

Comparison of an extracellular v. total DNA extraction approach for environmental DNA-based monitoring of sediment biota
Johan Pansu A B C D , Michelle B. Chapman C , Grant C. Hose C and Anthony A. Chariton C
A B C D , Michelle B. Chapman C , Grant C. Hose C and Anthony A. Chariton C
A Station Biologique de Roscoff, UMR 7144, CNRS, Sorbonne Université, Place Georges Teissier, F-29680 Roscoff, France.
B CSIRO Oceans & Atmosphere, New Illawarra Road, Lucas Heights, NSW 2234, Australia.
C Department of Biological Sciences, Macquarie University, Balaclava Road, Macquarie Park, NSW 2109, Australia.
D Corresponding author. Present address: Institut des Sciences de l’Évolution de Montpellier (ISEM, UMR 5554), IPHE, IRD, Université de Montpellier, Place Eugène Bataillon, F-34000 Montpellier, France. Email: johan.pansu@gmail.com
Marine and Freshwater Research - https://doi.org/10.1071/MF20269
Submitted: 2 September 2020 Accepted: 1 December 2020 Published online: 23 February 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
Monitoring sediment biota is an essential step for the quality assessment of aquatic ecosystems. Environmental DNA-based approaches for biomonitoring are increasing in popularity; yet, commercial kits and protocols for extracting total DNA from sediments remain expensive and time-consuming. Furthermore, they can accommodate only small amounts of sediments, potentially preventing an adequate representation of local biodiversity, especially for macro-organisms. Here, we assessed the reliability of a cost- and time-effective extracellular DNA extraction approach able to account for large volumes of starting material, for characterising bacterial, eukaryote and metazoan communities in three sedimentary environments. DNA concentrations extracted with the extracellular approach were at least similar to those obtained with the commercial kit. Local diversity estimates were not biased towards any particular extraction method, although specific responses were observed depending of the sediment type. Community composition and β-diversity patterns were moderately affected by the extraction approach and the initial amount of starting material; differences being more important for macro- than microorganisms. Thus, the extracellular DNA approach appears as robust and efficient as those based on the commercially available kit for biomonitoring sedimentary communities. Its low cost and fast processing time make it a promising alternative for large-scale ecological assessments of aquatic environments.
Keywords: benthic communities, biomonitoring, ecological assessment, eDNA protocols, metabarcoding, sample size.
Introduction
Sedimentary environments are a highly biodiverse component of aquatic ecosystems (Snelgrove 1997; Wang et al. 2012). They also play a key role in biogeochemical processes and are intrinsically coupled with the pelagic system (Alkhatib et al. 2012; Huettel et al. 2014). However, sedimentary environments are highly susceptible to anthropogenic activities because they ultimately act as a repository for many contaminants, resulting in contaminant concentrations often several orders of magnitude higher than that of the overlying water (Simpson and Batley 2016).
As a means of tempering the increasing influence of anthropogenic activities on aquatic systems, many jurisdictions have adopted regulations and guidelines for assessing, monitoring and restoring the ecological integrity of benthic environments (Lyons et al. 2010; Carere et al. 2012; Simpson et al. 2013). Although approaches vary, they generally involve the collection of chemical and physical data, along with ecological information on benthic communities residing in or interacting with sediments. These communities are essential for marine and freshwater ecosystems because they are involved in many ecosystem functions, including trophic transfer and biogeochemical processes (Freckman et al. 1997; Snelgrove 1997; Simčič 2005). Information about benthic community composition can further inform ecological risk assessments (Graham et al. 2019) and predictions of ecosystem vulnerability to biodiversity loss (Heilpern et al. 2018).
The most widespread approach for obtaining such ecological information is via the sorting, identification and enumeration of macrobenthic invertebrate communities (Canfield et al. 1994; Canfield et al. 1996; Dauer et al. 2000; Chariton et al. 2010), although other communities, for example diatoms, are also less commonly examined (Cattaneo et al. 2011; Desianti et al. 2017). Yet, these essential taxa represent only a minute fraction of benthic communities. As such, constraining ecological monitoring to macrobenthos not only provides a narrow view of biodiversity, but also excludes the crucial roles of meio- and micro-biota, with a growing body of evidence showing that they may be better indicators of disturbance and ecosystem health (Kennedy and Jacoby 1999; Gyedu-Ababio and Baird 2006; Chariton et al. 2014). Other major limitations of traditional macrobenthic surveys include the reliance of suitable taxonomic expertise, the high costs of sample processing, and the latency in producing the data (Chariton et al. 2010).
Environmental DNA (eDNA, i.e. the DNA of all organisms present in environmental samples; Pawlowski et al. 2020) has been increasingly seen as a viable alternative to traditional ‘capture and count’ methods for biomonitoring and environmental quality assessment (Chariton et al. 2010, 2015; Pochon et al. 2015; Pawlowski et al. 2018; Taberlet et al. 2018; Cordier et al. 2020; DiBattista et al. 2020). Environmental-DNA metabarcoding seeks to identify multiple taxa from DNA retrieved from an environmental matrix such as samples of sediment or water. From a single sample, numerous DNA (meta)barcodes, each characterising a particular taxon, can be amplified and sequenced simultaneously, virtually capturing all life forms. Thousands of samples can be processed in a single analysis, making the approach both time and cost efficient (Taberlet et al. 2012a). In addition, this approach can also be efficiently implemented over large spatial scales (Bohmann et al. 2014; Thomsen and Willerslev 2015), and is now considered as an ecological line of evidence for sediment and water-quality guidelines in an increasing number of jurisdictions (Simpson and Batley 2016; Hering et al. 2018).
Protocols for using eDNA-based approaches for environmental monitoring are continually being developed (Taberlet et al. 2018), with several studies also comparing intra-laboratory differences in protocols (e.g. Birer et al. 2017; Djurhuus et al. 2017; Majaneva et al. 2018). Consequently, there has been far greater conformity in approaches in recent years. This is critical, given that biomonitoring and assessment is founded on reproducibility. Environmental-DNA extraction is widely recognised as a critical and sensitive step of the process (Natarajan et al. 2016; Zinger et al. 2016, 2019; Ramírez et al. 2018a) and, in the case of sediment analysis, the method of eDNA extraction and the initial volume of the sample analysed are important considerations. Standard approaches using commercial kits result in the extraction of total DNA containing both intracellular (resulting from the lysis of living, dormant and dead cells during the extraction process) and extracellular DNA (previously released by lysis of damaged or dead cells or actively excreted into the surrounding environment; Torti et al. 2015; Ramírez et al. 2018b). Although widely used, these kits are expensive (Majaneva et al. 2018) and require time and suitable facilities, which make them often unsuitable for regular biodiversity monitoring (Zinger et al. 2016). Most importantly, commercial kits allow processing of only small amounts (≤10 g) of sample material, potentially constraining their ability to fully assess local biodiversity (Taberlet et al. 2012b). This is particularly relevant if macrobenthic invertebrates are being targeted.
More recently, protocols targeting only extracellular DNA have been developed as a cost-effective alternative to rapidly process larger (and potentially more representative) sample volumes of soil, with little to no facilities being required (Taberlet et al. 2012b; Zinger et al. 2016). Once adsorbed onto a mineral matrix (i.e. sediment particles), extracellular DNA is partially protected from degradation, enabling it to persist in the environment (Nielsen et al. 2007; Nagler et al. 2018). It is, therefore, expected to constitute a substantial fraction of the total eDNA retained within an aquatic ecosystem (Ceccherini et al. 2009; Pietramellara et al. 2009; Vuillemin et al. 2017). Desorption of extracellular DNA from the sedimentary matrix (of any amount) can be easily achieved using a saturated phosphate buffer (Taberlet et al. 2012b); the cost and processing time for its extraction have been estimated to be five and four times lower respectively than that required for extracting total DNA using a commercially available kit (Zinger et al. 2016). However, as the extracellular DNA fraction includes DNA derived from ancient and recent terrestrial and aquatic origins, the biodiversity signal inferred from extracellular DNA can differ from those obtained by total DNA, with some authors arguing that the extracellular DNA-based approaches inflate diversity (Alawi et al. 2014; Carini et al. 2017). A comparison of both approaches performed on tropical soil samples showed that, while local (α-) diversity was underestimated using extracellular DNA, β-diversity patterns were globally similar, with strong correlations in the relative abundances of clades observed between the two methods (Zinger et al. 2016). Despite its use in a few sediment eDNA metabarcoding studies (e.g. Pansu et al. 2015a; Guardiola et al. 2016a, 2016b), the efficiency of the extracellular DNA approach (hereafter called ‘extDNA’) has never been properly compared with the total DNA approach (hereafter called ‘totDNA’) for the purpose of sediment biomonitoring.
In this study, we evaluated the reliability of the extDNA approach for characterising sediment biota via eDNA metabarcoding. For this, we targeted taxonomic groups routinely used in sediment-quality assessments (prokaryote, eukaryote and metazoans) in three different sedimentary environments (freshwater pond, estuary and near-shore marine sands), and assessed the influence of two extraction methods (a bespoke method targeting only extDNA v. commercially available kit-based method for extracting totDNA) and the original volume of sampling material on α- and β-diversity patterns of those communities. We hypothesised that (1) the DNA concentration should be higher after totDNA extraction than extDNA extraction, and that (2) totDNA should provide higher estimates of local richness (α-diversity) than does extDNA. However, (3) assemblages based on totDNA and extDNA approaches should not differ significantly in terms of β-diversity patterns and the relative abundance of phyla, although (4) we expect an influence from the original amount of sampling material.
Material and methods
Study sites and sample collection
Sediments were sampled from the following three biomes located in eastern New South Wales, Australia: a freshwater pond at Macquarie University, Sydney (–33°46′9′′, 151°6′52′′); the Lane Cove River estuary at Cunninghams Reach, Lane Cove (–33°49′42′′, 151°8′44′′); and a sandy seashore at Umina Beach (–33°31′42′′, 151°18′55′′). Sampling occurred between 7 August and 12 September 2018. At each site, five individual samples were collected (total n = 15), each of them containing ~500 mL of the top ~10 cm of surficial sediment, the most biologically active part of the sediment (Simpson and Batley 2016). Samples were collected in sterile plastic containers that were rinsed five times in the water at each sample site before collection. Sediment samples were placed immediately on ice and transported to the laboratory where they were stored at –30°C before eDNA extractions.
eDNA extraction
The sediment samples were thawed for 24 h at 4°C and homogenised for 30 min using a roller-mixer before eDNA extractions. To simultaneously assess the influence of the extraction approach (i.e. extDNA v. totDNA) and of the initial volume of sediment material on the diversity patterns, we subsampled 1, 10 and 200 g from each sample for the extracellular DNA extraction (Taberlet et al. 2012b), and 1 and 10 g for the total DNA extraction. In total, each of the 15 samples was subjected to three protocol variants (with changing volume of starting material) for the extDNA extraction method and two for the totDNA method, leading to a total of 75 extractions (Fig. S1 of the Supplementary material).
Total DNA extractions, for both 1- and 10-g subsamples, were performed using the DNeasy PowerMax Soil Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. This kit is one of the most widely used in eDNA research, and is designed to allow totDNA extraction from up to 10 g of sediment. Extracellular DNA was extracted following the bespoke protocol described in Zinger et al. (2016), a modified version of the protocol proposed by Taberlet et al. (2012b). Briefly, equivalent amounts of phosphate buffer (Na2HPO4, 0.12 M, pH ~8) were added to each subsample and mixed for 15 min to extract extracellular DNA. For each extraction, a 2-mL aliquot was then centrifuged at 10 000g for 10 min at room temperature; the supernatant was then used in subsequent extraction steps, which were performed using the NucleoSpin Soil Kit (Macherey-Nagel, Düren, Germany), following manufacturer’s instructions and skipping the cell-lysis step (Taberlet et al. 2012b). For each site, at least two extraction controls were performed, one using phosphate buffer for extracellular DNA extraction and one using solution C2 for the total DNA extraction. In total, 45 extracellular DNA extractions (15 per site) plus four negative controls were conducted, as well as 30 total DNA extractions (10 per site) plus three negative controls. DNA extracts and negative controls (n = 82) were stored at –20°C before polymerase chain reaction (PCR) amplification.
PCR amplification and sequencing
Standard PCR methods were used to amplify specific gene regions targeting prokaryotes (bacteria and archaea; 16S rRNA; Caporaso et al. 2011), eukaryotes (18S rRNA; Hardy et al. 2010) and metazoans (COI mtDNA; Leray et al. 2013). So as to multiplex PCR products and sequence them together in a single high-throughput analysis, primers were tagged on their 5′ ends with a combination of eight-nucleotide labels differing by at least five nucleotides (Binladen et al. 2007; Valentini et al. 2009). In addition to samples and extraction controls, two blank PCR controls (using nuclease-free water) and positive controls (using saltwater crocodile, Crocodylus porosus, and foolish mussel, Mytilus trossulus, DNA for 18S and COI, and a synthetic microbial sequence for 16S) were included.
Each PCR reaction was performed in a total volume of 20 μL and contained 2 μL of DNA extract and 10 µL of AmpliTaq Gold 360 PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Primer concentration was 0.2 µM for prokaryote primers (515F: 5′-GTGYCAGCMGCCGCGGTAA-3′, Parada et al. 2016; 806R: 5′-GGACTACNVGGGTWTCTAAT-3′; Apprill et al. 2015), 0.4 µM for eukaryote primers (All18SF: 5′-GGTGCATGGCCGTTCTTAGT-3′; All18SR: 5′-CATCTAAGGGCATCACAGACC-3′; Hardy et al. 2010) and 0.5 µM for metazoan primers (mlCOIintF: 5′-GGWACWGGWTGAACWGTWTAYCCYCC-3′; jgHCO2198: 5′-TAIACYTCIGGRTGICCRAARAAYCA-3′; Leray et al. 2013). UltraPure RNase/DNase-free distilled water (Invitrogen, Carlsbad, CA, USA) was added to the mix to reach a final volume of 20 μL. Thermocycling conditions included an initial denaturing period of 10 min at 95°C, followed by 35 cycles of variable duration and temperature depending of the primer pair, as follows: denaturing at 94°C for 45 s, annealing at 50°C for 1 min and elongation at 72°C for 1.5 min for prokaryote (Caporaso et al. 2011); denaturing at 94°C for 1 min, annealing at 50°C for 1 min and elongation at 72°C for 1.5 min for eukaryote (Hardy et al. 2010); denaturing at 95°C for 30 s, annealing at 46°C for 30 s and elongation at 72°C for 45 s for metazoan (Deagle et al. 2018); and a final elongation at 72°C step for 10 min (prokaryote and eukaryote) or 5 min (metazoan).
Pre- and post-PCR DNA concentrations were determined using Quant-iT PicoGreen dsDNA Reagent assays (Invitrogen) on a PHERAstar FSX plate reader. Amplicon concentration was measured twice and averaged. To avoid overbiased sequencing depth among samples, PCR products were pooled in equimolarity (Harris et al. 2010; Leray et al. 2016; Lim et al. 2018), and PCR products with a low concentration (<2.5 ng µL–1) were excluded. Pooled PCR samples were purified using AMPure XP (Agencourt Bioscience, Beverly, MA, USA). Sequencing libraries were prepared using a TruSeq DNA PCR-free protocol and sequenced on an Illumina MiSeq 2500 platform (Illumina Australia and New Zealand, Melbourne, Vic., Australia; 2 × 250-bp paired-end reads) at the Ramaciotti Centre for Genomics (University of New South Wales, Sydney, NSW, Australia).
Data processing and filtering
Metabarcoding data were processed using the GHAP amplicon clustering and classification pipeline (see https://doi.org/10.4225/08/59f98560eba25). This pipeline combines tools from USearch (Edgar 2010) and the Ribosomal Database Project (RDP) classifier (Cole et al. 2007) with locally written scripts to conduct the following steps: (1) demultiplexing to assign reads to their original samples on the basis of the tag information attached to the primers, (2) reads trimming to remove poor-quality tail regions (with an Illumina base calls quality score of <25) by using a windowed quality-score based technique, (3) paired reads merging using the fastq_mergepairs command implemented in USearch, non-paired reads being discarded, (4) de-replication of reads with the fastx_uniques command, (5) trimming of sequences outside the expected length range of the marker (245–265 bp for 16S, 100–220 bp for 18S, 295–355 bp for COI), (6) operational taxonomic unit (OTU) clustering at 97% similarity using the cluster_otus command, which also performs chimera checking, (7) filtering out of non-target gene sequences, and (8) taxonomic assignment. For this latter step, representative sequences from each 18S and COI OTUs were taxonomically assigned against a respective curated reference set derived from the SILVA non-bacterial sequence (V128) reference set (Quast et al. 2013) and a custom-made mitochondrial COI reference dataset derived from GenBank (with >410 000 reference sequences and >127 000 species being represented), using the ublast command implemented in USearch (with the following parameters: e-value = 1E–5, query coverage = 0.8). For taxonomic assignment at the phylum, class, order and family levels, BLAST similarity cut-off values implemented by default in GHAP were used (0.77, 0.8, 0.85 and 0.9 respectively; see https://doi.org/10.4225/08/59f98560eba25); below these thresholds, the OTU was considered as unassigned. 16S OTUs were classified in two ways to improve confidence in the taxonomic assignment. Classification was undertaken against a set of curated 16S reference sequences, derived from the RDP 16S training set and supplemented by sequences from the RefSeq 16S set, using (1) the RDP Naïve Bayesian Classifier (RDP minimum confidence threshold for classification = 0.5) to assign a taxonomy, possibly down to species level, and (2) the usearch_global command to find the closest match to each OTU in the reference set. Finally, the accurate number of reads of each OTU in each sample was finally calculated by mapping all the merged reads back onto the final set of classified OTUs, using the usearch_global command, and an OTU × sample table was generated.
The following steps were conducted in R (ver. 3.5.3, R Foundation for Statistical Computing, Vienna, Austria, see https://www.R-project.org/) to further filter and de-noise the datasets. Operational taxonomic units (OTUs) that could not be assigned at the Domain level in 18S and 16S datasets were removed (Sutcliffe et al. 2017), as well as those that were not assigned to the ‘Metazoa’ Kingdom in the COI one. In the 16S dataset, we also filtered out OTUs that matched with known chloroplast sequences (Sutcliffe et al. 2019). In addition, to remove putative contaminant sequences, OTUs that had their maximal relative abundance in controls were discarded (Pansu et al. 2019). Then, only common OTUs representing >0.1% of sequences in at least one sample were retained to limit the inclusion of potentially artefact sequences resulting from PCR or sequencing errors (Deagle et al. 2018). Finally, in an effort to reduce the impact of low-abundance false positives resulting from ‘tag-jumps’ (Carlsen et al. 2012; Schnell et al. 2015), we considered an OTU as genuinely present in a sample if its abundance in this sample represented at least 0.5% of its total abundance across samples (Zinger et al. 2016). In the COI dataset, one cross-contaminated sample and two others with a low number of reads (<1000 reads) were discarded. Statistics about the metabarcoding data filter process are provided in Table S1 of the Supplementary material.
Statistical analysis
We first tested the effect of the extraction protocol variants on the DNA yield after extraction and after PCR amplification. Prior to statistical analysis of sequencing results, OTU data were rarefied on the basis of the minimum number of reads observed to 45 000, 7000 and 3000 reads per sample for eukaryotes, bacteria and metazoans respectively. We then investigated the α-diversity of bacterial, eukaryote and metazoan OTUs in relation to the extraction protocol variant used. Three different α-diversity indices (OTU richness, Shannon index and evenness) were estimated on the basis of the average values from 100 rarefactions (Sutcliffe et al. 2017). Differences in DNA concentration and OTU richness or diversity were assessed using Kruskal–Wallis tests and post hoc Dunn’s multiple-comparison tests (with Benjamini–Hochberg correction method) at a significance level (α) of 0.05.
To assess the influence of the extraction protocol variants on β-diversity patterns, rarefied communities were first analysed using correspondence analysis (Borcard et al. 2011); initially with all samples, followed by a second separate analysis for each sediment type. We further partitioned the total inertia of each correspondence analysis by sediment type to determine the portion due to the variation between extractions from a same sample and that due to the variation among samples (see the ‘Code for the inertia decomposition analysis’ section in the Supplementary material). Second, pairwise Bray–Curtis distances among samples were calculated, and permutational analyses of variance (PERMANOVA) were conducted on each full dataset by using the R package ‘vegan’ (J. Oksanen, F. G. Blanchet, M. Friendly, R. Kindt, P. Legendre, D. McGlinn, P. R. Minchin, R. B. O’Hara, G. L. Simpson, P. Solymos, M. H. H. Stevens, E. Szoecs, and H. Wagner, see https://CRAN.R-project.org/package=vegan) to assess the direct effects of the extraction protocol variants on these dissimilarity values (Anderson 2001). Similar analyses were performed for each sediment type to test for the effect of the DNA extraction approach (extDNA and totDNA). Bray–Curtis distances between samples were also compared between each pair of extraction protocol variants by using a Spearman rank-correlation test and a Mantel procedure with 999 permutations. Finally, for each sediment type, we tested whether the multivariate homogeneity of dispersion values differed between extraction protocol variants using the ‘betadisper’ function in ‘vegan’ with a bias correction for small sample size (Anderson 2006). We specifically expected the different samples from the same biome (sediment type) to be more homogeneous when extracted with larger amounts of sampling material.
To investigate the taxonomic composition of samples, the relative abundance of each phyla was averaged per sediment type for each extraction protocol variant. The fold-change ratio was then determined to quantify differences between pairs of protocol with similar amounts of starting material (Zinger et al. 2016). Operational taxonomic units and phylum relative abundance were then averaged per extraction approach (extDNA and totDNA), and we used Spearman rank-correlation tests to assess the congruence between OTUs and phylum relative abundances obtained from both approaches.
Results
DNA concentration
Many marine samples did not amplify with COI primers (9 of 25). This was particularly the case with samples extracted with the total DNA (totDNA) approach (6 of 10). Consequently, all marine COI amplicons were excluded from subsequent analyses (Fig. S2), resulting in a total of 46 COI samples being analysed for the pond and estuary environments. Samples that did not amplify for 18S (n = 1 of 75) and 16S (n = 4 of 75) were extracted using different extraction protocol variants and came from different sediment types; therefore, no particular bias related to the sediment type or extraction protocol variant was identified for these primers.
The DNA yield obtained from 1 g of sediment extracted with the totDNA approach was significantly lower than that from our other extraction protocol variants (i.e. all those based on the extDNA method and the totDNA one with 10 g of sediments), especially for pond and estuarine sediments (Dunn’s multiple-comparison test P < 0.05; Fig. 1, Table S2). The mean totDNA yield produced from 10 g of sediment did not differ significantly (P > 0.05) from the extDNA yields obtained from 1-, 10- or 200-g samples (Fig. 1). The initial amount of starting material did not have a significant effect on the quantity of extracellular DNA extracted (P > 0.6 for all pairwise comparisons; Table S2).
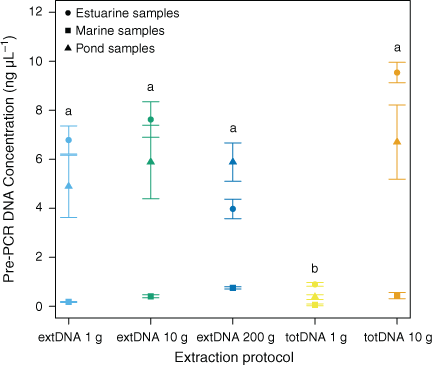
|
The sediment type (i.e. pond, estuary and marine) affected the total yield of DNA (Fig. 1). The DNA yield (mean ± s.e.m) was consistently lower in marine sediments (0.37 ± 0.06 ng µL–1) than in both estuarine (5.76 ± 0.65 ng µL–1) and pond (4.75 ± 0.66 ng µL–1) sediments, regardless of the extraction protocol variant used (all marine v. all estuary DNA extracts: Z = 6.01, P < 0.001; all marine v. all pond DNA extracts: Z = –4.70, P < 0.001; all pond v. all estuary DNA extracts: Z = 1.31, P = 0.19).
Alpha-diversity patterns
We found no consistent evidence that totDNA provides higher local richness estimates than does extDNA. There was no significant difference in OTU richness for prokaryotes between extraction protocol variants (Kruskall–Wallis χ2 = 2.04, d.f. = 4, P = 0.73; Table 1). The same pattern was observed for metazoan assemblages, although OTU richness was marginally greater with the totDNA protocol using 10 g of sediment (χ2 = 6.17, d.f. = 4, P = 0.19). By contrast, eukaryote richness using the totDNA extraction protocol with 1 g of sediment had a lower OTU richness than did protocols with larger amounts of starting material, regardless of the extraction approach (totDNA and extDNA alike, χ2 = 11.00, d.f. = 4, P = 0.027), although pairwise comparisons were marginally non-significant (Fig. S3, Table S3). However, there were sediment-specific responses; for example, OTU richness obtained from the pond samples using totDNA was overall higher than those obtained using extDNA, regardless of the primers pair used (Fig. S3). For eukaryotes and prokaryotes, no differences were observed in Shannon diversity (χ2 = 0.40, d.f. = 4, P = 0.98 and χ2 = 2.28, d.f. = 4, P = 0.69, for 18S and 16S respectively) and Pielou evenness (χ2 = 1.28, d.f. = 4, P = 0.87 and χ2 = 2.71, d.f. = 4, P = 0.61), indicating that potential differences observed in OTU richness are likely to be due to rare OTUs (Table 1). By contrast, we observed differences in Shannon and Pielou indices for metazoans (χ2 = 16.41, d.f. = 4, P < 0.003 and χ2 = 20.91, d.f. = 4, P < 0.001 respectively). In this case, the observed OTU richness was significantly higher in the totDNA protocol with 10 g of starting material than in all extDNA protocols.

|
Beta-diversity in community composition
Operational taxonomic unit composition samples clustered primarily by sediment type for all taxonomic groups (Fig. 2a–c). This suggests that the extraction protocol only moderately affects the overall β-diversity patterns, with discrimination being, here, predominately due to sediment type. When restricted by sediment type, the two first axes of the correspondence analyses discriminated samples according to the DNA extraction approach (extDNA v. totDNA), regardless of the taxonomic group and the sediment type (Fig. 2d–k). The inertia decomposition analysis performed per sediment type showed that, for all taxonomic groups, the inertia owing to the variation between extraction protocol variants was far greater than that owing to the variation among samples. For marine and estuarine sediments, 76.8–84.9 and 78.5–87.1% of the inertia respectively was due to differences in extraction protocol variants. The inertia was more evenly distributed between sample and extraction protocol variants for pond samples (58.5–65.0%). Although it was not possible to distinguish the respective effects of the extraction approach and the initial amount of sediment, results suggest that most of the variation was associated with the extraction approach. This was confirmed by the PERMANOVA analyses for each sediment type, which showed that Bray–Curtis distance was influenced by the DNA extraction approach (extDNA v. totDNA; Table S4). This was most evident in the estuarine sediments (r2 = 0.33–0.61, P < 0.001); in all other cases, r2 ranged between 0.16 and 0.36 (P < 0.002).

|
Overall, Bray–Curtis dissimilarities were unaffected by the DNA extraction protocol for prokaryotes (perMANOVA: Fpseudo = 0.65, r2 = 0.038, P = 0.79), eukaryotes (perMANOVA: Fpseudo = 1.02, r2 = 0.056, P = 0.42) or metazoans (perMANOVA: Fpseudo = 0.78, r2 = 0.071, P = 0.65). Beta-diversity patterns among samples were consistent among extraction protocol variants, especially for prokaryotes (Spearman’s ρ = 0.89–0.96, P = 0.001, for all pairwise comparisons in the prokaryote dataset; Spearman’s ρ = 0.78–0.93, P = 0.001 for eukaryotes; Spearman’s ρ = 0.81–0.93, P < 0.01 for metazoans; Fig. 3). Eukaryote and metazoan Bray–Curtis distances derived from the totDNA extraction approach using 1 g of sediment were overall more dissimilar to those derived from other protocols (Fig. 3a–d), except for prokaryotes.

|
The effect of the initial amount of sediment on β-diversity patterns was moderate. Bray–Curtis distances obtained using the same extraction approach with varying amount of sampling material were highly similar (Fig. 3a for totDNA, Spearman’s ρ = 0.94; and Fig. 3h–j for extDNA, Spearman’s ρ = 0.97–0.98). Eukaryote and metazoan communities from a same sedimentary environment tended to be more homogenous when a larger amount of sampling material was used (multivariate dispersion analysis; Fig. S4), but significant differences among extraction protocol variants were detected only for metazoan communities (ANOVA, P < 0.05). No consistent pattern emerged for prokaryote communities, although significant differences were observed (Fig. S4).
Taxonomic composition at the phylum level was very similar among extraction protocols, especially for prokaryotes (Fig. 4a). Relative abundances of prokaryote phyla were strongly correlated between extraction approaches (extDNA v. totDNA, Spearman’s ρ = 0.97, P < 0.001; Fig. S5). Although slightly weaker, this correlation was also high for eukaryotes and metazoans (Spearman’s ρ = 0.90 and 0.89 respectively; Fig. 4b, c, S5). To a lesser extent, this pattern was also observed at the OTU level (Spearman’s ρ = 0.55, 0.40 and 0.36 for prokaryotes, eukaryotes and metazoans respectively, P < 0.001). For a given amount of starting material, the average fold-change (AFC) ratio was always <2 for the 10 most abundant bacterial clades, suggesting that there were no marked differences in the relative abundance of bacterial phyla between the extraction methods (extDNA v. totDNA; Table S5). For eukaryotes and metazoan primers, marked changes were observed for arthropods and nematodes (both being overestimated with totDNA compared with extDNA), whereas annelids exhibited an opposite trend (Fig. 4, Table S5). Plant (Chlorophyta phylum) relative abundances determined with the 18S eukaryotic marker were substantially higher in the totDNA (AFC > 7) than extDNA, whereas diatoms (Bacillariophyta phylum) tended to be overestimated with extDNA.
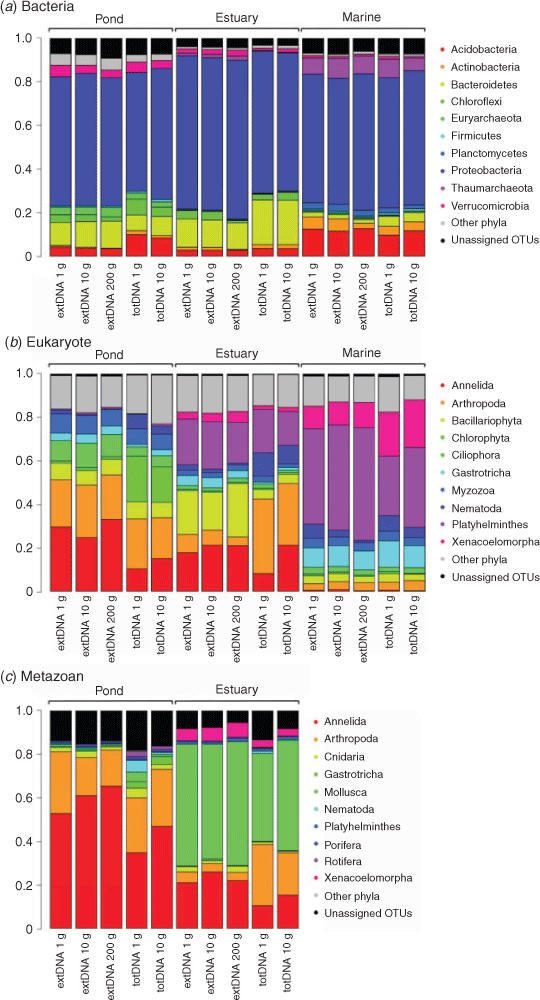
|
Discussion
DNA concentration and amplification success
In this study, we examined the potential suitability of extracellular DNA (extDNA) for eDNA-based biomonitoring of sedimentary communities, by comparing this approach with the traditional total DNA (totDNA) extraction approach. The fundamental difference between the two methods is in the eDNA release technique; extDNA is extracted by desorption from sediment matrix by using saturated phosphate buffer, whereas totDNA relies on the mechanical and chemical lysis of cells contained in the matrix, allowing the extraction of both intra- and extracellular DNA (Zinger et al. 2016). However, variations in techniques and buffers between extraction protocols are known to lead to slightly different outputs (Ramírez et al. 2018a; Dopheide et al. 2019), and we cannot rule out that factors other than the eDNA fraction targeted (e.g. buffers used) also played a role in differences observed between protocols.
We first compared the yields obtained from the different protocols (Hypothesis 1). Among the three sediment types examined, DNA concentration obtained from extDNA extractions was at least as high as (or even higher than) those obtained with the totDNA protocols (Fig. 1). Specifically, the totDNA concentration from 1 g of sediment was much lower than those obtained with any other protocol. One possible explanation for this result is technical; we used a PowerMax Soil DNA extraction kit (following manufacturer’s protocol) to extract totDNA, which is designed for a starting material mass between 5 and 10 g. As such, the protocol may have failed to deal with a smaller amount of starting material because the DNA would be more diluted in the buffer. Nevertheless, even with 10 g of sediment, we found no evidence that totDNA provided a higher DNA yield than did extDNA. This contrasts with Zinger et al. (2016) who found extDNA yields from tropical soils to be lower than those obtained using totDNA. Matrix composition and local environmental parameters are known to influence both the persistence and concentration of extDNA in soils and sediments (Pietramellara et al. 2009; Nagler et al. 2018). Given that aquatic systems have been shown to contain significant amounts of DNA derived from the surrounding catchments (Deiner and Altermatt 2014; Chariton et al. 2015), it is likely that extDNA constitutes the dominant fraction of environmental DNA in aquatic ecosystems (Dell’Anno and Danovaro 2005; Ceccherini et al. 2009; Vuillemin et al. 2017). This can explain the absence of significant differences in the yield obtained from extDNA and totDNA protocols.
In the case of both the 16S rDNA bacterial and 18S rDNA eukaryote primers, we found that the proportion of samples that successfully amplified by PCR was similar regardless of the extraction protocol variant or sediment matrix. By contrast, amplification success using the COI primers was considerably lower, especially for the marine sediments extracted with the totDNA extraction method where a majority of the samples failed to amplify (Fig. S2). One explanation is the higher sand content of the marine sediments that effectively operates as an abiotic stressor, limiting burrowing capacity of many taxa and, ultimately, leading to reduction in diversity and abundance of organisms (Gray 1974; Snelgrove and Butman 1994). This interpretation is supported by the overall lower DNA yield observed in marine samples (Fig. 1). However, results from the 18S dataset showed that metazoan DNA was present in marine samples and that metazoan phyla constituted a significant proportion of the eukaryote community (Fig. 4). Consequently, DNA yield is not the only factor contributing to the discrepancy between the COI and 18S datasets; PCR amplification biases are also likely to play a role. This is a known pitfall with the COI marker (Clarke et al. 2014; Deagle et al. 2014). The size range of amplicons can partly explain this pattern; the average length of COI amplicons is approximately twice that of 18S amplicons. Yet, DNA in environmental samples is degraded and shorter fragments are more common, making them more easily amplifiable than are longer fragments (Yoccoz et al. 2012).
Alpha-diversity patterns
Contrary to our expectation (Hypothesis 2), we found no evidence, overall, that estimates of local richness were higher with totDNA (containing both extDNA and intracellular DNA) than with extDNA (Tables 1, S3). This result is in line with the supposition that extDNA constitutes most of eDNA found in aquatic sediment (Dell’Anno and Danovaro 2005; Ceccherini et al. 2009). In aquatic environments, part of the sedimentary extDNA is of allogenous origin (i.e. coming from the surrounding environment, including terrestrial ones; Torti et al. 2015), or from cells that are no longer alive (‘relic DNA’; Carini et al. 2017). Our results suggest that the totDNA extraction protocol effectively captures this extDNA fraction, and that, in turn, the extDNA extraction protocol effectively integrates the diversity associated with the active communities that continuously release extDNA through biomass turnover. However, sediment- (and taxonomic-) specific responses to the extraction protocol variants in term of richness were observed (Fig. S3), which may reflect the strong influence of the matrix composition on both biological community richness (McLachlan 1996; Seiderer and Newell 1999) and DNA preservation (Ogram et al. 1987; Pietramellara et al. 2009). Furthermore, community-diversity metrics accounting for both richness and relative abundance of OTUs (i.e. Shannon index) exhibited moderate differences between protocols (excepted for metazoans), suggesting that potential differences observed in OTU richness concern mostly rare OTUs. In the context of biomonitoring, this is unlikely to be of concern, because the primary goal is to obtain representative and reproducible samples of the community, rather than a comprehensive inventory, as in the case of biodiversity studies (Chariton et al. 2016).
Beta-diversity patterns
The two extraction approaches used here were tailored to capture different components of sediment eDNA. Although the totDNA undoubtedly includes extracellular DNA (Chariton et al. 2015), the approach is designed to maximise the extraction of DNA residing within cells, be it active, dead or dormant. By contrast, the extDNA approach is unlikely to capture recalcitrant DNA within whole cell structures, but rather provides a temporally integrated biodiversity signal, including the presence of ‘relic’ DNA. Although this can lead to differences in the identity of OTUs retrieved between extraction methods (Taberlet et al. 2012b; Wagner et al. 2015; Zinger et al. 2016; Nagler et al. 2018), our findings support our hypothesis that the extraction approach has only a moderate effect on β-diversity patterns (Hypothesis 3; Fig. 2a–c, 3). These results are in line with those by Ramírez et al. (2018b), who found only minimum differences between total and intercellular bacterial communities extracted from marine sediments.
Overall, compositional profiles based on the relative abundances of phyla were highly congruent among extraction protocol variants (Fig. 4), and strong correlations in the relative abundances of most phyla were observed between the two extraction approaches (Fig. S5). This was particularly the case for prokaryotes, whereas some eukaryote and metazoan phyla tended to be biased towards a particular extraction approach (Fig. 4, S5, Table S5). Two main distinctions in the methods can explain these differences; in contrast to the totDNA extraction protocol, the extDNA protocol does not include any cell lysis step; active communities with recalcitrant DNA could, thus, be better extracted by the totDNA protocol. However, extDNA is expected to be less biased by the presence of macroremains in the original samples (e.g. rootlets; Pansu et al. 2015a), which can release massive quantity of DNA when those cells are lysed. In that sense, estimates of macro-organism biodiversity made from totDNA can be more sensitive to ‘subsampling’ effects than are those based on extDNA.
In general, the mass of the extracted sediment had only a moderate effect on community composition (Hypothesis 4), and played a much less important role than the extraction approach. However, the initial starting volume of sediment had a more pronounced effect in the samples extracted using totDNA (Fig. 3a, h–j, 4), with eukaryote and metazoan Bray–Curtis distances derived from the totDNA extraction of 1 g of sediment being more dissimilar to those derived from other extraction protocol variants (Fig. 3a–d). Interestingly, this was not observed for prokaryotes, which is likely to be a reflection of their size and density within a sample, and, thus, the capacity to capture a representative sample using a small amount of sediment. Because of their porosity and water movements, the genetic signal in sediments can be more homogenous than in soils. Here, community composition patterns from samples extracted using the extDNA protocols with 10 and 200 g of sediments (and, to a lower extent, 1 g) were very similar. These results indicated that even small amounts of sediment (<10 g of an original sample of 500 g) extracted with the extDNA protocol can efficiently represent local biodiversity, assuming that the original sample was well mixed. However, even though results were not necessarily significant, replicate samples tended to be more homogenous when a larger amount of sampling material was used for eukaryote and metazoan primers (Fig. S4), suggesting that a larger sample size is necessary to get relevant and consistent patterns of larger organisms (Ranjard et al. 2003; Taberlet et al. 2012b). Given the large body of evidence on how macrofauna (e.g. insects, crustacea, annelids) respond to natural and anthropogenic stressors, they are likely to remain the focus of biomonitoring studies for the foreseeable future. It is imperative, therefore, that biomonitoring approaches using eDNA consider the importance of the starting sample volume.
Conclusions
Extracellular DNA extraction from aquatic sediment provides reliable estimates of community diversity and composition, qualitatively comparable to those obtained with totDNA. Although both methods may provide marginally different ‘views’ of composition, other parts of the metabarcoding pipeline, including bioinformatics, primer choice and filtering, can also profoundly shape compositional profiles (Clarke et al. 2014; Alberdi et al. 2018; Taberlet et al. 2018; Pauvert et al. 2019). Hence, each extraction method has its own trade-off, and the choice of the most appropriate approach ultimately depends of the study objectives, taxa of interest along with financial and technical constraints. In the context of routine biomonitoring programs, which aims at providing a comprehensive view of the local diversity in a reproducible way, the extracellular DNA approach offers many benefits; namely, it is easy to implement, and cheaper and faster than commercial kits dedicated to total DNA (Taberlet et al. 2012b; Pansu et al. 2015b; Zinger et al. 2016). It allows the processing of a larger number of samples in a reduced amount of time, and, thus, potentially increased sampling intensity or the extent of the study area for a limited financial and quality cost. In doing so, targeting extDNA provides opportunities for biomonitoring in situations where it may have been excluded because of cost, lack of taxonomic expertise and latency in producing data. In addition, extracellular DNA can be extracted from a greater mass of sediment, which can promote the sampling replicability of larger taxa, such as polychaetes, whose responses to a range of environmental stressors have been well documented. We argue that the advantages and the relevance of this approach will contribute to the increased use of sediment eDNA-based biomonitoring for ecological assessment of aquatic environments.
Authors’ contribution
A. A. Chariton and G. C. Hose designed the study. A. Chariton and M. B. Chapman collected samples. M. B. Chapman and J. Pansu conducted DNA analyses. J. Pansu processed the data and analysed the results. J. Pansu and A. A. Chariton drafted the manuscript. All authors contributed to manuscript revisions and approved the submitted version.
Data accessibility
Datasets have been deposited in the Dryad depository (https://doi.org/10.5061/dryad.kwh70rz34) and the CSIRO data access portal (see https://data.csiro.au).
Conflicts of interest
J. Pansu and A. A. Chariton are Guest Editors of the Environmental DNA special issue. Despite this relationship, they took no part in the review and acceptance of this or any other manuscript in this issue that they authored. The authors declare that they have no further conflicts of interest.
Declaration of funding
This study was supported collectively by Macquarie University, CSIRO Ocean & Atmosphere, and the European Union Horizon H2020 program through the Marie Skłodowska-Curie actions grant awarded to J. Pansu (TEAM-Coast project, MSCA-GF 750570).
Acknowledgements
Authors are grateful to Natalie Caulfield for her help in the laboratory and to James McLaughlin (CSIRO), Stuart Simpson (CSIRO) and Fréderique Viard (CNRS) for logistical support.
References
Alawi, M., Schneider, B., and Kallmeyer, J. (2014). A procedure for separate recovery of extra- and intracellular DNA from a single marine sediment sample. Journal of Microbiological Methods 104, 36–42.| A procedure for separate recovery of extra- and intracellular DNA from a single marine sediment sample.Crossref | GoogleScholarGoogle Scholar | 24955890PubMed |
Alberdi, A., Aizpurua, O., Gilbert, M. T. P., and Bohmann, K. (2018). Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods in Ecology and Evolution 9, 134–147.
| Scrutinizing key steps for reliable metabarcoding of environmental samples.Crossref | GoogleScholarGoogle Scholar |
Alkhatib, M., Lehmann, M. F., and del Giorgio, P. A. (2012). The nitrogen isotope effect of benthic remineralization–nitrification–denitrification coupling in an estuarine environment. Biogeosciences 9, 1633–1646.
| The nitrogen isotope effect of benthic remineralization–nitrification–denitrification coupling in an estuarine environment.Crossref | GoogleScholarGoogle Scholar |
Anderson, M. J. (2001). A new method for nonâ€parametric multivariate analysis of variance. Austral Ecology 26, 32–46.
| A new method for nonâ€parametric multivariate analysis of variance.Crossref | GoogleScholarGoogle Scholar |
Anderson, M. J. (2006). Distance-based tests for homogeneity of multivariate dispersions. Biometrics 62, 245–253.
| Distance-based tests for homogeneity of multivariate dispersions.Crossref | GoogleScholarGoogle Scholar | 16542252PubMed |
Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Microbial Ecology 75, 129–137.
| Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton.Crossref | GoogleScholarGoogle Scholar |
Binladen, J., Gilbert, M. T. P., Bollback, J. P., Panitz, F., Bendixen, C., Nielsen, R., and Willerslev, E. (2007). The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing. PLoS One 2, e197.
| The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing.Crossref | GoogleScholarGoogle Scholar | 17299583PubMed |
Birer, C., Tysklind, N., Zinger, L., and Duplais, C. (2017). Comparative analysis of DNA extraction methods to study the body surface microbiota of insects: a case study with ant cuticular bacteria. Molecular Ecology Resources 17, e34–e45.
| Comparative analysis of DNA extraction methods to study the body surface microbiota of insects: a case study with ant cuticular bacteria.Crossref | GoogleScholarGoogle Scholar | 28477337PubMed |
Bohmann, K., Evans, A., Gilbert, M. T. P., Carvalho, G. R., Creer, S., Knapp, M., Yu, D. W., and de Bruyn, M. (2014). Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology & Evolution 29, 358–367.
| Environmental DNA for wildlife biology and biodiversity monitoring.Crossref | GoogleScholarGoogle Scholar |
Borcard, D., Gillet, F., and Legendre, P. (2011). ‘Numerical Ecology with R’, 1st edn. (Springer: New York, NY, USA.)
Canfield, T. J., Kemble, N. E., Brumbaugh, W. G., Dwyer, F. J., Ingersoll, C. G., and Fairchild, J. F. (1994). Use of benthic invertebrate community structure and the sediment quality triad to evaluate metal-contaminated sediment in the upper Clark Fork river, Montana. Environmental Toxicology and Chemistry 13, 1999–2012.
| Use of benthic invertebrate community structure and the sediment quality triad to evaluate metal-contaminated sediment in the upper Clark Fork river, Montana.Crossref | GoogleScholarGoogle Scholar |
Canfield, T. J., James Dwyer, F., Fairchild, J. F., Haverland, P. S., Ingersoll, C. G., Kemble, N. E., Mount, D. R., La Point, T. W., Allen Burton, G., and Swift, M. C. (1996). Assessing contamination in Great Lakes sediments using benthic invertebrate communities and the sediment quality triad approach. Journal of Great Lakes Research 22, 565–583.
| Assessing contamination in Great Lakes sediments using benthic invertebrate communities and the sediment quality triad approach.Crossref | GoogleScholarGoogle Scholar |
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., and Turnbaugh, P. J. Fierer, Nand Knight, R. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences of the United States of America 108, 4516–4522
Carere, M., Dulio, V., Hanke, G., and Polesello, S. (2012). Guidance for sediment and biota monitoring under the Common Implementation Strategy for the Water Framework Directive. Trends in Analytical Chemistry 36, 15–24.
| Guidance for sediment and biota monitoring under the Common Implementation Strategy for the Water Framework Directive.Crossref | GoogleScholarGoogle Scholar |
Carini, P., Marsden, P. J., Leff, J. W., Morgan, E. E., Strickland, M. S., and Fierer, N. (2017). Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nature Microbiology 2, 16242.
| Relic DNA is abundant in soil and obscures estimates of soil microbial diversity.Crossref | GoogleScholarGoogle Scholar |
Carlsen, T., Aas, A. B., Lindner, D., Vrålstad, T., Schumacher, T., and Kauserud, H. (2012). Don’t make a mista(g)ke: is tag switching an overlooked source of error in amplicon pyrosequencing studies? Fungal Ecology 5, 747–749.
| Don’t make a mista(g)ke: is tag switching an overlooked source of error in amplicon pyrosequencing studies?Crossref | GoogleScholarGoogle Scholar |
Cattaneo, A., Couillard, Y., Wunsam, S., and Fortin, C. (2011). Littoral diatoms as indicators of recent water and sediment contamination by metals in lakes. Journal of Environmental Monitoring 13, 572–582.
| Littoral diatoms as indicators of recent water and sediment contamination by metals in lakes.Crossref | GoogleScholarGoogle Scholar | 21184001PubMed |
Ceccherini, M. T., Ascher, J., Agnelli, A., Borgogni, F., Pantani, O. L., and Pietramellara, G. (2009). Experimental discrimination and molecular characterization of the extracellular soil DNA fraction. Antonie van Leeuwenhoek 96, 653–657.
| Experimental discrimination and molecular characterization of the extracellular soil DNA fraction.Crossref | GoogleScholarGoogle Scholar | 19533410PubMed |
Chariton, A. A., Court, L. N., Hartley, D. M., Colloff, M. J., and Hardy, C. M. (2010). Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA. Frontiers in Ecology and the Environment 8, 233–238.
| Ecological assessment of estuarine sediments by pyrosequencing eukaryotic ribosomal DNA.Crossref | GoogleScholarGoogle Scholar |
Chariton, A. A., Ho, K. T., Proestou, D., Bik, H., Simpson, S. L., Portis, L. M., Cantwell, M. G., Baguley, J. G., Burgess, R. M., Pelletier, M. M., Perron, M., Gunsch, C., and Matthews, R. A. (2014). A molecular-based approach for examining responses of eukaryotes in microcosms to contaminant-spiked estuarine sediments: DNA comparison of assemblages exposed to spiked sediments. Environmental Toxicology and Chemistry 33, 359–369.
| A molecular-based approach for examining responses of eukaryotes in microcosms to contaminant-spiked estuarine sediments: DNA comparison of assemblages exposed to spiked sediments.Crossref | GoogleScholarGoogle Scholar | 24399368PubMed |
Chariton, A. A., Stephenson, S., Morgan, M. J., Steven, A. D. L., Colloff, M. J., Court, L. N., and Hardy, C. M. (2015). Metabarcoding of benthic eukaryote communities predicts the ecological condition of estuaries. Environmental Pollution 203, 165–174.
| Metabarcoding of benthic eukaryote communities predicts the ecological condition of estuaries.Crossref | GoogleScholarGoogle Scholar | 25909325PubMed |
Chariton, A. A., Pettigrove, V. J., and Baird, D. J. (2016). Ecological assessment. Chapter 7. In ‘Sediment Quality Assessment: a Practical Guide’. (Eds S. L. Simpson and G. E. Batley.) pp. 195–236. (CSIRO Publishing: Melbourne, Vic., Australia.) Available at http://hdl.handle.net/102.100.100/91025?index=1 [Verified 8 January 2021].
Clarke, L. J., Soubrier, J., Weyrich, L. S., and Cooper, A. (2014). Environmental metabarcodes for insects: in silico PCR reveals potential for taxonomic bias. Molecular Ecology Resources 14, 1160–1170.
| Environmental metabarcodes for insects: in silico PCR reveals potential for taxonomic bias.Crossref | GoogleScholarGoogle Scholar | 24751203PubMed |
Cole, J. R., Chai, B., Farris, R. J., Wang, Q., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., Bandela, A. M., Cardenas, E., Garrity, G. M., and Tiedje, J. M. (2007). The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Research 35, D169–D172.
| The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data.Crossref | GoogleScholarGoogle Scholar | 17090583PubMed |
Cordier, T., Alonso‐Sáez, L., Apothéloz‐Perret‐Gentil, L., Aylagas, E., Bohan, D. A., Bouchez, A., Chariton, A., Creer, S., Frühe, L., Keck, F., Keeley, N., Laroche, O., Leese, F., Pochon, X., Stoeck, T., Pawlowski, J., and Lanzén, A. (2020). Ecosystems monitoring powered by environmental genomics: a review of current strategies with an implementation roadmap. Molecular Ecology , mec.15472.
| Ecosystems monitoring powered by environmental genomics: a review of current strategies with an implementation roadmap.Crossref | GoogleScholarGoogle Scholar | 33070453PubMed |
Dauer, D. M., Weisberg, S. B., and Ranasinghe, J. A. (2000). Relationships between benthic community condition, water quality, sediment quality, nutrient loads, and land use patterns in Chesapeake Bay. Estuaries 23, 80.
| Relationships between benthic community condition, water quality, sediment quality, nutrient loads, and land use patterns in Chesapeake Bay.Crossref | GoogleScholarGoogle Scholar |
Deagle, B. E., Jarman, S. N., Coissac, E., Pompanon, F., and Taberlet, P. (2014). DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biology Letters 10, 20140562.
| DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match.Crossref | GoogleScholarGoogle Scholar | 25209199PubMed |
Deagle, B. E., Clarke, L. J., Kitchener, J. A., Polanowski, A. M., and Davidson, A. T. (2018). Genetic monitoring of open ocean biodiversity: an evaluation of DNA metabarcoding for processing continuous plankton recorder samples. Molecular Ecology Resources 18, 391–406.
| Genetic monitoring of open ocean biodiversity: an evaluation of DNA metabarcoding for processing continuous plankton recorder samples.Crossref | GoogleScholarGoogle Scholar | 29171158PubMed |
Deiner, K., and Altermatt, F. (2014). Transport distance of invertebrate environmental DNA in a natural river. PLoS One 9, e88786.
| Transport distance of invertebrate environmental DNA in a natural river.Crossref | GoogleScholarGoogle Scholar | 24523940PubMed |
Dell’Anno, A., and Danovaro, R. (2005). Extracellular DNA plays a key role in deep-sea ecosystem functioning. Science 309, 2179.
| Extracellular DNA plays a key role in deep-sea ecosystem functioning.Crossref | GoogleScholarGoogle Scholar | 16195451PubMed |
Desianti, N., Potapova, M., Enache, M., Belton, T. J., Velinsky, D. J., Thomas, R., and Mead, J. (2017). Sediment diatoms as environmental indicators in New Jersey coastal lagoons. Journal of Coastal Research 78, 127–140.
| Sediment diatoms as environmental indicators in New Jersey coastal lagoons.Crossref | GoogleScholarGoogle Scholar |
DiBattista, J. D., Reimer, J. D., Stat, M., Masucci, G. D., Biondi, P., De Brauwer, M., Wilkinson, S. P., Chariton, A. A., and Bunce, M. (2020). Environmental DNA can act as a biodiversity barometer of anthropogenic pressures in coastal ecosystems. Scientific Reports 10, 8365.
| Environmental DNA can act as a biodiversity barometer of anthropogenic pressures in coastal ecosystems.Crossref | GoogleScholarGoogle Scholar | 32433472PubMed |
Djurhuus, A., Port, J., Closek, C. J., Yamahara, K. M., Romero-Maraccini, O., Walz, K. R., Goldsmith, D. B., Michisaki, R., Breitbart, M., Boehm, A. B., and Chavez, F. P. (2017). Evaluation of filtration and DNA extraction methods for environmental DNA biodiversity assessments across multiple trophic levels. Frontiers in Marine Science 4, 314.
| Evaluation of filtration and DNA extraction methods for environmental DNA biodiversity assessments across multiple trophic levels.Crossref | GoogleScholarGoogle Scholar |
Dopheide, A., Xie, D., Buckley, T. R., Drummond, A. J., and Newcomb, R. D. (2019). Impacts of DNA extraction and PCR on DNA metabarcoding estimates of soil biodiversity. Methods in Ecology and Evolution 10, 120–133.
| Impacts of DNA extraction and PCR on DNA metabarcoding estimates of soil biodiversity.Crossref | GoogleScholarGoogle Scholar |
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461.
| Search and clustering orders of magnitude faster than BLAST.Crossref | GoogleScholarGoogle Scholar | 20709691PubMed |
Freckman, D. W., Blackburn, T. H., Brussaard, L., Hutchings, P., Palmer, M. A., and Snelgrove, P. V. R. (1997). Linking biodiversity and ecosystem functioning of soils and sediments. Ambio 26, 556–562.
Graham, S. E., Chariton, A. A., and Landis, W. G. (2019). Using Bayesian networks to predict risk to estuary water quality and patterns of benthic environmental DNA in Queensland. Integrated Environmental Assessment and Management 15, 93–111.
| Using Bayesian networks to predict risk to estuary water quality and patterns of benthic environmental DNA in Queensland.Crossref | GoogleScholarGoogle Scholar | 30117283PubMed |
Gray, J. S. (1974). Animal–sediment relationships. Oceanography and Marine Biology – an Annual Review 12, 223–261.
Guardiola, M., Uriz, M. J., Taberlet, P., Coissac, E., Wangensteen, O. S., and Turon, X. (2016a). Deep-sea, deep-sequencing: metabarcoding extracellular DNA from sediments of marine canyons. PLoS One 10, e0139633.
| Deep-sea, deep-sequencing: metabarcoding extracellular DNA from sediments of marine canyons.Crossref | GoogleScholarGoogle Scholar |
Guardiola, M., Wangensteen, O. S., Taberlet, P., Coissac, E., Uriz, M. J., and Turon, X. (2016b). Spatio-temporal monitoring of deep-sea communities using metabarcoding of sediment DNA and RNA. PeerJ 4, e2807.
| Spatio-temporal monitoring of deep-sea communities using metabarcoding of sediment DNA and RNA.Crossref | GoogleScholarGoogle Scholar | 28028473PubMed |
Gyedu-Ababio, T. K., and Baird, D. (2006). Response of meiofauna and nematode communities to increased levels of contaminants in a laboratory microcosm experiment. Ecotoxicology and Environmental Safety 63, 443–450.
| Response of meiofauna and nematode communities to increased levels of contaminants in a laboratory microcosm experiment.Crossref | GoogleScholarGoogle Scholar | 16406597PubMed |
Hardy, C. M., Krull, E. S., Hartley, D. M., and Oliver, R. L. (2010). Carbon source accounting for fish using combined DNA and stable isotope analyses in a regulated lowland river weir pool. Molecular Ecology 19, 197–212.
| Carbon source accounting for fish using combined DNA and stable isotope analyses in a regulated lowland river weir pool.Crossref | GoogleScholarGoogle Scholar | 19912537PubMed |
Harris, J. K., Sahl, J. W., Castoe, T. A., Wagner, B. D., Pollock, D. D., and Spear, J. R. (2010). Comparison of normalization methods for construction of large, multiplex amplicon pools for next-generation sequencing. Applied and Environmental Microbiology 76, 3863–3868.
| Comparison of normalization methods for construction of large, multiplex amplicon pools for next-generation sequencing.Crossref | GoogleScholarGoogle Scholar | 20418443PubMed |
Heilpern, S. A., Weeks, B. C., and Naeem, S. (2018). Predicting ecosystem vulnerability to biodiversity loss from community composition. Ecology 99, 1099–1107.
| Predicting ecosystem vulnerability to biodiversity loss from community composition.Crossref | GoogleScholarGoogle Scholar | 29569236PubMed |
Hering, D., Borja, A., Jones, J. I., Pont, D., Boets, P., Bouchez, A., Bruce, K., Drakare, S., Hänfling, B., Kahlert, M., Leese, F., Meissner, K., Mergen, P., Reyjol, Y., Segurado, P., Vogler, A., and Kelly, M. (2018). Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive. Water Research 138, 192–205.
| Implementation options for DNA-based identification into ecological status assessment under the European Water Framework Directive.Crossref | GoogleScholarGoogle Scholar | 29602086PubMed |
Huettel, M., Berg, P., and Kostka, J. E. (2014). Benthic exchange and biogeochemical cycling in permeable sediments. Annual Review of Marine Science 6, 23–51.
| Benthic exchange and biogeochemical cycling in permeable sediments.Crossref | GoogleScholarGoogle Scholar | 23987916PubMed |
Kennedy, A. D., and Jacoby, C. A. (1999). Biological indicators of marine environmental health: meiofauna – a neglected benthic component? Environmental Monitoring and Assessment 54, 47–68.
| Biological indicators of marine environmental health: meiofauna – a neglected benthic component?Crossref | GoogleScholarGoogle Scholar |
Leray, M., Yang, J. Y., Meyer, C. P., Mills, S. C., Agudelo, N., Ranwez, V., Boehm, J. T., and Machida, R. J. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Frontiers in Zoology 10, 34.
| A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents.Crossref | GoogleScholarGoogle Scholar | 23767809PubMed |
Leray, M., Haenel, Q., and Bourlat, S. J. (2016). Preparation of amplicon libraries for metabarcoding of marine eukaryotes using Illumina MiSeq: the adapter ligation method. In ‘Marine Genomics’. (Ed. S. J. Bourlat.) pp. 209–218. (Humana Press: New York, NY, USA.)
Lim, M. Y., Song, E. J., Kim, S. H., Lee, J., and Nam, Y. D. (2018). Comparison of DNA extraction methods for human gut microbial community profiling. Systematic and Applied Microbiology 41, 151–157.
| Comparison of DNA extraction methods for human gut microbial community profiling.Crossref | GoogleScholarGoogle Scholar | 29305057PubMed |
Lyons, B. P., Thain, J. E., Stentiford, G. D., Hylland, K., Davies, I. M., and Vethaak, A. D. (2010). Using biological effects tools to define good environmental status under the European Union Marine Strategy Framework Directive. Marine Pollution Bulletin 60, 1647–1651.
| Using biological effects tools to define good environmental status under the European Union Marine Strategy Framework Directive.Crossref | GoogleScholarGoogle Scholar | 20609451PubMed |
Majaneva, M., Diserud, O. H., Eagle, S. H. C., Boström, E., Hajibabaei, M., and Ekrem, T. (2018). Environmental DNA filtration techniques affect recovered biodiversity. Scientific Reports 8, 4682.
| Environmental DNA filtration techniques affect recovered biodiversity.Crossref | GoogleScholarGoogle Scholar | 29549344PubMed |
McLachlan, A. (1996). Physical factors in benthic ecology: effects of changing sand particle size on beach fauna. Marine Ecology Progress Series 131, 205–217.
| Physical factors in benthic ecology: effects of changing sand particle size on beach fauna.Crossref | GoogleScholarGoogle Scholar |
Nagler, M., Insam, H., Pietramellara, G., and Ascher-Jenull, J. (2018). Extracellular DNA in natural environments: features, relevance and applications. Applied Microbiology and Biotechnology 102, 6343–6356.
| Extracellular DNA in natural environments: features, relevance and applications.Crossref | GoogleScholarGoogle Scholar | 29858957PubMed |
Natarajan, V. P., Zhang, X., Morono, Y., Inagaki, F., and Wang, F. (2016). A modified SDS-based DNA extraction method for high quality environmental DNA from seafloor environments. Frontiers in Microbiology 7, 986.
| A modified SDS-based DNA extraction method for high quality environmental DNA from seafloor environments.Crossref | GoogleScholarGoogle Scholar | 27446026PubMed |
Nielsen, K. M., Johnsen, P. J., Bensasson, D., and Daffonchio, D. (2007). Release and persistence of extracellular DNA in the environment. Environmental Biosafety Research 6, 37–53.
| Release and persistence of extracellular DNA in the environment.Crossref | GoogleScholarGoogle Scholar | 17961479PubMed |
Ogram, A., Sayler, G. S., and Barkay, T. (1987). The extraction and purification of microbial DNA from sediments. Journal of Microbiological Methods 7, 57–66.
| The extraction and purification of microbial DNA from sediments.Crossref | GoogleScholarGoogle Scholar |
Pansu, J., Giguet-Covex, C., Ficetola, G. F., Gielly, L., Boyer, F., Zinger, L., Arnaud, F., Poulenard, J., Taberlet, P., and Choler, P. (2015a). Reconstructing long-term human impacts on plant communities: an ecological approach based on lake sediment DNA. Molecular Ecology 24, 1485–1498.
| Reconstructing long-term human impacts on plant communities: an ecological approach based on lake sediment DNA.Crossref | GoogleScholarGoogle Scholar | 25735209PubMed |
Pansu, J., De Danieli, S., Puissant, J., Gonzalez, J.-M., Gielly, L., Cordonnier, T., Zinger, L., Brun, J.-J., Choler, P., Taberlet, P., and Cécillon, L. (2015b). Landscape-scale distribution patterns of earthworms inferred from soil DNA. Soil Biology & Biochemistry 83, 100–105.
| Landscape-scale distribution patterns of earthworms inferred from soil DNA.Crossref | GoogleScholarGoogle Scholar |
Pansu, J., Guyton, J. A., Potter, A. B., Atkins, J. L., Daskin, J. H., Wursten, B., Kartzinel, T. R., and Pringle, R. M. (2019). Trophic ecology of large herbivores in a reassembling African ecosystem. Journal of Ecology 107, 1355–1376.
| Trophic ecology of large herbivores in a reassembling African ecosystem.Crossref | GoogleScholarGoogle Scholar |
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples: primers for marine microbiome studies. Environmental Microbiology 18, 1403–1414.
| Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples: primers for marine microbiome studies.Crossref | GoogleScholarGoogle Scholar | 26271760PubMed |
Pauvert, C., Buée, M., Laval, V., Edel-Hermann, V., Fauchery, L., Gautier, A., Lesur, I., Vallance, J., and Vacher, C. (2019). Bioinformatics matters: the accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline. Fungal Ecology 41, 23–33.
| Bioinformatics matters: the accuracy of plant and soil fungal community data is highly dependent on the metabarcoding pipeline.Crossref | GoogleScholarGoogle Scholar |
Pawlowski, J., Kelly-Quinn, M., Altermatt, F., Apothéloz-Perret-Gentil, L., Beja, P., Boggero, A., Borja, A., Bouchez, A., Cordier, T., Domaizon, I., Feio, M. J., Filipe, A. F., Fornaroli, R., Graf, W., Herder, J., van der Hoorn, B., Iwan Jones, J., Sagova-Mareckova, M., Moritz, C., Barquín, J., Piggott, J. J., Pinna, M., Rimet, F., Rinkevich, B., Sousa-Santos, C., Specchia, V., Trobajo, R., Vasselon, V., Vitecek, S., Zimmerman, J., Weigand, A., Leese, F., and Kahlert, M. (2018). The future of biotic indices in the ecogenomic era: integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems. The Science of the Total Environment 637–638, 1295–1310.
| The future of biotic indices in the ecogenomic era: integrating (e)DNA metabarcoding in biological assessment of aquatic ecosystems.Crossref | GoogleScholarGoogle Scholar | 29801222PubMed |
Pawlowski, J., Apothéloz‐Perret‐Gentil, L., and Altermatt, F. (2020). Environmental DNA: what’s behind the term? Clarifying the terminology and recommendations for its future use in biomonitoring. Molecular Ecology 29, 4258–4264.
| Environmental DNA: what’s behind the term? Clarifying the terminology and recommendations for its future use in biomonitoring.Crossref | GoogleScholarGoogle Scholar | 32966665PubMed |
Pietramellara, G., Ascher, J., Borgogni, F., Ceccherini, M. T., Guerri, G., and Nannipieri, P. (2009). Extracellular DNA in soil and sediment: fate and ecological relevance. Biology and Fertility of Soils 45, 219–235.
| Extracellular DNA in soil and sediment: fate and ecological relevance.Crossref | GoogleScholarGoogle Scholar |
Pochon, X., Wood, S. A., Keeley, N. B., Lejzerowicz, F., Esling, P., Drew, J., and Pawlowski, J. (2015). Accurate assessment of the impact of salmon farming on benthic sediment enrichment using foraminiferal metabarcoding. Marine Pollution Bulletin 100, 370–382.
| Accurate assessment of the impact of salmon farming on benthic sediment enrichment using foraminiferal metabarcoding.Crossref | GoogleScholarGoogle Scholar | 26337228PubMed |
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., Peplies, J., and Glöckner, F. O. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41, D590–D596.
| The SILVA ribosomal RNA gene database project: improved data processing and web-based tools.Crossref | GoogleScholarGoogle Scholar | 23193283PubMed |
Ramírez, G. A., Graham, D., and D’Hondt, S. (2018a). Influence of commercial DNA extraction kit choice on prokaryotic community metrics in marine sediment. Limnology and Oceanography, Methods 16, 525–536.
| Influence of commercial DNA extraction kit choice on prokaryotic community metrics in marine sediment.Crossref | GoogleScholarGoogle Scholar |
Ramírez, G. A., Jørgensen, S. L., Zhao, R., and D’Hondt, S. (2018b). Minimal influence of extracellular DNA on molecular surveys of marine sedimentary communities. Frontiers in Microbiology 9, 2969.
| Minimal influence of extracellular DNA on molecular surveys of marine sedimentary communities.Crossref | GoogleScholarGoogle Scholar | 30564217PubMed |
Ranjard, L., Lejon, D. P. H., Mougel, C., Schehrer, L., Merdinoglu, D., and Chaussod, R. (2003). Sampling strategy in molecular microbial ecology: influence of soil sample size on DNA fingerprinting analysis of fungal and bacterial communities. Environmental Microbiology 5, 1111–1120.
| Sampling strategy in molecular microbial ecology: influence of soil sample size on DNA fingerprinting analysis of fungal and bacterial communities.Crossref | GoogleScholarGoogle Scholar | 14641591PubMed |
Schnell, I. B., Bohmann, K., and Gilbert, M. T. P. (2015). Tag jumps illuminated: reducing sequence-to-sample misidentifications in metabarcoding studies. Molecular Ecology Resources 15, 1289–1303.
| Tag jumps illuminated: reducing sequence-to-sample misidentifications in metabarcoding studies.Crossref | GoogleScholarGoogle Scholar | 25740652PubMed |
Seiderer, L., and Newell, R. C. (1999). Analysis of the relationship between sediment composition and benthic community structure in coastal deposits: implications for marine aggregate dredging. ICES Journal of Marine Science 56, 757–765.
| Analysis of the relationship between sediment composition and benthic community structure in coastal deposits: implications for marine aggregate dredging.Crossref | GoogleScholarGoogle Scholar |
Simčič, T. (2005). The role of plankton, zoobenthos, and sediment in organic matter degradation in oligotrophic and eutrophic mountain lakes. Hydrobiologia 532, 69–79.
| The role of plankton, zoobenthos, and sediment in organic matter degradation in oligotrophic and eutrophic mountain lakes.Crossref | GoogleScholarGoogle Scholar |
Simpson, S. L., and Batley, G. E. (Eds) (2016). ‘Sediment Quality Assessment: a Practical Guide’, 2nd edn. (CSIRO Publishing: Melbourne, Vic., Australia.)
Simpson, S. L., Batley, G. E., and Chariton, A. A. (2013). Revision of the ANZECC/ARMCANZ sediment quality guidelines. (CSIRO.) Available at https://publications.csiro.au/publications/publication/PIlegacy:965 [Verified 8 January 2020].
Snelgrove, P. V. R. (1997). The importance of marine sediment biodiversity in ecosystem processes. Ambio 26, 578–583.
Snelgrove, P. V. R., and Butman, C. A. (1994). Animal–sediment relationships revisited: cause versus effect. Oceanography and Marine Biology – an Annual Review 32, 111–177.
Sutcliffe, B., Chariton, A. A., Harford, A. J., Hose, G. C., Greenfield, P., Elbourne, L. D. H., Oytam, Y., Stephenson, S., Midgley, D. J., and Paulsen, I. T. (2017). Effects of uranium concentration on microbial community structure and functional potential. Environmental Microbiology 19, 3323–3341.
| Effects of uranium concentration on microbial community structure and functional potential.Crossref | GoogleScholarGoogle Scholar | 28631400PubMed |
Sutcliffe, B., Hose, G. C., Harford, A. J., Midgley, D. J., Greenfield, P., Paulsen, I. T., and Chariton, A. A. (2019). Microbial communities are sensitive indicators for freshwater sediment copper contamination. Environmental Pollution 247, 1028–1038.
| Microbial communities are sensitive indicators for freshwater sediment copper contamination.Crossref | GoogleScholarGoogle Scholar | 30823331PubMed |
Taberlet, P., Coissac, E., Hajibabaei, M., and Rieseberg, L. H. (2012a). Environmental DNA. Molecular Ecology 21, 1789–1793.
| Environmental DNA.Crossref | GoogleScholarGoogle Scholar | 22486819PubMed |
Taberlet, P., Prud’Homme, S. M., Campione, E., Roy, J., Miquel, C., Shehzad, W., Gielly, L., Rioux, D., Choler, P., Clément, J.-C., Melodelima, C., Pompanon, F., and Coissac, E. (2012b). Soil sampling and isolation of extracellular DNA from large amount of starting material suitable for metabarcoding studies: extraction of extracellular DNA from soil. Molecular Ecology 21, 1816–1820.
| Soil sampling and isolation of extracellular DNA from large amount of starting material suitable for metabarcoding studies: extraction of extracellular DNA from soil.Crossref | GoogleScholarGoogle Scholar | 22300434PubMed |
Taberlet, P., Bonin, A., Zinger, L., and Coissac, E. (2018). ‘Environmental DNA: for Biodiversity Research and Monitoring.’ (Oxford University Press: Oxford, UK.)
Thomsen, P. F., and Willerslev, E. (2015). Environmental DNA: an emerging tool in conservation for monitoring past and present biodiversity. Biological Conservation 183, 4–18.
| Environmental DNA: an emerging tool in conservation for monitoring past and present biodiversity.Crossref | GoogleScholarGoogle Scholar |
Torti, A., Lever, M. A., and Jørgensen, B. B. (2015). Origin, dynamics, and implications of extracellular DNA pools in marine sediments. Marine Genomics 24, 185–196.
| Origin, dynamics, and implications of extracellular DNA pools in marine sediments.Crossref | GoogleScholarGoogle Scholar | 26452301PubMed |
Valentini, A., Miquel, C., Nawaz, M. A., Bellemain, E., Coissac, E., Pompanon, F., Gielly, L., Cruaud, C., Nascetti, G., Wincker, P., Swenson, J. E., and Taberlet, P. (2009). New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Molecular Ecology Resources 9, 51–60.
| New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach.Crossref | GoogleScholarGoogle Scholar | 21564566PubMed |
Vuillemin, A., Horn, F., Alawi, M., Henny, C., Wagner, D., Crowe, S. A., and Kallmeyer, J. (2017). Preservation and significance of extracellular DNA in ferruginous sediments from Lake Towuti, Indonesia. Frontiers in Microbiology 8, 1440.
| Preservation and significance of extracellular DNA in ferruginous sediments from Lake Towuti, Indonesia.Crossref | GoogleScholarGoogle Scholar | 28798742PubMed |
Wagner, A. O., Praeg, N., Reitschuler, C., and Illmer, P. (2015). Effect of DNA extraction procedure, repeated extraction and ethidium monoazide (EMA)/propidium monoazide (PMA) treatment on overall DNA yield and impact on microbial fingerprints for bacteria, fungi and archaea in a reference soil. Applied Soil Ecology 93, 56–64.
| Effect of DNA extraction procedure, repeated extraction and ethidium monoazide (EMA)/propidium monoazide (PMA) treatment on overall DNA yield and impact on microbial fingerprints for bacteria, fungi and archaea in a reference soil.Crossref | GoogleScholarGoogle Scholar | 26339125PubMed |
Wang, Y., Sheng, H.-F., He, Y., Wu, J.-Y., Jiang, Y.-X., Tam, N. F.-Y., and Zhou, H.-W. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags. Applied and Environmental Microbiology 78, 8264–8271.
| Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags.Crossref | GoogleScholarGoogle Scholar | 23001654PubMed |
Yoccoz, N. G., Bråthen, K. A., Gielly, L., Haile, J., Edwards, M. E., Goslar, T., Von Stedingk, H., Brysting, A. K., Coissac, E., Pompanon, F., Sønstebø, J. H., Miquel, C., Valentini, A., De Bello, F., Chave, J., Thuiller, W., Wincker, P., Cruaud, C., Gavory, F., Rasmussen, M., Gilbert, M. T. P., Orlando, L., Brochmann, C., Willerslev, E., and Taberlet, P. (2012). DNA from soil mirrors plant taxonomic and growth form diversity: DNA from soils mirrors plant diversity. Molecular Ecology 21, 3647–3655.
| DNA from soil mirrors plant taxonomic and growth form diversity: DNA from soils mirrors plant diversity.Crossref | GoogleScholarGoogle Scholar | 22507540PubMed |
Zinger, L., Chave, J., Coissac, E., Iribar, A., Louisanna, E., Manzi, S., Schilling, V., Schimann, H., Sommeria-Klein, G., and Taberlet, P. (2016). Extracellular DNA extraction is a fast, cheap and reliable alternative for multi-taxa surveys based on soil DNA. Soil Biology & Biochemistry 96, 16–19.
| Extracellular DNA extraction is a fast, cheap and reliable alternative for multi-taxa surveys based on soil DNA.Crossref | GoogleScholarGoogle Scholar |
Zinger, L., Bonin, A., Alsos, I. G., Bálint, M., Bik, H., Boyer, F., Chariton, A. A., Creer, S., Coissac, E., Deagle, B. E., De Barba, M., Dickie, I. A., Dumbrell, A. J., Ficetola, G. F., Fierer, N., Fumagalli, L., Gilbert, M. T. P., Jarman, S., Jumpponen, A., Kauserud, H., Orlando, L., Pansu, J., Pawlowski, J., Tedersoo, L., Thomsen, P. F., Willerslev, E., and Taberlet, P. (2019). DNA metabarcoding: need for robust experimental designs to draw sound ecological conclusions. Molecular Ecology 28, 1857–1862.
| DNA metabarcoding: need for robust experimental designs to draw sound ecological conclusions.Crossref | GoogleScholarGoogle Scholar | 31033079PubMed |