponding acetonide derivative 71. A RCM reaction of the diene 71 afforded the 2,5-dihydropyrrole 72 (97% yield). A DH reaction of 72 furnished the tetrol 73 as a single diastereomer (Scheme 20) due to the stereodirecting effect of the C-2 pyrrolidine substituent in 72. The tetrol 73 was then converted into its per-O-benzyl derivative 74 which upon acid hydrolysis resulted in the amino diol 75 in 81% yield. Regioselective O-silylation of 75 gave the primary silyl ether 76 in 85% yield. In our earlier synthesis of (+)-uniflorine A,[52] the compound ent-76 underwent cyclization under Mitsunobu reaction conditions to give a 4:1 mixture of the desired pyrrolizidine ent-77 and an undesired indolizidine product (structure not shown) in poor yield (30%). The undesired product arose from an initial base catalyzed O-TBS migration to the secondary hydroxy group in ent-76 followed by Mitsunobu cyclization onto the primary carbon of the butyl side chain. However, by buffering the reaction mixture with Et3N·HCl[54] we found that the yield of 77 could be enhanced to 76% with undetectible amounts, by NMR analysis, of the undesired product. Acid hydrolysis of 77 gave the primary alcohol 78 (90% yield) which upon exposure to PdCl2/H2 gave uniflorine A (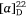 −3.7 (~1.2, H2O), lit.
−3.7 (~1.2, H2O), lit. 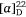 −4.4 (~1.2, H2O)),[53] in 87% yield in a total of 11 synthetic steps and 13% overall yield from l-xylose The 1H NMR spectroscopic data of our synthetic uniflorine A and those of the natural isolate were nearly identical (ΔδH = 0.00–0.02 ppm). The 13C NMR signals of our synthetic uniflorine A and that of the natural product showed consistent differences (Δδc = 2.1–2.2 ppm) which we suggested were due to alternative referencing between the two samples.
−4.4 (~1.2, H2O)),[53] in 87% yield in a total of 11 synthetic steps and 13% overall yield from l-xylose The 1H NMR spectroscopic data of our synthetic uniflorine A and those of the natural isolate were nearly identical (ΔδH = 0.00–0.02 ppm). The 13C NMR signals of our synthetic uniflorine A and that of the natural product showed consistent differences (Δδc = 2.1–2.2 ppm) which we suggested were due to alternative referencing between the two samples.
|
Total synthesis of casuarine
Casuarine was prepared from the 2,5-dihydropyrrole 72 as summarized in Scheme 21. The strategy employed required a modification to that used for uniflorine A to secure the desired 6α,7β-configuration. This was achieved via the regioselective ring-opening reaction of the epoxide 80 with NaHSO4 and then O-benzyl deprotection.[53]
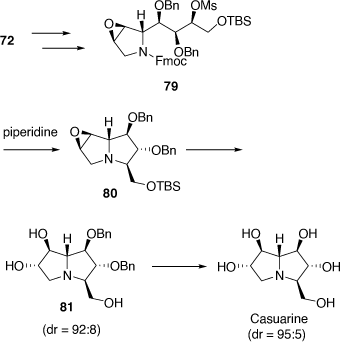
|
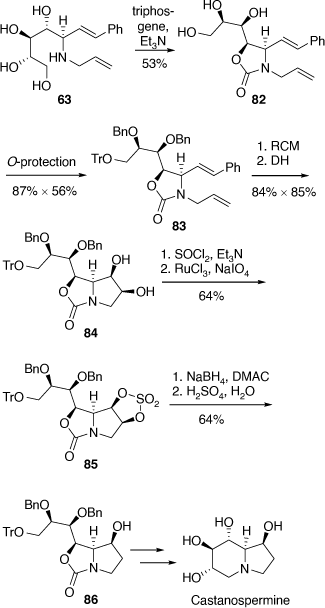
Total synthesis of castanospermine
A related synthetic strategy was applied to our synthesis of castanospermine where the amino tetrol 63 was converted into the oxazolidinone 82 which upon O-protection, RCM and then DH gave the diol 84 which was converted into the cyclic sulfate 85.[55] Regioselective ring-opening of the cyclic sulfate moiety by hydride provided the alcohol 86 which was then transformed to castanospermine. This diastereoselective and regioselective synthesis once again demonstrated the utility of pyrrolo[1,2-c]oxazol-3-one intermediates (Scheme 22).
|
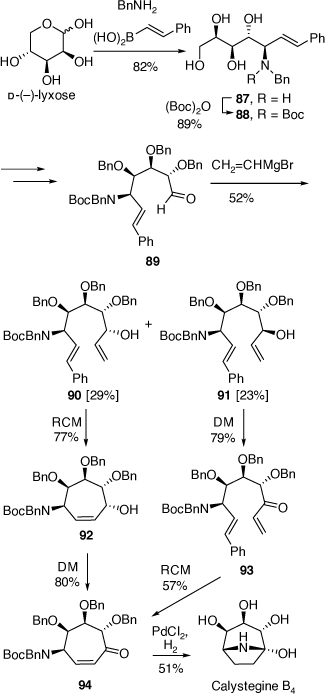
Total synthesis of calystegine B4
The calystegine alkaloids include thirteen different polyhydroxylated, 1-hydroxy-nortropanes and one trihydroxylated 1-amino-nortropane (calystegine N1). The N-methyl derivatives of calystegine B2 and C1 have also been isolated along with their glycoside derivatives.[56] A number of successful syntheses of these alkaloids have involved a protected di- or tri-hydroxylated 4-aminocycloheptene intermediate.[56] Hydroboration and then oxidation gives the corresponding 4-aminocycloheptanone which upon deprotection gives a 1-hydroxynorptropane structure. Unfortunately, these hydroboration reactions are poorly regioselective, leading, after oxidation to mixtures of 4-aminocycloheptanone products. We developed a synthetic strategy for the synthesis of calystegine B4 that avoids the possibility of formation of regioisomeric ketones (Scheme 23).[57]
|
This synthesis starts with the PBMR reaction of d-(−)-lyxose with benzylamine and trans-2-phenylvinylboronic acid which gave the amino tetrol 87 in 82% yield (Scheme 23). After a series of functional group protections and a deprotection reaction this compound was converted into the aldehyde 89 which upon treatment with vinylmagnesium bromide gave a mixture (~1:1) of the alcohols 90 and 91. This mixture was not a concern since this carbinol center was to be oxidized to the corresponding ketone in future steps. However, these diastereomers were chromatographically separated to examine their individual chemistries. The RCM reaction of 90 gave the cycloheptenol 92 in 77% yield which upon oxidation with Dess-Martin (DM) periodinane gave the protected aminocycloheptenone 94. In contrast, the RCM reaction of the diastereomer 91 was much less efficient under similar reaction conditions. However, a reversal of operations involving first oxidation of 91 to the ketone 93 and then RCM was more successful and produced the same protected aminocycloheptenone 94. Finally, treatment of 94 over PdCl2/H2 gave calystegine B4 in 51% yield.
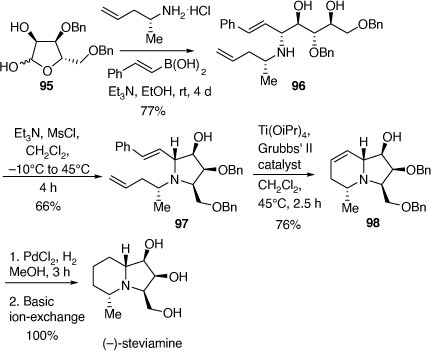
Total synthesis of (−)-steviamine
(−)-Steviamine is the first polyhydroxylated indolizidine to have a C-5 methyl and a C-3 hydroxymethyl substituent. It was isolated from the leaves of Stevia rebaudiana (Asteraceae) and the absolute configuration of its HBr salt was determined by X-ray crystallographic analysis.[58] In 2013 we reported the first synthesis of steviamine as outlined in Scheme 24.[59]
|
Our synthesis commenced with a PBMR between the l-β-ribofuranose derivative 95, (R)-4-penten-2-amine and E-styrylboronic acid (Scheme 24) which gave the 1,2-AA 96 in 77% yield. Treatment of 96 with methanesulfonyl chloride (1.07 equiv) and triethylamine (3.5 equiv) at −10°C gave the corresponding O-mesylate intermediate which cyclized to the fully substituted pyrrolidine 97 upon warming to 40–45°C. Column chromatography then gave 97 in 66% yield. A RCM reaction of diene 97 using Grubbs’ 2nd generation catalyst (18 mol%), in the presence of Ti(OiPr)4 (0.2 equiv) gave the indolizidine 98 in 76% yield. Treatment of 98, over PdCl2/H2 gave (−)-steviamine in quantitative yield. The NMR spectroscopic data of synthetic steviamine and its specific rotation matched closely with those of the natural isolate.[58] Thus, we achieved the first synthesis of (−)-steviamine in four synthetic steps from compound 95, which was prepared in four steps (45% overall yield) from commercially available β-l-ribofuranose-1,2,3,5-tetra-O-acetate. Thus, our synthesis of steviamine, involves an eight-step total synthesis in 17% overall yield, from commercially available starting materials.[59]
Total synthesis of (−)-swainsonine
We have successfully employed the PBMR using either sugars or their partially protected derivatives as the chiral α-hydroxyaldehyde component to alkalid synthesis, however, to extend the generality of this process we required a more general method of preparing such aldehydes. To this end we examined the PBMR of in situ generation of α-hydroxyaldehydes prepared from the Sharpless ADH reactions of vinyl sulfones as originally described by Evans.[60] This method has allowed much more rapid access to valuable anti-1,2-AA chiral building blocks. The derived diene products, obtained using allylamine, allowed for a short, formal synthesis of the important natural product (−)-swainsonine (Scheme 25).[61] For example, the ADH reaction of the vinyl sulfone 99 gave the α-hydroxyaldehyde 100 (formed as its cyclic hemiacetal dimer) which upon reaction with allyl amine and E-styrylboronic acid gave the 1,2-AA 101 in 93% ee and 38% overall yield. This was converted into the known indolizidine 102 which has been converted into (−)-swainsonine in two synthetic steps.
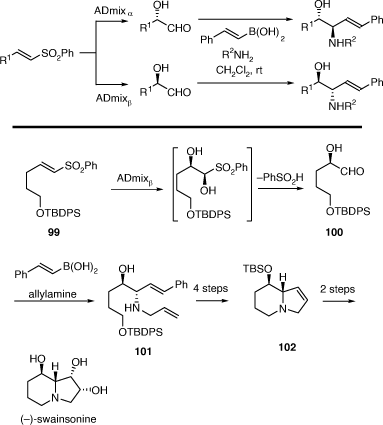
|
Total synthesis of the hyacinthacine alkaloids
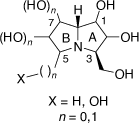
The hyacinthacine group of alkaloids comprise nineteen polyhydroxylated 3-hydroxymethylpyrrolizidine natural products of general structure shown in Scheme 26. These have been isolated from extracts of Hyacinthoides nonscripta (the common bluebell), Muscari armeniacum, Scilla campanulata, S. sibirica and S. sociali.[62] These alkaloids have been classified as hyacinthacines A1–7, B1–7 and C1–5 based on their total number of hydroxy and hydroxymethyl groups in the ring B.[62] Their structures have been assigned based primarily on NMR spectroscopic analysis. Synthetic chemistry studies have revealed, however, that the purported structures of hyacinthacines C3 and C5 are incorrect.[62]
|
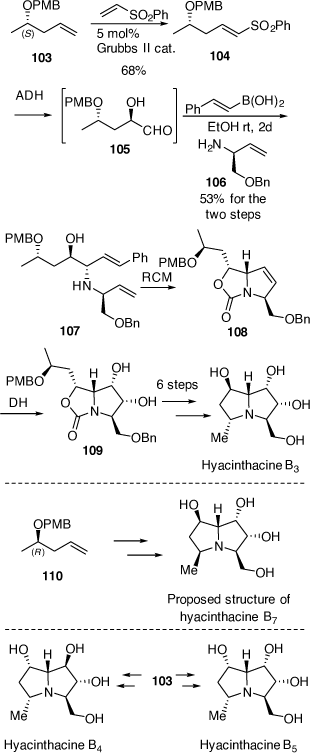
In 2010 we reported the first synthesis of hyacinthacine B3[63] which confirmed the structure and absolute configuration of this alkaloid and the synthesis of the C-5 epimer of hyacinthacine B3, hyacinthacine B7, which revealed that the purported structure of the natural isolate was incorrect.[63]
The syntheses of hyacinthacine B3 and hyacinthacine B7 from the O-PMB derivatives of (S)- and (R)-4-penten-2-ol, 103 and 110, are outlined in Scheme 27. For the synthesis of hyacinthacine B3, 103 (ee > 98%) was converted into the (E)-vinyl sulfone 104 employing a cross-metathesis reaction. Vinyl sulfone 104 reacted very sluggishly under standard ADH conditions,[60] however, using the less hindered DHQD-IND chiral ligand, 104 was converted into the corresponding α-hydroxyaldehyde 105, as a mixture of acetal derivatives, at rt in 24 h.[63] This mixture was treated with the (S)-allylic amine 106 and (E)-styrenylboronic acid, to provide the anti-1,2-AA 107 in 53% overall yield from 104. Compound 107 was converted via a RCM reaction into the pyrrolo[1,2-c]oxazol-3-one 108 and then into the diol 109 by DH with OsO4/NMO. This high level of diastereoselectivity in the DH reaction of 108 was explained based on stereoelectronic effects and nature of the HOMO of 108 about the alkene moiety where the non-bonding orbital of the N-atom overlaps more efficiently with the π-system of the alkene moiety on the α-face making this face more favorable to DH.[64,65] Importantly, the pyrrolo[1,2-c]oxazol-3-one 108 allowed us to obtain the target alkaloid with the desired 2,3-diol configuration, on essentially a trans-2,5-disubstituted-2,5-dihydropyrrole A-ring precursor, that would otherwise be expected to show poor diastereofacial selectivity. The diol 109 was then converted into hyachinthacine B3 using methods described above in earlier schemes.
|
The purported structure of hyacinthacine B7 was prepared from the O-PMB derivative of (R)-4-penten-2-ol (ee > 98%) using related chemistry (Scheme 27). Of import was that the NMR spectra of our synthetic compound did not match with that reported for hyacinthacine B7. NOESY NMR analysis of our synthetic compound clearly indicated that it had the correct relative configuration, with a significant correlation observed between H-5 and H-7. This correlation was not reported for the original isolate. Unfortunately, natural hyacinthacine B7 was no longer available to make a direct comparison, however the same Scilla socialis plants that were used in the original isolation paper were. GC-MS analysis of a fresh plant extract indicated no hyacinthacine corresponding to the retention time of 10.71 min of our synthetic compound. The tetra-TMS derivative of our synthetic compound gave a characteristic mass spectrum with a base ion peak at m/z 388. Four hyacinthacines in the new plant extract showed the same fragmentation pattern indicating they were epimers of synthetic hyachinthacine B7. One major hyacinthacine with the m/z 388 base ion peak had a retention time of 11.31 min by GC-MS which was the same retention time as a standard of hyacinthacine B5. Another epimer was also observed at 10.97 min. This GC-MS analysis indicated that our synthesized hyachinthacine B7 is not a natural product and that epimers of this compound are. We thus concluded that the structure purported for hyacinthacine B7 is incorrect. Later we reported the synthesis of hyacinthacines B4 and B5 from (2S)-4-penten-2-ol[66] which confirmed the structures and absolute configurations of these natural products. By comparing the NMR spectroscopic data of all our synthetic B-type hyacinthacines we proposed that naturally occurring hyacinthacine B7 is actually hyacinthacine B5. Unfortunately, the unavailability of these natural alkaloids did not allow us to be unequivocal about this structural reassignment.
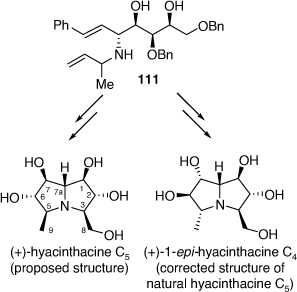
Structurally, the hyacinthacine C-type subclass of these alkaloids is the most complex and can contain up to seven possible stereogenic centers. Consequently, there are 128 unique possible diastereomers (together with their enantiomers) containing a 3-hydroxymethyl-5-methylpyrrolizidine-1,2,6,7-tetraol core that can be potentially synthesized.[67] In 2018 we reported the total synthesis of natural (+)-hyacinthacine C5, which allowed correction of its initially proposed structure to that of (+)-1-epi-hyacinthacine C4, as well as its putative structure and five additional hyacinthacine C-type alkaloids.[68] These compounds were prepared from the PBMR product 111, prepared using either (R)-2-amino-3-butene or rac-2-amino-3-butene as the amine component (Scheme 28).
|
The 1H and 13C NMR spectroscopic data of our synthetic 1-epi-hyacinthacine C4 (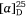 +5.2 (~1.00, H2O)) were found to be identical with those of the isolate titled hyacinthacine C5 (lit.[α]D +1.5 (~0.22, H2O)).[68] Thus the correct structure of hyacinthacine C5 has the opposite configuration at the three contiguous stereogenic centers, C-5, C-6 and C-7, to that of the originally proposed structure (Scheme 28).
+5.2 (~1.00, H2O)) were found to be identical with those of the isolate titled hyacinthacine C5 (lit.[α]D +1.5 (~0.22, H2O)).[68] Thus the correct structure of hyacinthacine C5 has the opposite configuration at the three contiguous stereogenic centers, C-5, C-6 and C-7, to that of the originally proposed structure (Scheme 28).
Hyacinthacine C1 and hyacinthacine C4 were isolated from plant extracts of Hyacinthoides non-scripta and Scilla socialis in 1999 and 2007, respectively. These alkaloids were assigned the same structures (Scheme 29),[67] even though they had different 1H and 13C NMR spectroscopic data.
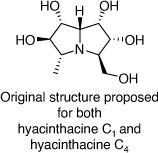
|
In 2019 we were able to correct the structure of hyacinthacine C1 through its total synthesis as shown in Scheme 30.[69] Our advanced intermediate 112 was subjected to a Swern oxidation, followed by a stereoselective reduction with l-selectride. This approach led to the synthesis of (+)-5-epi-hyacinthacine C1 which was identical to the original isolate named hyacinthacine C1.
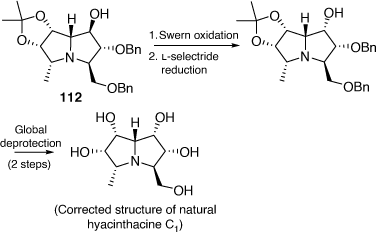
|
Other aza-sugar alkaloids
The PBMR has also been employed in a diastereoselective concise syntheses of the polyhydroxylated alkaloids DMDP and DAB[70] and progress toward the total synthesis of 9β-hydroxyvertine.[71]
Synthesis and structural correction to glyphaeaside C
The natural product glyphaeaside C was originally reported to be a derivative of the piperidine natural product 1-deoxynojirimycin. Through total synthesis of its antipode we revised its structure to a derivative of 2,5-dideoxy-2,5-imino-l-mannitol.[72] This revised l-DMDP-derived configuration is the first of its kind to be observed in Nature. The synthesis involved ring opening of the novel epoxide 113 with a Gilman-like diorganocopper reagent to give the alcohol 114 which upon a cross-metathesis reaction with 115 gave 116 as a 4.8:1 mixture of E and Z isomers, respectively. ADH of 116 with ADmix-α or ADmix-β gave their respective 8′S,9′S-syn diols and 8′R,9′R-syn diols as the major diastereomeric products, with the anti-8′,9′-diols arising from ADH of the minor Z alkene of 116. Deprotection and then separation by HPLC allowed the isolation of the four possible 8′,9′-diol diastereoisomers of the target alkaloid. The major 8′S,9′S-syn and 8′R,9′R-syn diols had almost identical NMR spectra in CD3OD as each other and as glyphaeaside C, with the specific rotation of the 8′S,9′S-syn diol closer in magnitude, but opposite in sign to that of the natural product. Consequently, we concluded that the natural product was the enantiomer of our synthetic compound, however, without access to a sample of the natural material we could not unambiguously confirm this claim. We are currently exploiting the epoxide 113 in the synthesis of the related broussonetine alkaloids (Scheme 31).
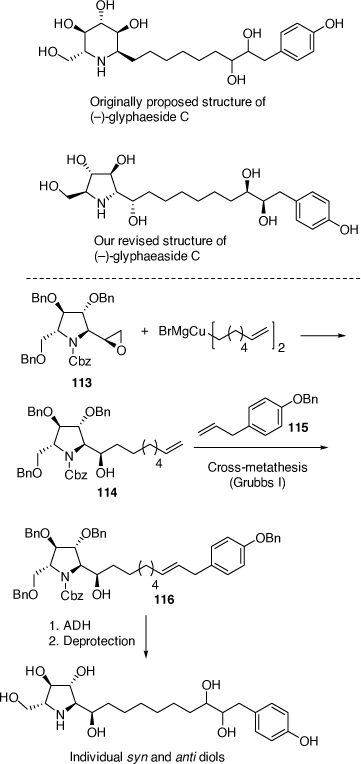
|
Other developments on the PBMR
Other developments of the PBMR include the regioselective and diastereoselective PBMR using α-hydroxyaldehydes and pinacol allenylboronate (Scheme 32a) leading to highly functionalized β-allenyl-β-amino alcohols (e.g. 117).[73] In a related study using ethyl glyoxylate as the aldehyde component, α-propargylglycinates were obtained.[74] An indium chloride catalyzed addition reaction of allenyl potassium trifluoroborate with the (S)-N-tert-butylsulfinyl imine of ethyl glyoxylate gave, after N-deprotection, (S)-ethyl α-propargylglycinate, in 97% ee.[74] In collaboration with Chris Hyland, the β-amino alcohols 117, after first O-TBS protection, were acylated using propiolic acid, DCC and DMAP to give the α-allenyl propiolamides 118 (Scheme 32b). These substrates were found to undergo intramolecular Alder-ene reactions upon heating at 110°C to give α-methylene-γ-lactams 119 in yields ranging from 69 to 92% with dr values ranging from 4.2:1 to 4.9:1.[75] These reactions occurred via a mechanism distinct from previous [2 + 2] cycloisomerization reactions of related systems. The mechanism involves an unusual transfer of the allene-hydrogen and is favored by the electron-deficient nature of the propiolamide moiety.
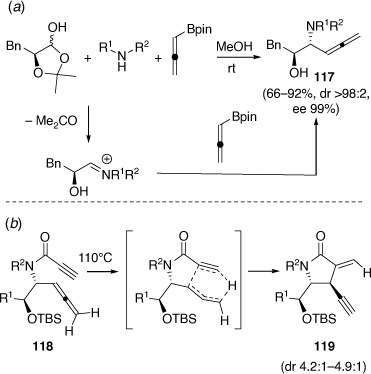
|
In related studies with Chris Hyland we prepared the α-allenyl-β-amino alcohols 121 and 122, which are isomeric with 117.[76] These were prepared in a regioselective and diastereoselective manner via the Et2Zn-catalyzed allenylation reactions of chiral N-protected l-α-amino aldehydes 120 with pinacol allenylboronate (Scheme 33a). The N-Boc protected l-α-amino aldehydes 120 (R2 = H) gave the syn products 121, consistent with a Cram-chelation like transition state (intramolecular H-bonding between the NH and aldehyde carbonyl) while the N-Boc, N-Bn protected l-α-amino aldehydes 120 (R2 = Bn) gave the anti-products 122, consistent with the Felkin-Ahn like transition state model. Treatment of 122 with an excess amount of NaH in THF gave the eneynes 123 in good yield which were converted into the ene-diynes 124 via Sonagashira coupling with aryl iodides (R2I) and then an amide coupling reaction with propiolic acid. Treatment of these ene-diynes 124 with 2.5 mol% of the basic trigold oxo complex [(Ph3PAu)3O]BF4 gave the isoindolinones 125 via a novel cycloaromatization process that involved a dual gold-catalyzed reaction pathway (Scheme 33b).[77] Other collaborative efforts have also uncovered a novel RhI-catalyzed dehydro Diels–Alder reaction of enediynes 126 proceeding via a rhodium-stabilized cyclic allene (Scheme 33c)[78] and the Pd-catalyzed asymmetric allylic alkylation reactions of sulfamidate imines.[79] In other studies on metal-promoted or metal-catalyzed cyclization reactions we have reported cyclization-cyanation and cyclization-halogenation reactions of β-hydroxyalkynes and o-alkynyl-anilines and phenols, leading to valuable 3-cyano-indoles and benzofurans,[80] and cycloisomerization reactions leading to furo[3,2-b]pyridines and furo[3,2-b]pyrroles.[81]
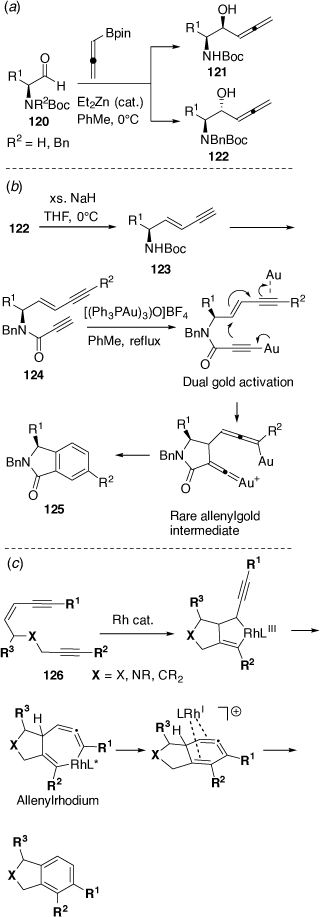
|
Further studies on the PBMR resulted in the development of a highly diastereoselective synthesis of enantioenriched anti-α-allyl-β-fluoroamines from in situ prepared chiral α-fluoroaldehydes leading to 3-, 5- and 6-membered ring heterocycles, with the latter two types having an exo-cyclic alkylfluoro-substituent (Scheme 34a);[82] and the synthesis of enantioenriched anti-β-amino alcohols (Scheme 34b) via allylation reactions of in situ prepared chiral, O-protected α-hydroxyaldehydes.[83]
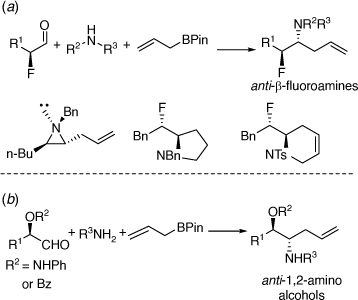
|
Synthesis of other heterocycles
My research group also pioneered the discovery of cyclization reactions of α,β-unsaturated N-acyliminium ions with tethered bisnucleophiles to provide access to novel spirocyclic and bridged heterocycles and 5,5-disubstituted pyrrolidines (Scheme 35),[84–86] including those shown in Scheme 36, that have promising biological activities. The analogous reactions of α-cyclopropyl N-acyliminium ions with tethered bisnucleophiles were also developed leading to novel spirocycles (Scheme 37).[87] Diastereoselective Ritter reactions of chiral cyclic N-acyliminium ions were also developed to allow, after hydrolysis, rapid access to chiral, substituted, 5-N-acylamino pyrrolidines and 6-N-acylamino-piperidines.[88]
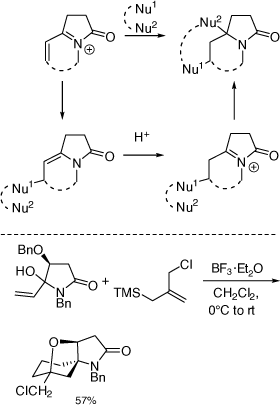
|
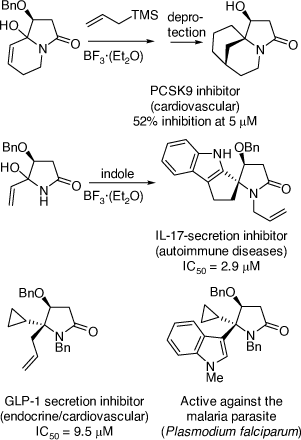
|
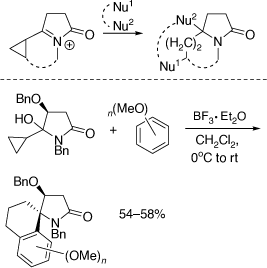
|
We have used chiral oxazolidinones in [4 + 2][89,90] and [3 + 2][91–93] cycloaddition reactions to prepare conformationally restricted amino acids, including conformationally restricted glutamates (Scheme 38a) and the carbocyclic analogue of the proherbicide hydantocidin using the related heterocyclic substrate, N,N-dibenzyl-5-methylenehydantoin (Scheme 38b).[94] These methods were extended to the synthesis of 2-azaspiro[4.4]nonan-1-ones,[95] and chiral proline derivatives[96] and to reactions involving additions of 1,3-dipolar[97,98] or free radicals.[99] Along with Paul Keller we have had a productive collaboration on the preparation of mono and disubstituted fullerenes (C60 derivatives) functionalized with amino acid derivatives, including dihydrofullerenylpyrroles.[100,101] A peptide system comprising two tethered dihydrofullerenylpyrroles allowed for the important discovery of fullerene van der Waals oligomers as electron traps.[102]
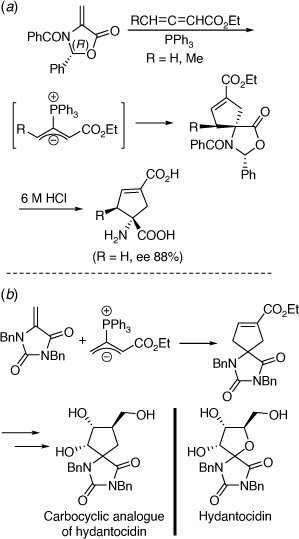
|
Isolation of natural heterocycles from Nature (alkaloids)
In the area of natural products chemistry research, our group has developed strong and very productive research collaborations with chemists and biologists in Thailand,[103,104] Malaysia,[105] Bhutan[106] and Nigeria,[107] resulting in the isolation of a number of biologically active alkaloids. A highlight of this research was the discovery of the first pyrido[1,2-a]azepine Stemona alkaloid, stemocurtisine (Scheme 39), in my lab at UOW in 2003 through a collaboration with Thai scientists.[42] We also developed a successful synthesis of the tricyclic A-B-C ring structure of stemocurtisine, however, we were not able to complete the total synthesis.[108] A study of Bhutanese traditional medicinal plant Aconitum laciniatum led to the isolation of the diterpenoid alkaloid 14-O-acetylneoline (Scheme 39) which demonstrated mitigation of inflammation in a murine model of ulcerative colitis.[109] Work on a more recent project concerned with Nigerian traditional medicinal plants that are used in the treatment of secretory diarrhea in domestic animals and humans, led to the isolation of CFTR and TMEM16A inhibitors (chloride channel blockers) from Neorautanenia mitis (A. Rich) Verdcourt which validated the traditional medicinal use of this plant.[110] The most potent isolate was dolineone (Scheme 39).
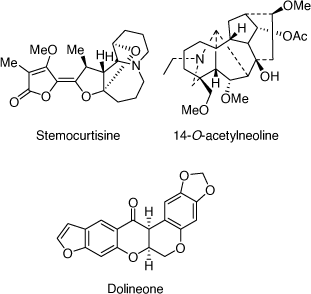
|
Conclusions
In conclusion, this account has highlighted work from my laboratory at UOW from 1985 to 2022, concerning the synthesis of nitrogen heterocycles and alkaloids starting from the use of chiral auxiliaries (sulfoxides and sulfoximines) to chiral vinyl epoxides and the PBMR using chiral α-hydroxyaldehydes. This has resulted in the synthesis of a number of alkaloids, from different structural groups, which have often confirmed the structures and configurations of these natural products and in several cases, corrected their structures. This work has also contributed a better understanding of the stereochemical outcomes of these diastereoselective reactions. Furthermore, we have developed a number of diastereoselective reactions based on other PBMR, cycloaddition chemistry, thermal and metal-catalyzed cycloisomerization reactions and N-acyl iminium ion chemistry. Working in collaboration with academics in Thailand, and through PhD students from Malaysia, Bhutan and Nigeria we have isolated and identified many novel natural products, and in some cases, verified the use of their plant source in traditional medicine.
Data availability
This is an Account of our work published in peer-reviewed journals and as such no supporting or supplementary data is provided. This information can be found within the original publications.
Conflicts of interest
The author declares no conflicts of interest.
Declaration of funding
The author thanks the Australian Research Council (DP130101968 and DP180101332) and the University of Wollongong for funding this research.
Acknowledgements
I acknowledge my academic collaborators who made valuable contributions to the work described here, Leon Kane-Maguire, Paul Keller, John Bremner, Alison Ung, Chis Hyland, Ute Wille, Alireza Ariafard, Renate Griffith, Thunwadee Limtharakul (nee Ritthiwigrom), Pitchaya Mungkornasawakul, Araya Jatisatienr, Surat Laphookhieo, Somdej Kanokmedhakul, Kwanjai Kanokmedhakul, Prapairat Seephonkai, Sumalee Kamchonwongpaisan, Pornngarm Limtrakul, Boonsom Liawruangrath, Saisunee Liawruangrath, Phurpa Wangchuk, Atsushi Kato, Robert Nash, Graham Ball, Brian Skelton, Allan White, Tony Willis, Chris Richardson and Wilford Lie. I also acknowledge the hard work of my many PhD students and post-doctoral fellows whose names are cited in the reference section. I thank the Australian Research Council and UOW for continuous support and the Royal Australian Chemical Institute for the recognition of my work through the recent award of a distinguished fellow at the national conference in Brisbane in July 2022.
References
[1] PJ Babidge, RA Massy-Westropp, SG Pyne, D Shiengthong, A Ungphakorn, G Veerachat, The synthesis and stereochemistry of odorine. Aust J Chem 1980, 33, 1841.| The synthesis and stereochemistry of odorine.Crossref | GoogleScholarGoogle Scholar |
[2] LN Mander, SG Pyne, A new strategy for gibberellin synthesis. J Am Chem Soc 1979, 101, 3373.
| A new strategy for gibberellin synthesis.Crossref | GoogleScholarGoogle Scholar |
[3] SG Pyne, MJ Hensel, SR Byrn, AT McKenzie, PL Fuchs, Cytochalasin support studies. 2. Chiral and stereochemical control via an intramolecular Diels–Alder reaction of a (Z)-diene. J Am Chem Soc 1980, 102, 5960.
| Cytochalasin support studies. 2. Chiral and stereochemical control via an intramolecular Diels–Alder reaction of a (Z)-diene.Crossref | GoogleScholarGoogle Scholar |
[4] SG Pyne, MJ Hensel, PL Fuchs, Chiral and stereochemical control via intramolecular Diels–Alder reaction of Z dienes. J Am Chem Soc 1982, 104, 5719.
| Chiral and stereochemical control via intramolecular Diels–Alder reaction of Z dienes.Crossref | GoogleScholarGoogle Scholar |
[5] SG Pyne, DC Spellmeyer, S Chen, PL Fuchs, Cytochalasin support studies. 5. Conjugate addition of .beta.-oxo ester dianions to vinyl sulfones: a new procedure for seven-ring annulation. Synthesis of a chiral cytochalasin C intermediate via an intramolecular Diels–Alder reaction of a chiral Z diene. J Am Chem Soc 1982, 104, 5728.
| Cytochalasin support studies. 5. Conjugate addition of .beta.-oxo ester dianions to vinyl sulfones: a new procedure for seven-ring annulation. Synthesis of a chiral cytochalasin C intermediate via an intramolecular Diels–Alder reaction of a chiral Z diene.Crossref | GoogleScholarGoogle Scholar |
[6] EJ Corey, SG Pyne, AI Schafer, Synthesis of a new series of potent inhibitors of thromboxane A2 biosynthesis. Tetrahedron Lett 1983, 24, 3291.
| Synthesis of a new series of potent inhibitors of thromboxane A2 biosynthesis.Crossref | GoogleScholarGoogle Scholar |
[7] EJ Corey, SG Pyne, Conversion of ketones having δ, ϵ-π-functions to cyclopentanols by zinc-trimethylchlorosilane. Tetrahedron Lett 1983, 24, 2821.
| Conversion of ketones having δ, ϵ-π-functions to cyclopentanols by zinc-trimethylchlorosilane.Crossref | GoogleScholarGoogle Scholar |
[8] EJ Corey, SG Pyne, W Su, Total synthesis of leukotriene B5. Tetrahedron Lett 1983, 24, 4883.
| Total synthesis of leukotriene B5.Crossref | GoogleScholarGoogle Scholar |
[9] AG Leitch, TH Lee, EW Ringel, JD Prickett, DR Robinson, SG Pyne, EJ Corey, JM Drazen, KF Austen, RA Lewis, Immunologically induced generation of tetraene and pentaene leukotrienes in the peritoneal cavities of menhaden-fed rats. J Immunol 1984, 132, 2559.
[10] SG Pyne, Asymmetric conjugate addition of organometallic reagents to chiral vinyl sulfoximines. J Org Chem 1986, 51, 81.
| Asymmetric conjugate addition of organometallic reagents to chiral vinyl sulfoximines.Crossref | GoogleScholarGoogle Scholar |
[11] SG Pyne, Asymmetric intramolecular conjugate addition of amines to chiral vinyl sulphoximides. Total synthesis of (R)-(+)- and (S)-(–)-carnegine. J Chem Soc Chem Commun 1986, 1986, 1686.
| Asymmetric intramolecular conjugate addition of amines to chiral vinyl sulphoximides. Total synthesis of (R)-(+)- and (S)-(–)-carnegine.Crossref | GoogleScholarGoogle Scholar |
[12] SG Pyne, SL Chapman, Asymmetric intramolecular conjugate addition of amines to chiral vinyl sulphoxides. Total synthesis of (R)-(+)-carnegine. J Chem Soc Chem Commun 1986, 1986, 1688.
| Asymmetric intramolecular conjugate addition of amines to chiral vinyl sulphoxides. Total synthesis of (R)-(+)-carnegine.Crossref | GoogleScholarGoogle Scholar |
[13] SG Pyne, Intramolecular addition of amines to chiral vinyl sulfoxides, total synthesis of ()-(+)-canadine. Tetrahedron Lett 1987, 28, 4737.
| Intramolecular addition of amines to chiral vinyl sulfoxides, total synthesis of ()-(+)-canadine.Crossref | GoogleScholarGoogle Scholar |
[14] SG Pyne, P Bloem, SL Chapman, CE Dixon, R Griffith, Chiral sulfur compounds. 9. Stereochemistry of the intermolecular and intramolecular conjugate additions of amines and anions to chiral (E)- and (Z)-vinyl sulfoxides. Total syntheses of (R)-(+)-carnegine and (+)- and (-)-sedamine. J Org Chem 1990, 55, 1086.
| Chiral sulfur compounds. 9. Stereochemistry of the intermolecular and intramolecular conjugate additions of amines and anions to chiral (E)- and (Z)-vinyl sulfoxides. Total syntheses of (R)-(+)-carnegine and (+)- and (-)-sedamine.Crossref | GoogleScholarGoogle Scholar |
[15] SG Pyne, B Dikic, Diastereoselective kinetically and thermodynamically controlled additions of (R)-(+)-methyl p-tolyl sulphoxide anion to imines (tolyl = C6H4Me). J Chem Soc Chem Commun 1989, 1989, 826.
| Diastereoselective kinetically and thermodynamically controlled additions of (R)-(+)-methyl p-tolyl sulphoxide anion to imines (tolyl = C6H4Me).Crossref | GoogleScholarGoogle Scholar |
[16] SG Pyne, P Bloem, R Griffith, Conjugate addition of amines to (Rs)-10-isobornyl vinyl sulfoxides. Tetrahedron 1989, 45, 7013.
| Conjugate addition of amines to (Rs)-10-isobornyl vinyl sulfoxides.Crossref | GoogleScholarGoogle Scholar |
[17] SG Pyne, G Boche, Chiral sulfur compounds. 7. Stereoselective reactions of lithium and zinc tert-butyl phenylmethyl sulfoxide with carbonyl compounds and imines. J Org Chem 1989, 54, 2663.
| Chiral sulfur compounds. 7. Stereoselective reactions of lithium and zinc tert-butyl phenylmethyl sulfoxide with carbonyl compounds and imines.Crossref | GoogleScholarGoogle Scholar |
[18] SG Pyne, B Dikic, Chiral sulfur compounds. II. Diastereoselective additions of (R)-(+)-methyl p-tolyl sulfoxide anion to imines. Asymmetric synthesis of (R)-(+)-tetrahydropalmatine. J Org Chem 1990, 55, 1932.
| Chiral sulfur compounds. II. Diastereoselective additions of (R)-(+)-methyl p-tolyl sulfoxide anion to imines. Asymmetric synthesis of (R)-(+)-tetrahydropalmatine.Crossref | GoogleScholarGoogle Scholar |
[19] SG Pyne, B Dikic, BW Skelton, AH White, Diastereoselective additions of lithiated N-t-butyldiphenylsilyl-S-benzyl-S-methylsulphoximine to imines. J Chem Soc Chem Commun 1990, 1990, 1376.
| Diastereoselective additions of lithiated N-t-butyldiphenylsilyl-S-benzyl-S-methylsulphoximine to imines.Crossref | GoogleScholarGoogle Scholar |
[20] SG Pyne, B Dikic, Diastereoselective additions of lithiated N-tertbutyldiphenylsilyl- S-methyl-S-phenylsulfoximine to imines and aldehydes. Tetrahedron Lett 1990, 31, 5231.
| Diastereoselective additions of lithiated N-tertbutyldiphenylsilyl- S-methyl-S-phenylsulfoximine to imines and aldehydes.Crossref | GoogleScholarGoogle Scholar |
[21] SG Pyne, B Dikic, BW Skelton, AH White, Chiral sulfur compounds. XV. Diastereoselective additions of lithiated S-benzyl-N-t-butyldiphenylsilyl-S-methyl-sulfoximine to imines and aldehydes. Aust J Chem 1992, 45, 807.
| Chiral sulfur compounds. XV. Diastereoselective additions of lithiated S-benzyl-N-t-butyldiphenylsilyl-S-methyl-sulfoximine to imines and aldehydes.Crossref | GoogleScholarGoogle Scholar |
[22] SG Pyne, AR Hajipour, Stereochemistry of the addition of lithiated methyl phenyl sulfoxide to nitrones. Tetrahedron 1992, 48, 9385.
| Stereochemistry of the addition of lithiated methyl phenyl sulfoxide to nitrones.Crossref | GoogleScholarGoogle Scholar |
[23] SG Pyne, Z Dong, BW Skelton, AH White, Diastereoselective reductions of β-substituted-γ-keto sulfoximines and a novel palladium(0)-catalysed allylic sulfoximine to allylic sulfinamide rearrangement. J Chem Soc Chem Commun 1995, 1995, 445.
| Diastereoselective reductions of β-substituted-γ-keto sulfoximines and a novel palladium(0)-catalysed allylic sulfoximine to allylic sulfinamide rearrangement.Crossref | GoogleScholarGoogle Scholar |
[24] SG Pyne, Z Dong, Palladium (0) catalysed allylic sulfoximine to allylic sulfinamide rearrangement. Tetrahedron Lett 1995, 36, 3029.
| Palladium (0) catalysed allylic sulfoximine to allylic sulfinamide rearrangement.Crossref | GoogleScholarGoogle Scholar |
[25] SG Pyne, Z Dong, Palladium-catalyzed rearrangement of allylic sulfoximines: application to the asymmetric synthesis of chiral allylic amines. J Org Chem 1996, 61, 5517.
| Palladium-catalyzed rearrangement of allylic sulfoximines: application to the asymmetric synthesis of chiral allylic amines.Crossref | GoogleScholarGoogle Scholar |
[26] SG Pyne, Z Dong, BW Skelton, AH White, Diasteoselective conjugate additions reactions of a lithiated allylic sulfoximine to acyclic enones. J Chem Soc Chem Commun 1994, 1994, 751.
| Diasteoselective conjugate additions reactions of a lithiated allylic sulfoximine to acyclic enones.Crossref | GoogleScholarGoogle Scholar |
[27] SG Pyne, Z Dong, BW Skelton, AH White, Cyclopropanation reactions of enones with lithiated sulfoximines: application to the asymmetric synthesis of chiral cyclopropanes. J Org Chem 1997, 62, 2337.
| Cyclopropanation reactions of enones with lithiated sulfoximines: application to the asymmetric synthesis of chiral cyclopropanes.Crossref | GoogleScholarGoogle Scholar |
[28] KB Lindsay, M Tang, SG Pyne, Diastereoselective synthesis of polyfunctional-pyrrolidines via vinyl epoxide aminolysis/ring-closing metathesis: synthesis of chiral 2,5-dihydropyrroles and (1R,2S,7R,7aR)-1,2,7-trihydroxypyrrolizidine. Synlett 2002, 2002, 0731.
| Diastereoselective synthesis of polyfunctional-pyrrolidines via vinyl epoxide aminolysis/ring-closing metathesis: synthesis of chiral 2,5-dihydropyrroles and (1R,2S,7R,7aR)-1,2,7-trihydroxypyrrolizidine.Crossref | GoogleScholarGoogle Scholar |
[29] AS Davis, NJ Gates, KB Lindsay, M Tang, SG Pyne, A new strategy for the diastereoselective synthesis of polyfunctionalized pyrrolidines. Synlett 2004, 2004, 49.
| A new strategy for the diastereoselective synthesis of polyfunctionalized pyrrolidines.Crossref | GoogleScholarGoogle Scholar |
[30] SG Pyne, AS Davis, NJ Gates, JP Hartley, KB Lindsay, T Machan, M Tang, Asymmetric synthesis of polyfunctionalized pyrrolidines and related alkaloids. Synlett 2004, 2004, 2670.
| Asymmetric synthesis of polyfunctionalized pyrrolidines and related alkaloids.Crossref | GoogleScholarGoogle Scholar |
[31] KB Lindsay, SG Pyne, Asymmetric synthesis of (−)-swainsonine, (+)-1,2-di-epi-swainsonine, and (+)-1,2,8-tri-epi-swainsonine. J Org Chem 2002, 67, 7774.
| Asymmetric synthesis of (−)-swainsonine, (+)-1,2-di-epi-swainsonine, and (+)-1,2,8-tri-epi-swainsonine.Crossref | GoogleScholarGoogle Scholar |
[32] KB Lindsay, SG Pyne, Asymmetric synthesis of (–)-swainsonine. Aust J Chem 2004, 57, 669.
| Asymmetric synthesis of (–)-swainsonine.Crossref | GoogleScholarGoogle Scholar |
[33] M Tang, SG Pyne, Asymmetric synthesis of (−)-7-epiaustraline and (+)-1,7-diepiaustraline. J Org Chem 2003, 68, 7818.
| Asymmetric synthesis of (−)-7-epiaustraline and (+)-1,7-diepiaustraline.Crossref | GoogleScholarGoogle Scholar |
[34] M Tang, SG Pyne, Asymmetric synthesis of (+)-1-epiaustraline and attempted synthesis of australine. Tetrahedron 2004, 60, 5759.
| Asymmetric synthesis of (+)-1-epiaustraline and attempted synthesis of australine.Crossref | GoogleScholarGoogle Scholar |
[35] KB Lindsay, SG Pyne, Synthesis of (+)-(1R,2S,9S,9aR)-octahydro-1H-pyrrolo[1,2-a]azepine-1,2,9-triol: a potential glycosidase inhibitor. Tetrahedron 2004, 60, 4173.
| Synthesis of (+)-(1R,2S,9S,9aR)-octahydro-1H-pyrrolo[1,2-a]azepine-1,2,9-triol: a potential glycosidase inhibitor.Crossref | GoogleScholarGoogle Scholar |
[36] KB Lindsay, SG Pyne, Studies on the synthesis of croomine: synthesis of the tricyclic B,C,D-ring core structure. Synlett 2004, 2004, 0779.
| Studies on the synthesis of croomine: synthesis of the tricyclic B,C,D-ring core structure.Crossref | GoogleScholarGoogle Scholar |
[37] SG Pyne, Recent developments on the synthesis of (-)-swainsonine and analogues. Curr Org Syn 2005, 2, 39.
| Recent developments on the synthesis of (-)-swainsonine and analogues.Crossref | GoogleScholarGoogle Scholar |
[38] SG Pyne, M Tang, The structure, biological activities and synthesis of 3-hydroxylpyrrolizidine alkaloids and related compounds. Curr Org Syn 2005, 9, 1393.
| The structure, biological activities and synthesis of 3-hydroxylpyrrolizidine alkaloids and related compounds.Crossref | GoogleScholarGoogle Scholar |
[39] H Greger, Structural classification and biological activities of Stemona alkaloids. Phytochem Rev 2019, 18, 463.
| Structural classification and biological activities of Stemona alkaloids.Crossref | GoogleScholarGoogle Scholar |
[40] T Iwata, M Shindo, Synthesis, stereochemical stability, and biological activity of stemonamine and its related Stemona alkaloids. Heterocycles 2019, 98, 349.
| Synthesis, stereochemical stability, and biological activity of stemonamine and its related Stemona alkaloids.Crossref | GoogleScholarGoogle Scholar |
[41] SG Pyne, A Jatisatienr, P Mungkornasawakul, AT Ung, P Limtrakul, T Sastraruji, K Sastraruji, S Chaiyong, S Umsumarng, MC Baird, XD Dau, RA Ramli, Phytochemical, synthetic and biological studies on Stemona and Stichoneuron plants and alkaloids: a personal perspective. Nat Prod Commun 2017, 12, 1365.
| Phytochemical, synthetic and biological studies on Stemona and Stichoneuron plants and alkaloids: a personal perspective.Crossref | GoogleScholarGoogle Scholar |
[42] P Mungkornasawakul, SG Pyne, A Jatisatienr, D Supyen, W Lie, AT Ung, BW Skelton, AH White, Stemocurtisine, the first pyrido[1,2-a]azapine Stemona alkaloid. J Nat Prod 2003, 66, 980.
| Stemocurtisine, the first pyrido[1,2-a]azapine Stemona alkaloid.Crossref | GoogleScholarGoogle Scholar |
[43] P Mungkornasawakul, SG Pyne, A Jatisatienr, D Supyen, C Jatisatienr, W Lie, AT Ung, BW Skelton, AH White, Phytochemical and larvicidal studies on Stemona curtisii: structure of a new pyrido[1,2-a]azepine Stemona alkaloid. J Nat Prod 2004, 67, 675.
| Phytochemical and larvicidal studies on Stemona curtisii: structure of a new pyrido[1,2-a]azepine Stemona alkaloid.Crossref | GoogleScholarGoogle Scholar |
[44] SG Pyne, AT Ung, A Jatisatienr, P Mungkornasawakul, The pyrido[1,2-a]azepine Stemona alkaloids. Maejo Int J Sci Tech 2007, 1, 157.
[45] S Umsumarng, P Pitchakarn, K Sastraruji, S Yodkeeree, AT Ung, SG Pyne, P Limtrakul, Reversal of human multi-drug resistance leukaemic cells by stemofoline derivatives via inhibition of P-glycoprotein function. Basic Clin Pharmacol Toxicol 2015, 116, 390.
| Reversal of human multi-drug resistance leukaemic cells by stemofoline derivatives via inhibition of P-glycoprotein function.Crossref | GoogleScholarGoogle Scholar |
[46] S Umsumarng, P Pitchakarn, S Yodkeeree, W Punfa, S Mapoung, RA Ramli, SG Pyne, P Limtrakul, Modulation of P-glycoprotein by Stemona alkaloids in human multidrug resistance leukemic cells and structural relationships. Phytomedicine 2017, 34, 182.
| Modulation of P-glycoprotein by Stemona alkaloids in human multidrug resistance leukemic cells and structural relationships.Crossref | GoogleScholarGoogle Scholar |
[47] S Umsumarng, S Mapoung, S Yodkeeree, SG Pyne, P Limtrakul (Dejkriengkraikul), A pharmacological strategy using stemofoline for more efficacious chemotherapeutic treatments against human multidrug resistant leukemic cells. Asian Pac J Cancer Prev 2018, 19, 3533.
| A pharmacological strategy using stemofoline for more efficacious chemotherapeutic treatments against human multidrug resistant leukemic cells.Crossref | GoogleScholarGoogle Scholar |
[48] NA Petasis, IA Zavialov, Highly stereocontrolled one-step synthesis of anti-β-amino alcohols from organoboronic acids, amines, and α-hydroxy aldehydes. J Am Chem Soc 1998, 120, 11798.
| Highly stereocontrolled one-step synthesis of anti-β-amino alcohols from organoboronic acids, amines, and α-hydroxy aldehydes.Crossref | GoogleScholarGoogle Scholar |
[49] T Matsumura, M Kasai, T Hayashi, M Arisawa, Y Momose, I Arai, S Amagaya, Y Komatsu, a-glucosidase inhibitors from Paraguayan natural medicine, Ñangapiry, the leaves of Eugenia uniflora. Pharm Biol 2000, 38, 302.
| a-glucosidase inhibitors from Paraguayan natural medicine, Ñangapiry, the leaves of Eugenia uniflora.Crossref | GoogleScholarGoogle Scholar |
[50] AS Davis, SG Pyne, BW Skelton, AH White, Synthesis of putative uniflorine A. J Org Chem 2004, 69, 3139.
| Synthesis of putative uniflorine A.Crossref | GoogleScholarGoogle Scholar |
[51] AS Davis, T Ritthiwigrom, SG Pyne, Synthetic and spectroscopic studies on the structures of uniflorines A and B: structural revision to 1,2,6,7-tetrahydroxy-3-hydroxymethylpyrrolizidine alkaloids. Tetrahedron 2008, 64, 4868.
| Synthetic and spectroscopic studies on the structures of uniflorines A and B: structural revision to 1,2,6,7-tetrahydroxy-3-hydroxymethylpyrrolizidine alkaloids.Crossref | GoogleScholarGoogle Scholar |
[52] T Ritthiwigrom, SG Pyne, Synthesis of (+)-uniflorine A: a structural reassignment and a configurational assignment. Org Lett 2008, 10, 2769.
| Synthesis of (+)-uniflorine A: a structural reassignment and a configurational assignment.Crossref | GoogleScholarGoogle Scholar |
[53] T Ritthiwigrom, AC Willis, SG Pyne, Total synthesis of uniflorine A, casuarine, australine, 3-epi-australine, and 3,7-di-epi-australine from a common precursor. J Org Chem 2010, 75, 815.
| Total synthesis of uniflorine A, casuarine, australine, 3-epi-australine, and 3,7-di-epi-australine from a common precursor.Crossref | GoogleScholarGoogle Scholar |
[54] JC Anderson, HA Chapman, Regiochemical switching of Mitsunobu cyclisation mode of vicinal diamines with pendant hydroxyl group. Org Biomol Chem 2007, 5, 2413.
| Regiochemical switching of Mitsunobu cyclisation mode of vicinal diamines with pendant hydroxyl group.Crossref | GoogleScholarGoogle Scholar |
[55] T Machan, AS Davis, B Liawruangrath, SG Pyne, Synthesis of castanospermine. Tetrahedron 2008, 64, 2725.
| Synthesis of castanospermine.Crossref | GoogleScholarGoogle Scholar |
[56] B Dräger, Chemistry and biology of calystegines. Nat Prod Rep 2004, 21, 211.
| Chemistry and biology of calystegines.Crossref | GoogleScholarGoogle Scholar |
[57] P Moosophon, MC Baird, S Kanokmedhakul, SG Pyne, Total synthesis of calystegine B4. Eur J Org Chem 2010, 2010, 3337.
| Total synthesis of calystegine B4.Crossref | GoogleScholarGoogle Scholar |
[58] A Michalik, J Hollinshead, L Jones, GWJ Fleet, C-Y Yu, X-G Hu, R van Well, G Horne, FX Wilson, A Kato, SF Jenkinson, RJ Nash, Steviamine, a new indolizidine alkaloid from Stevia rebaudiana. Phytochem Lett 2010, 3, 136.
| Steviamine, a new indolizidine alkaloid from Stevia rebaudiana.Crossref | GoogleScholarGoogle Scholar |
[59] N Jiangseubchatveera, ME Bouillon, B Liawruangrath, S Liawruangrath, RJ Nash, SG Pyne, Concise synthesis of (−)-steviamine and analogues and their glycosidase inhibitory activities. Org Biomol Chem 2013, 11, 3826.
| Concise synthesis of (−)-steviamine and analogues and their glycosidase inhibitory activities.Crossref | GoogleScholarGoogle Scholar |
[60] P Evans, M Leffray, Asymmetric dihydroxylation of vinyl sulfones: routes to enantioenriched α-hydroxyaldehydes and the enantioselective syntheses of furan-2(5H)-ones. Tetrahedron 2003, 59, 7973.
| Asymmetric dihydroxylation of vinyl sulfones: routes to enantioenriched α-hydroxyaldehydes and the enantioselective syntheses of furan-2(5H)-ones.Crossref | GoogleScholarGoogle Scholar |
[61] CWG Au, SG Pyne, Asymmetric synthesis of anti-1,2-amino alcohols via the Borono–Mannich reaction: a formal synthesis of (−)-swainsonine. J Org Chem 2006, 71, 7097.
| Asymmetric synthesis of anti-1,2-amino alcohols via the Borono–Mannich reaction: a formal synthesis of (−)-swainsonine.Crossref | GoogleScholarGoogle Scholar |
[62] T Ritthiwigrom, CWG Au, SG Pyne, Structure, biological activities and synthesis of hyacinthacine alkaloids and their stereoisomers. Curr Org Syn 2012, 9, 583.
| Structure, biological activities and synthesis of hyacinthacine alkaloids and their stereoisomers.Crossref | GoogleScholarGoogle Scholar |
[63] CWG Au, RJ Nash, SG Pyne, Synthesis of hyacinthacine B3 and purported hyacinthacine B7. Chem Commun 2010, 46, 713.
| Synthesis of hyacinthacine B3 and purported hyacinthacine B7.Crossref | GoogleScholarGoogle Scholar |
[64] AJ Murray, PJ Parsons, ES Greenwood, EME Viseux, Novel routes to the kainates: stereoselectivity in addition reactions to pyrrole [1,2c]-oxazol-3-one. Synlett 2004, 2004, 1589.
| Novel routes to the kainates: stereoselectivity in addition reactions to pyrrole [1,2c]-oxazol-3-one.Crossref | GoogleScholarGoogle Scholar |
[65] AJ Murray, PJ Parsons, P Hitchcock, The combined use of stereoelectronic control and ring closing metathesis for the synthesis of (−)-8-epi-swainsonine. Tetrahedron 2007, 63, 6485.
| The combined use of stereoelectronic control and ring closing metathesis for the synthesis of (−)-8-epi-swainsonine.Crossref | GoogleScholarGoogle Scholar |
[66] K Savaspun, CWG Au, SG Pyne, Total synthesis of hyacinthacines B3, B4, and B5 and purported hyacinthacine B7, 7-epi-hyacinthacine B7, and 7a-epi-hyacinthacine B3 from a common precursor. J Org Chem 2014, 79, 4569.
| Total synthesis of hyacinthacines B3, B4, and B5 and purported hyacinthacine B7, 7-epi-hyacinthacine B7, and 7a-epi-hyacinthacine B3 from a common precursor.Crossref | GoogleScholarGoogle Scholar |
[67] AW Carroll, SG Pyne, The history of the glycosidase inhibiting hyacinthacine C-type alkaloids: from discovery to synthesis. Curr Org Syn 2019, 16, 498.
| The history of the glycosidase inhibiting hyacinthacine C-type alkaloids: from discovery to synthesis.Crossref | GoogleScholarGoogle Scholar |
[68] AW Carroll, K Savaspun, AC Willis, M Hoshino, A Kato, SG Pyne, Total synthesis of natural hyacinthacine C5 and six related hyacinthacine C5 epimers. J Org Chem 2018, 83, 5558.
| Total synthesis of natural hyacinthacine C5 and six related hyacinthacine C5 epimers.Crossref | GoogleScholarGoogle Scholar |
[69] AW Carroll, AC Willis, M Hoshino, A Kato, SG Pyne, Corrected structure of natural hyacinthacine C1 via total synthesis. J Nat Prod 2019, 82, 358.
| Corrected structure of natural hyacinthacine C1 via total synthesis.Crossref | GoogleScholarGoogle Scholar |
[70] ME Bouillon, SG Pyne, Diastereoselective concise syntheses of the polyhydroxylated alkaloids DMDP and DAB. Tetrahedron Lett 2014, 55, 475.
| Diastereoselective concise syntheses of the polyhydroxylated alkaloids DMDP and DAB.Crossref | GoogleScholarGoogle Scholar |
[71] T Thaima, AC Willis, SG Pyne, Progress toward the total synthesis of 9β-hydroxyvertine: construction of an advanced quinolizidine intermediate. Tetrahedron 2019, 75, 130476.
| Progress toward the total synthesis of 9β-hydroxyvertine: construction of an advanced quinolizidine intermediate.Crossref | GoogleScholarGoogle Scholar |
[72] BJ Byatt, A Kato, SG Pyne, Synthesis and structural revision of glyphaeaside C. Org Lett 2021, 23, 4029.
| Synthesis and structural revision of glyphaeaside C.Crossref | GoogleScholarGoogle Scholar |
[73] T Thaima, SG Pyne, Regioselective and diastereoselective Borono–Mannich reactions with pinacol allenylboronate. Org Lett 2015, 17, 778.
| Regioselective and diastereoselective Borono–Mannich reactions with pinacol allenylboronate.Crossref | GoogleScholarGoogle Scholar |
[74] RK Chambers, N Chaipukdee, T Thaima, K Kanokmedhakul, SG Pyne, Synthesis of α-propargylglycinates using the Borono–Mannich reaction with pinacol allenylboronate and potassium allenyltrifluoroborate. Eur J Org Chem 2016, 2016, 3765.
| Synthesis of α-propargylglycinates using the Borono–Mannich reaction with pinacol allenylboronate and potassium allenyltrifluoroborate.Crossref | GoogleScholarGoogle Scholar |
[75] LM Joyce, MA Drew, AJ Tague, T Thaima, A Gouranourimi, A Ariafard, SG Pyne, CJT Hyland, A rare Alder-ene cycloisomerization of 1,6-allenynes. Chem Eur J 2022, 28, e202104022.
| A rare Alder-ene cycloisomerization of 1,6-allenynes.Crossref | GoogleScholarGoogle Scholar |
[76] F Zamani, SG Pyne, CJT Hyland, Oxazolidinones and 2,5-dihydrofurans via zinc-catalyzed regioselective allenylation reactions of l-α-amino aldehydes. J Org Chem 2017, 82, 6819.
| Oxazolidinones and 2,5-dihydrofurans via zinc-catalyzed regioselective allenylation reactions of l-α-amino aldehydes.Crossref | GoogleScholarGoogle Scholar |
[77] F Zamani, R Babaahmadi, BF Yates, MG Gardiner, A Ariafard, SG Pyne, CJT Hyland, Dual gold-catalyzed cycloaromatization of unconjugated (E)-enediynes. Angew Chem Int Ed 2019, 58, 2114.
| Dual gold-catalyzed cycloaromatization of unconjugated (E)-enediynes.Crossref | GoogleScholarGoogle Scholar |
[78] S Thadkapally, K Farshadfar, MA Drew, C Richardson, A Ariafard, SG Pyne, CJT Hyland, Rhodium-catalysed tetradehydro-Diels–Alder reactions of enediynes via a rhodium-stabilized cyclic allene. Chem Sci 2020, 11, 10945.
| Rhodium-catalysed tetradehydro-Diels–Alder reactions of enediynes via a rhodium-stabilized cyclic allene.Crossref | GoogleScholarGoogle Scholar |
[79] QH Pham, AJ Tague, C Richardson, CJT Hyland, SG Pyne, The Pd-catalysed asymmetric allylic alkylation reactions of sulfamidate imines. Chem Sci 2021, 12, 12695.
| The Pd-catalysed asymmetric allylic alkylation reactions of sulfamidate imines.Crossref | GoogleScholarGoogle Scholar |
[80] NK Swamy, A Yazici, SG Pyne, Copper-mediated cyclization−halogenation and cyclization−cyanation reactions of β-hydroxyalkynes and o-alkynylphenols and anilines. J Org Chem 2010, 75, 3412.
| Copper-mediated cyclization−halogenation and cyclization−cyanation reactions of β-hydroxyalkynes and o-alkynylphenols and anilines.Crossref | GoogleScholarGoogle Scholar |
[81] JC Jury, NK Swamy, A Yazici, AC Willis, SG Pyne, Metal-catalyzed cycloisomerization reactions of cis-4-hydroxy-5-alkynylpyrrolidinones and cis-5-hydroxy-6-alkynylpiperidinones: synthesis of furo[3,2-b]pyrroles and furo[3,2-b]pyridines. J Org Chem 2009, 74, 5523.
| Metal-catalyzed cycloisomerization reactions of cis-4-hydroxy-5-alkynylpyrrolidinones and cis-5-hydroxy-6-alkynylpiperidinones: synthesis of furo[3,2-b]pyrroles and furo[3,2-b]pyridines.Crossref | GoogleScholarGoogle Scholar |
[82] PJ Chevis, S Wangngae, T Thaima, AW Carroll, AC Willis, M Pattarawarapan, SG Pyne, Highly diastereoselective synthesis of enantioenriched anti-α-allyl-β-fluoroamines. Chem Commun 2019, 55, 6050.
| Highly diastereoselective synthesis of enantioenriched anti-α-allyl-β-fluoroamines.Crossref | GoogleScholarGoogle Scholar |
[83] PJ Chevis, T Promchai, C Richardson, T Limtharakul, SG Pyne, Synthesis of syn- and enantioenriched anti-β-amino alcohols by highly diastereoselective Borono–Mannich allylation reactions. Chem Commun 2022, 58, 2220.
| Synthesis of syn- and enantioenriched anti-β-amino alcohols by highly diastereoselective Borono–Mannich allylation reactions.Crossref | GoogleScholarGoogle Scholar |
[84] A Yazici, SG Pyne, Sequential 1,4- and 1,2-addition reactions to α,β-unsaturated N-acyliminium ions: a new strategy for the synthesis of spiro and bridged heterocycles. Org Lett 2013, 15, 5878.
| Sequential 1,4- and 1,2-addition reactions to α,β-unsaturated N-acyliminium ions: a new strategy for the synthesis of spiro and bridged heterocycles.Crossref | GoogleScholarGoogle Scholar |
[85] A Yazici, U Wille, SG Pyne, Synthesis of bridged heterocycles via sequential 1,4- and 1,2-addition reactions to α,β-unsaturated N-acyliminium ions: mechanistic and computational studies. J Org Chem 2016, 81, 1434.
| Synthesis of bridged heterocycles via sequential 1,4- and 1,2-addition reactions to α,β-unsaturated N-acyliminium ions: mechanistic and computational studies.Crossref | GoogleScholarGoogle Scholar |
[86] T Thaima, A Yazici, C Auranwiwat, AC Willis, U Wille, T Limtharakul, SG Pyne, Synthesis of spirocyclic heterocycles from α,β-unsaturated N-acyliminium ions. Org Biomol Chem 2021, 19, 259.
| Synthesis of spirocyclic heterocycles from α,β-unsaturated N-acyliminium ions.Crossref | GoogleScholarGoogle Scholar |
[87] GM Ryder, U Wille, AC Willis, SG Pyne, 1,2-Addition versus homoconjugate addition reactions of indoles and electron-rich arenes to α-cyclopropyl N-acyliminium ions: synthetic and computational studies. Org Biol Chem 2019, 17, 7025.
| 1,2-Addition versus homoconjugate addition reactions of indoles and electron-rich arenes to α-cyclopropyl N-acyliminium ions: synthetic and computational studies.Crossref | GoogleScholarGoogle Scholar |
[88] IR Morgan, A Yazici, SG Pyne, BW Skelton, Diastereoselective Ritter reactions of chiral cyclic N-acyliminium ions: synthesis of pyrido- and pyrrolo[2,3-d]oxazoles and 4-hydroxy-5-N-acylaminopyrrolidines and 5-hydroxy-6-N-acylaminopiperidines. J Org Chem 2008, 73, 2943.
| Diastereoselective Ritter reactions of chiral cyclic N-acyliminium ions: synthesis of pyrido- and pyrrolo[2,3-d]oxazoles and 4-hydroxy-5-N-acylaminopyrrolidines and 5-hydroxy-6-N-acylaminopiperidines.Crossref | GoogleScholarGoogle Scholar |
[89] SG Pyne, B Dikic, PA Gordon, BW Skelton, AH White, Highly exo-diastereoselective Diels–Alder reactions of (2S)-N-benzoyl-2-tert-butyl-4-methylene-1,3-oxazolidin-5-one. J Chem Soc Chem Commun 1991, 1991, 1505.
| Highly exo-diastereoselective Diels–Alder reactions of (2S)-N-benzoyl-2-tert-butyl-4-methylene-1,3-oxazolidin-5-one.Crossref | GoogleScholarGoogle Scholar |
[90] SG Pyne, B Dikic, P Gordon, BW Skelton, AH White, Asymmetric\ synthesis of chiral cyclic amino acids by Diels–Alder reactions of (2S)- and (2R)-4-methyleneoxazolidin-5-ones. Aust J Chem 1993, 46, 73.
| Asymmetric\ synthesis of chiral cyclic amino acids by Diels–Alder reactions of (2S)- and (2R)-4-methyleneoxazolidin-5-ones.Crossref | GoogleScholarGoogle Scholar |
[91] SG Pyne, K Schafer, BW Skelton, AH White, Synthesis of novel conformationally restricted L-glutamate analogues. Chem Commun 1997, 1997, 2267.
| Synthesis of novel conformationally restricted L-glutamate analogues.Crossref | GoogleScholarGoogle Scholar |
[92] AT Ung, K Schafer, KB Lindsay, SG Pyne, K Amornraksa, R Wouters, I Van der Linden, I Biesmans, ASJ Lesage, BW Skelton, AH White, Synthesis and biological activities of conformationally restricted cyclopentenyl–glutamate analogues. J Org Chem 2002, 67, 227.
| Synthesis and biological activities of conformationally restricted cyclopentenyl–glutamate analogues.Crossref | GoogleScholarGoogle Scholar |
[93] AT Ung, SG Pyne, U Batenburg-Nguyen, AS Davis, A Sherif, F Bischoff, ASJ Lesage, Synthesis and antagonist activities of 4-aryl-substituted conformationally restricted cyclopentenyl and cyclopentanyl–glutamate analogues. Tetrahedron 2005, 61, 1803.
| Synthesis and antagonist activities of 4-aryl-substituted conformationally restricted cyclopentenyl and cyclopentanyl–glutamate analogues.Crossref | GoogleScholarGoogle Scholar |
[94] TQ Pham, SG Pyne, BW Skelton, AH White, Synthesis of carbocyclic hydantocidins via regioselective and diastereoselective phosphine-catalyzed [3 + 2]-cycloadditions to 5-methylenehydantoins. J Org Chem 2005, 70, 6369.
| Synthesis of carbocyclic hydantocidins via regioselective and diastereoselective phosphine-catalyzed [3 + 2]-cycloadditions to 5-methylenehydantoins.Crossref | GoogleScholarGoogle Scholar |
[95] SR Yong, MC Williams, SG Pyne, AT Ung, BW Skelton, AH White, P Turner, Synthesis of 2-azaspiro[4.4]nonan-1-ones via phosphine-catalysed [3+2]-cycloadditions. Tetrahedron 2005, 61, 8120.
| Synthesis of 2-azaspiro[4.4]nonan-1-ones via phosphine-catalysed [3+2]-cycloadditions.Crossref | GoogleScholarGoogle Scholar |
[96] SG Pyne, A Javidan, BW Skelton, AH White, Asymmetric synthesis of proline derivatives from (2R) and (2S)-2-tert-butyl-3-benzoyl-4-methyleneoxazolidin-5-one. Tetrahedron 1995, 51, 5157.
| Asymmetric synthesis of proline derivatives from (2R) and (2S)-2-tert-butyl-3-benzoyl-4-methyleneoxazolidin-5-one.Crossref | GoogleScholarGoogle Scholar |
[97] SG Pyne, J Safaei-G, F Koller, Exo-Diastereoselective 1,3-dipolar cycloadditions of azomethine ylides to (2R)-3-Benzoyl-4-methylene-2-phenyloxazolidin-5-one. Tetrahedron Lett 1995, 36, 2511.
| Exo-Diastereoselective 1,3-dipolar cycloadditions of azomethine ylides to (2R)-3-Benzoyl-4-methylene-2-phenyloxazolidin-5-one.Crossref | GoogleScholarGoogle Scholar |
[98] SG Pyne, J Safaei-G, BW Skelton, AH White, 1,3-Dipolar cycloadditions of a chiral oxazolidinone with nitrones and nitrile oxides. Aust J Chem 1995, 48, 1511.
| 1,3-Dipolar cycloadditions of a chiral oxazolidinone with nitrones and nitrile oxides.Crossref | GoogleScholarGoogle Scholar |
[99] SG Pyne, K Schafer, Diastereoselective addition of α-hydroxyalkyl and α-alkoxyalkyl radicals to chiral 4-methyleneoxazolidin-5-ones. Tetrahedron 1998, 54, 5709.
| Diastereoselective addition of α-hydroxyalkyl and α-alkoxyalkyl radicals to chiral 4-methyleneoxazolidin-5-ones.Crossref | GoogleScholarGoogle Scholar |
[100] GE Ball, GA Burley, L Chaker, BC Hawkins, JR Williams, PA Keller, SG Pyne, Structural reassignment of the mono- and bis-addition products from the addition reactions of N-(diphenylmethylene)glycinate esters to [60]fullerene under Bingel conditions. J Org Chem 2005, 70, 8572.
| Structural reassignment of the mono- and bis-addition products from the addition reactions of N-(diphenylmethylene)glycinate esters to [60]fullerene under Bingel conditions.Crossref | GoogleScholarGoogle Scholar |
[101] R Thayumanavan, BC Hawkins, PA Keller, SG Pyne, GE Ball, A mild and general method for the synthesis of 5-substituted and 5,5-disubstituted fulleroprolines. Org Lett 2008, 10, 1315.
| A mild and general method for the synthesis of 5-substituted and 5,5-disubstituted fulleroprolines.Crossref | GoogleScholarGoogle Scholar |
[102] TE Shubina, DI Sharapa, C Schubert, D Zahn, M Halik, PA Keller, SG Pyne, S Jennepalli, DM Guldi, T Clark, Fullerene Van der Waals oligomers as electron traps. J Am Chem Soc 2014, 136, 10890.
| Fullerene Van der Waals oligomers as electron traps.Crossref | GoogleScholarGoogle Scholar |
[103] W Jaidee, RJ Andersen, BO Patrick, SG Pyne, C Muanprasate, S Borwornpinyo, S Laphookhieo, Alkaloids and styryllactones from Goniothalamus cheliensis. Phytochemistry 2019, 157, 8.
| Alkaloids and styryllactones from Goniothalamus cheliensis.Crossref | GoogleScholarGoogle Scholar |
[104] T Promchai, A Jaidee, S Cheenpracha, K Trisuwan, R Rattanajak, S Kamchonwongpaisan, S Laphookhieo, SG Pyne, T Ritthiwigrom, Antimalarial oxoprotoberberine alkaloids from the leaves of Miliusa cuneata. J Nat Prod 2016, 79, 978.
| Antimalarial oxoprotoberberine alkaloids from the leaves of Miliusa cuneata.Crossref | GoogleScholarGoogle Scholar |
[105] RA Ramli, W Lie, SG Pyne, Alkaloids from the roots of Stichoneuron caudatum and their acetylcholinesterase inhibitory activities. J Nat Prod 2014, 77, 894.
| Alkaloids from the roots of Stichoneuron caudatum and their acetylcholinesterase inhibitory activities.Crossref | GoogleScholarGoogle Scholar |
[106] P Wangchuk, PA Keller, SG Pyne, AC Willis, S Kamchonwongpaisan, Antimalarial alkaloids from a Bhutanese traditional medicinal plant Corydalis dubia. J Ethnopharm 2012, 143, 310.
[107] CJ Dawurung, JG Gotep, JG Usman, IL Elisha, LH Lombin, SG Pyne, Antidiarrheal activity of some selected Nigerian plants used in traditional medicine. Pharmacogn Res 2019, 11, 371.
| Antidiarrheal activity of some selected Nigerian plants used in traditional medicine.Crossref | GoogleScholarGoogle Scholar |
[108] XD Dau, AC Willis, SG Pyne, Diastereoselective synthesis of the A-B-C tricyclic ring structure of stemocurtisine. Eur J Org Chem 2015, 2015, 7682.
| Diastereoselective synthesis of the A-B-C tricyclic ring structure of stemocurtisine.Crossref | GoogleScholarGoogle Scholar |
[109] P Wangchuk, S Navarro, C Shepherd, PA Keller, SG Pyne, A Loukas, Diterpenoid alkaloids of Aconitum laciniatum and mitigation of inflammation by 14-O-acetylneoline in a murine model of ulcerative colitis. Sci Rep 2015, 5, 12845.2015
| Diterpenoid alkaloids of Aconitum laciniatum and mitigation of inflammation by 14-O-acetylneoline in a murine model of ulcerative colitis.Crossref | GoogleScholarGoogle Scholar |
[110] CJ Dawurung, R Noitem, R Rattanajak, R Bunyong, C Richardson, AC Willis, S Kamchonwongpaisan, C Yimnual, C Muanprasat, SG Pyne, Isolation of CFTR and TMEM16A inhibitors from Neorautanenia mitis (A. Rich) Verdcourt: potential lead compounds for treatment of secretory diarrhea. Phytochemistry 2020, 179, 112464.
| Isolation of CFTR and TMEM16A inhibitors from Neorautanenia mitis (A. Rich) Verdcourt: potential lead compounds for treatment of secretory diarrhea.Crossref | GoogleScholarGoogle Scholar |