

Metalloligand Strategies for Assembling Heteronuclear Nanocages – Recent Developments*
Feng Li A C and Leonard F. Lindoy B CA School of Science and Health, Western Sydney University, Locked Bag 1797, Penrith, NSW 2751, Australia.
B School of Chemistry, F11, The University of Sydney, Sydney, NSW 2006, Australia.
C Corresponding authors. Email: feng.li@westernsydney.edu.au; len.lindoy@sydney.edu.au

Feng Li obtained his B.Sc. (1999) and M.Sc. (2002) at Zhengzhou University, China. In 2006, he completed his Ph.D. in the Instituto de Tecnologia Química e Biológica (ITQB) at the Universidade Nova de Lisboa (UNL), Portugal. In 2007, he took up a post-doctoral position at the University of Sydney (2007–2012), where he carried out research in the areas of metallo-supramolecular chemistry and inorganic materials chemistry. In 2012, he was appointed to a permanent lecturership in chemistry at Western Sydney University (WSU). In 2015, he was promoted to Senior Lecturer. His expertise spans experimental and theoretical supramolecular chemistry, synthetic organic chemistry, and coordination chemistry with emphasis on molecular recognition and magnetism. |

Leonard F. Lindoy is an Emeritus Professor at the University of Sydney. He obtained his B.Sc. (1964), Ph.D. (1968), and D.Sc. (1992) from the University of New South Wales. Following a post-doctoral appointment at Ohio State University, he joined James Cook University (1970) and in 1996 moved to the Chair of Inorganic Chemistry at Sydney. Len is a Fellow of the Australian Academy of Science, a Senior Member of Robinson College, Cambridge, and has been awarded honorary doctorates by the Universities of Wollongong (NSW) and Edinburgh (UK). He holds Honorary/Guest Professorships at East China University of Science and Technology, Guizhou Normal University, and Guizhou University. |
Australian Journal of Chemistry 72(10) 731-741 https://doi.org/10.1071/CH19279
Submitted: 19 June 2019 Accepted: 29 July 2019 Published: 20 August 2019
Abstract
The use of metalloligands as building blocks for the assembly of metallo-organic cages has received increasing attention over the past two decades or so. In part, the popularity of this approach reflects its stepwise nature that lends itself to the predesigned construction of metallocages and especially heteronuclear metallocages. The focus of the present discussion is on the use of metalloligands for the construction of discrete polyhedral cages, very often incorporating heterometal ions as structural elements. The metalloligand approach uses metal-bound multifunctional ligand building blocks that display predesigned structural properties for coordination to a second metal ion such that the rational design and construction of both homo- and heteronuclear metal–organic cages are facilitated. The present review covers published literature in the area from early 2015 to early 2019.
Introduction
Nanoscale metal–organic cages have now been investigated for more than three decades, with aspects of their synthesis and properties being the subject of recent reviews.[1–19] Such discrete self-assembled species incorporating well-defined cavities have been demonstrated to display a range of unusual magnetic, optical, and other properties as well as a wide variety of interesting host–guest behaviour.[4,12,20–24] Potential applications, among others, include molecule storage and separation,[25] drug delivery,[26–28] stabilisation of reactive species within the cage’s cavity,[29,30] and catalysis in which the cage serves as a ‘molecular flask’ for promoting a range of reactions involving an encapsulated guest (or guests).[31–49] With respect to this, cage cavity sizes have been tailored to accommodate reactants, leading to a range of catalytic reactions (including stereoselective as well as bimolecular reactions) occurring in the cavity, with holes in the cavity walls that allow the ingress and egress of reactants and products.
Over recent years, strategies involving the use of metalloligands have increasingly been applied to the synthesis of a wide variety of metallosupramolecular assemblies.[38,46–50] In 2015, the present authors published a review covering the use of the metalloligand approach solely for the synthesis of nanocages.[51] The present review represents an update of this earlier work.
Scope of the Review
Metalloligands may be divided into two categories.[51] Category one involves metal-containing ligand systems incorporating free donor functions in which a primary metal ion is bound in the metalloligand but does not play a direct role in orienting the coordination vectors of the associated additional (free) donor sites present. This typically occurs for rigid (often highly conjugated) ligand systems such as, for example, metal porphyrin derivatives with additional donor functions appended to the porphyrin skeleton[7,52,53] or for metalloligands in which the primary metal-binding site is located on the ligand backbone in a manner that does not directly influence the positioning of the donor sites employed for cage assembly.[54] However, with respect to this it needs to be noted that, while not directly involved in promoting cage assembly, such primary metal centres may still serve to provide additional functionality to the product cage.
The second metalloligand category consists of systems that incorporate a bound (primary) metal ion that acts to direct additional unbound ligand donor sites such that they are suitably oriented for further coordination to (secondary) metal centres.[1,55–57] The present review is restricted to a discussion of cages derived from metalloligands belonging to the latter category, with such cage formation often taking place in the presence of suitable coligands needed to satisfy the coordination requirements of the particular metal ions involved.
Our earlier review was concerned with the use of metalloligands for discrete cage synthesis covering the available literature up to the end of 2014.[51] We now discuss further developments in the area stemming from this time, with earlier studies being mentioned only where it seemed appropriate to put the more recent studies into context. The discussion is restricted to homo- and heterometallic closed cage systems incorporating d- and f-block metal centres; in general, expanded cage and capsule-like systems displaying large apertures are not discussed. Metalloligand-derived cages of an organometallic nature are also excluded as are metalloligand-derived polymeric cage derivatives. Clathrochelate metalloligand systems were the subject of a recent review by Jansze and Severin[58] and have been otherwise discussed,[4,59–62] and are also excluded from the present discussion. Similarly, a detailed survey covering the synthesis and structures of photoactive supramolecular cages incorporating ruthenium(ii) and iridium(iii), including systems obtained employing metalloligands, appeared in early 2019;[63] accordingly only selected (representative) examples of metalloligand-derived cages of this type are presented in the following discussion.
Use of Metalloligands for Constructing Nanocages
Heterometallic [(M′)2(M″)3]n+ Trigonal Bipyramidal Cage Derivatives
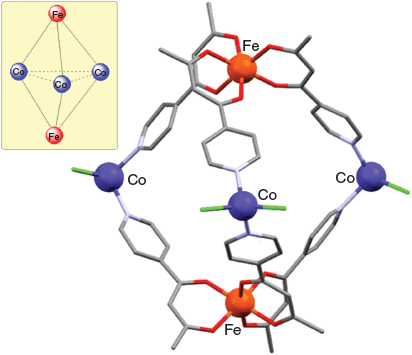
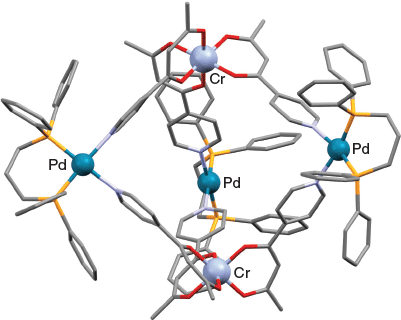
Crystalline trigonal bipyramidal cage derivatives of type [Fe2Co3L6Cl6], [Fe2Zn3L6Br6], [Cr2Zn3L6Br6], [Cr2Pd3L6(dppp)3](OTf)6, and [Al2Pd3L6(dppp)3](OTf)6 (where dppp = 1,3-bis(diphenylphosphino)propane and OTf is triflate) have been synthesised.[64]
The first three (neutral) cages were obtained on reacting the required fac-[MIIIL3] (M = Cr or Fe; HL = 1-(4-pyridyl) butane-1,3-dione, 1) (Chart 1) octahedrally coordinated metalloligand, initially present as an equilibrium mixture of its facial (fac) and meridional (mer) forms, with cobalt(ii) chloride or zinc(ii) bromide in a 2 : 3 molar ratio. In each structure, three of these divalent metal ions act as tetrahedrally coordinated linking units between two apical fac-[MIIIL3] metalloligands in which the equatorially positioned divalent metal centres are each bound to two pyridyl nitrogens from different metalloligands as well as to two halide ligands. The synthesis of the remaining two (charged) complexes [Cr2Pd3L6(dppp)3](OTf)6 and [Al2Pd3L6(dppp)3](OTf)6 involved the reaction of three cis-protected square-planar [PdII(dppp)(OTf2)2] units with two [MIIIL3] (MIII = Cr3+ or Al3+; HL = 1) metalloligands. In forming these cages, each palladium(ii) centre exchanges its two labile triflate ligands for two pyridyl groups, one from each of the two (apical) [MIIIL3] metalloligands. The locations of the five metal centres present in each of the above products define their respective trigonal bipyramidal geometries; that is, the two trivalent ions occupy the axial positions while the three divalent ions are positioned equatorially. This cage type represents the smallest assembly that employs all available donor sites present in the two [MIIIL3] metalloligands as well as all available metal coordination sites in the three cis-protected [Pd(dppp)(OTf)2] complexes (following loss of their labile cis-positioned triflate ligands).
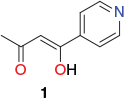
|
The assembly of the above cages occurs stereoselectively: each cage molecule incorporates either ΔΔ- or ΛΛ-octahedral metal centres, with each bulk product crystallising as a ΔΔ + ΛΛ racemic mixture. The X-ray structure of [FeIII2CoII3L6Cl6] (HL = 1) is given in Fig. 1, and that of [CrIII2PdII3L6(dppp)3](OTf)6 is shown in Fig. 2. [FeIII2CoII3L6Cl6] (HL = 1) exhibits antiferromagnetic exchange between its paramagnetic iron(iii) and cobalt(ii) ions.
|
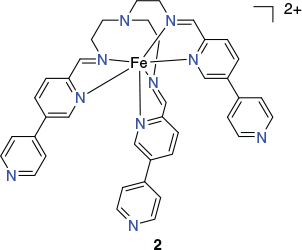
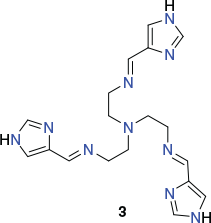
In a subsequent study, Lützen et al. prepared the low-spin iron(ii)-containing C3-symmetric metalloligand 2 (Chart 2), isolated as its tetrafluoroborate salt, by reaction of (3,4′-bipyridine)-4-carboxaldehyde (3 equiv.) with tris(2-aminoethyl)amine (1 equiv.) and iron(ii) tetrafluoroborate (1 equiv.) in acetonitrile.[65] The tripodal structure of this metalloligand was confirmed by X-ray diffraction.
|
Reaction of [FeIIL](BF4)2 (where [FeIIL]2+ = 2) (2 equiv.) with cis-protected [Pd(dppp)(OTf)2] (3 equiv.) in acetonitrile yielded the trigonal bipyramidal cage [(FeL)2{Pd(dppp)}3](BF4)4(OTf)6 as a blue crystalline product. The X-ray structure of the [(FeL)2{Pd(dppp)}3]10+ cation (Fig. 3) confirmed its trigonal bipyramidal geometry. In this structure, each of the palladium(ii) centres maintains a cis square-planar coordination geometry, being bound to two 4-pyridyl moieties from different iron(ii) metalloligands and a bidentate dppp ligand.
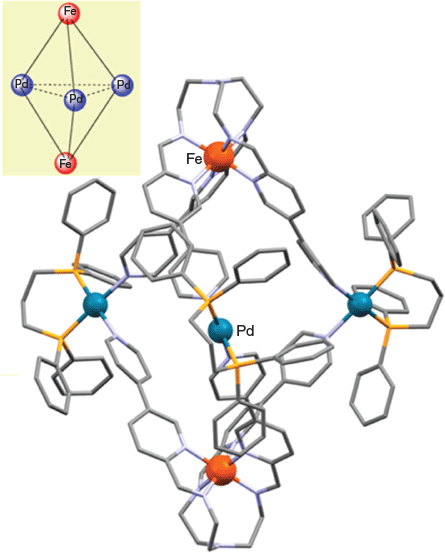
Key to the formation of the above trigonal bipyramidal [(M′)2(M″)3]n+ cages (as opposed to larger cubic cages of the type discussed in the next section) was considered to be the use of a tetrahedral or a cis-protected square-planar metal species to connect the tripodal metalloligands rather than employing non-cis-protected, square-planar metal species (sometimes with further coligands occupying the axial sites).[65] For example, if the cis-protected square-planar complex [Pd(dppp)(OTf)2], with its two available coordination sites, is replaced in the above trigonal bipyramidal cage synthesis by the square-planar complex [Pd(acetonitrile)4](BF4)2, incorporating four available coordination sites, then the geometry of the resulting cage is changed from trigonal bipyramidal to cubic. Namely, when [FeL](BF4)2 ([FeL]2+ = 2) was reacted with [Pd(acetonitrile)4](BF4)2, a corresponding [(FeL)8Pd6]36+ cubic cage of the general type discussed in the next section was generated.[65]
In related syntheses to those above, Murkerjee et al. reacted the neutral tripodal metalloligands ML (M = FeIII, CoIII; where H3L is 3) (Chart 3), with cis-protected cis-[Pd(tmen)(NO3)2] (tmen = tetramethylethylenediamine) in methanol in the required 2 : 3 ratio to yield the corresponding trigonal bipyramidal products; the structure of the [CoIII2PdII3]-containing cage was confirmed by single-crystal X-ray analysis.[66]
|
There are two reported examples of trigonal bipyramidal cage syntheses where the above-mentioned synthetic considerations have been essentially bypassed.[67]
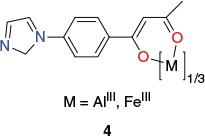
The [Al2Pd3(L)6Cl6] and [Fe2Pd3(L)6Cl6] cages (where HL is 1-4-(1H-imidazol-1-yl)phenylbutane-2,3-dione) with structures related to those discussed above have been synthesised employing tripodal metalloligands of type 4 (with M = Al3+ or Fe3+) (Chart 4) in combination with PdCl2 or Pd(MeCN)2Cl2. Both metalloligand isomerisation and homochiral assembly were found to occur spontaneously during cage formation. In each cage product, the M3+ ions occupy the two axial sites – adopting ΔΔ or ΛΛ configurations – while the three Pd2+ ions, with trans-planar coordination geometries, occupy the three equatorial positions. It appears that the successful synthesis of these cages reflects the extremely mild conditions employed. Crystals of [Al2Pd3(L)6Cl6] were obtained simply by layering a DMSO/CH2Cl2 solution over a CH2Cl2 solution of Al(L)3, with a solution of PdCl2 in DMSO then being layered over the DMSO/CH2Cl2 layer. On standing for 10 days at ambient temperature, yellowish crystals of [Al2Pd3(L)6Cl6] had formed. A related mild ‘layering’ procedure was employed to obtain crystals of [Al2Pd3(L)6Cl6].
|
Heterometallic [(M′)8(M″)6]n+ Cubic and Rhombododecahedral Cage Derivatives
In several studies before 2015, metalloligand strategies were used to assemble heterometallic [(M′)8(M″)6]n+ (where M′ and M″ are selected mono-, di-, and trivalent metal ions) face-centred cube derivatives in which the M′ ions occupy the corners of the cube while the M″ ions are located on or above the centre of each face (in the latter case to yield a distorted face-centred cubic or rhombododecahedral arrangement – the latter defined by the positions of the 14 metal centres present in each case).[68–74] Commonly (but not always – see later), M′ represents an octahedrally coordinated metal whereas M″ can be a square-planar, square-pyramidal or octahedral metal species.
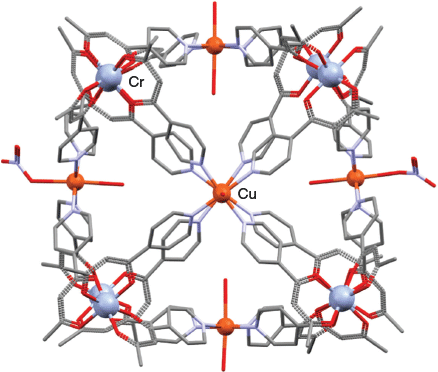
Following an earlier report of the synthesis of a diamagnetic [AlIII8PdII6L24]12+ cubic cage (where HL = 1-(4-pyridyl)butane-1,3-dione, 1) by Wu et al.,[69] Lusby, Brechin et al. demonstrated the versatility of the general strategy by employing the octahedral, kinetically inert metalloligand fac-[CrIIIL3] (HL = 1), to prepare a range of new paramagnetic [CrIII8MII6L24] cages.[75,76] The facial configuration of fac-[CrIIIL3] (HL = 1), which had been previously separated from its fac-/mer-isomeric mixture, was confirmed by an X-ray diffraction study. It was then reacted with different cobalt(ii) and copper(ii) salts to yield the structurally related cubic cages [CrIII8CoII6L24(H2O)12](ClO4)12,[76] [CrIII8CuII6L24(H2O)10(NO3)2](NO3)10,[76] [CrIII8CoII6L24X12] (X = Cl; NCS),[75] [CrIII8CuII6L24(H2O)24](SO4)6,[75] and [CrIII8CuII6L24Cl12].[75] As expected, the eight corners of each cube are occupied by chromium(iii) ions while the divalent cobalt(ii) or copper(ii) ions, which are axially coordinated by either water molecules or anions, are located at the centres of the faces.
The X-ray structure of [CrIII8CuII6L24(H2O)10(NO3)2]10+ is shown in Fig. 4;[76] in this case, the six copper(ii) centres show Jahn–Teller-distorted (axially elongated) six-coordinate geometries with four of these centres having solely water ligands in their axial sites while the remaining two (opposite) centres have a water and a nitrato ligand in these sites. The authors noted that the possibility of varying the divalent and trivalent metal centres in such structures provides a means for generating a family of cages whose properties (for example, their magnetic properties) can be tuned while maintaining a similar cube-like structural framework.
As an extension of the above studies, the syntheses of the [FeIII8MII6] (MII = Co, Ni, Cu) cages [FeIII8PdII6L24]Cl12, [FeIII8CuII6L24(H2O)4Br4]Br8, [FeIII8CuII6L24(H2O)10](NO3)12, [FeIII8NiII6L24(SCN)11Cl], and [FeIII8CoII6L24(SCN)10(H2O)2]Cl2 (HL = 1) were undertaken.[77] A related synthetic strategy to that employed for the preparation of the [CrIII8MII6L24] (HL = 1) cages just discussed was employed, but in this case, the synthesis started from the tripodal iron(iii) metalloligand [FeIIIL3] (HL = 1). However, unlike in the case of [CrIIIL3] (HL = 1), where the kinetically stable facial isomer was separated before its use for cage synthesis, [FeIIIL3] (HL = 1) was present in solution as an equilibrium mixture of its facial and meridional isomers and this mixture was employed for the assembly of the above FeIII-containing cages. During this process, the fac-isomer is selectively sequestered, disturbing the equilibrium and consequently increasing its availability for cage formation. Once again, the cubic structures of the resulting cages parallel in general terms those derived employing the fac-[CrIIIL3] (HL = 1) metalloligand, with in each case the iron(iii) metal centres occupying the corners of the respective cubes and being octahedrally coordinated. For the FeIII/PdII derivative, each palladium(ii) centre has a square-planar geometry whereas the remaining cages incorporating cobalt(ii), nickel(ii) or copper(ii) have these metal centres present in either square-pyramidal or octahedral geometries.
As mentioned earlier, and as commonly occurs for other cage systems, all five structures crystallise as homochiral racemates, namely with the eight [FeIIIL3] centres in individual cubes adopting either all-Λ or all-Δ configurations. Magnetic susceptibility measurements on [FeIII8CoII6L24(SCN)10(H2O)2]Cl2, [FeIII8NiII6L24(SCN)11Cl], and [FeIII8CuII6L24(H2O)10](NO3)12 revealed the presence of weak antiferromagnetic exchange between the metal centres in these cages.
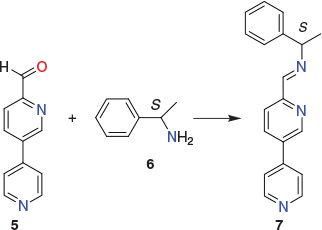
The direct synthesis and isolation of resolved homochiral [(M′)8(M″)6L24]n+ cubic cages (where L is a single enantiomer of the ditopic ligand 6) has been described by Wang et al.[78] Each of the syntheses involved the in situ generation of the required chiral metalloligand from its metal-free enantiomeric (ditopic) ligand precursor. For example, in the case of the S-isomer, this involved generation of the chiral heterotopic Schiff base ligand 7 via condensation of the dipyridyl-aldehyde 5 with the chiral amine S-1-phenylethylamine 6 in acetonitrile (Fig. 5). Heterometal complexation was then carried out without prior isolation of the metalloligand; that is, zinc(ii) then palladium(ii) ions were added directly to 7 in acetonitrile, with the overall Zn : Pd : 7 reaction ratio being 8 : 6 : 24. This led to isolation of the corresponding homochiral cubic cage of type ΔΔΔΔΔΔΔΔ-[Zn8Pd6(L)24](BF4)28 (L = 7).
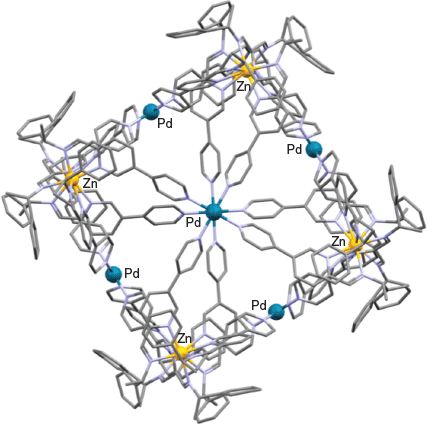
The X-ray structure of the above cage product (Fig. 6) confirmed that each of the eight corners of the resulting cube is occupied by a C3-symmetric [Zn(pyridylimine)3]2+ unit, with the respective octahedrally bound zinc(ii) centres each displaying a Δ configuration. The palladium(ii) ions (with their propensity to adopt a square-planar coordination geometry) are positioned on each of the six faces, with each palladium centre being bound to four pendent pyridyl nitrogens derived from four adjacent zinc-bound 7 ligands. Substitution of R-1-phenylethylamine for S-1-phenylethylamine in the above reaction sequence led to isolation of the corresponding ΛΛΛΛΛΛΛΛ-diastereoisomer.
|
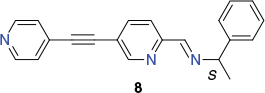
As part of this study, it was also demonstrated that a similar synthetic strategy employing the extended chiral ligand 8 yielded the corresponding larger cube [Zn8Pd6(L)24](BF4)28 (L = 8) (Chart 5).[78] Although no X-ray structure determination proved possible, this cage was assigned a homochiral ΔΔΔΔΔΔΔΔ-arrangement on the basis of its circular dichroism spectrum.
|
In a subsequent study, the Wang group also employed a related in situ procedure to obtain two further enantiopure cubic cages of type [MII8CuII6L24(MeCN)12](ClO4)28 (M = Ni or Co; L = the R- or S-enantiomer of 7, shown in Fig. 5 as the S-enantiomer).[79] Once again, these cages display controllable chirality by switching the hand of the chiral amine employed for their synthesis. Both products exhibit weak antiferromagnetism.
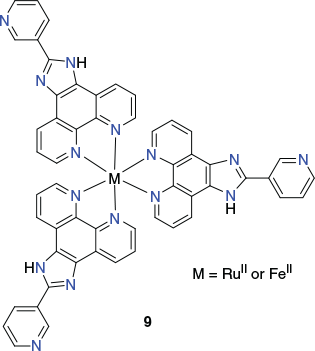
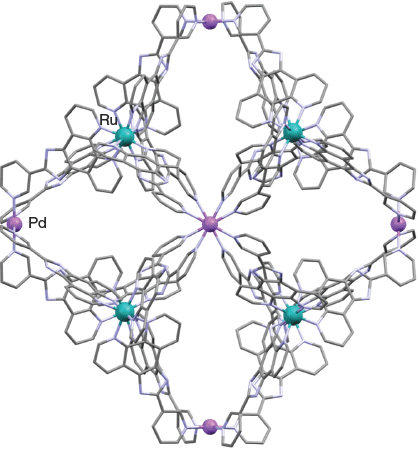
In 2014, Su et al. reported that the kinetically inert ruthenium(ii) C3-symmetric metalloligand 9 reacts with [Pd(CH3CN)4](BF4)2 in a 4 : 3 ratio in DMSO to form a coordination cage of type [Pd6(RuL3)8]28+ (RuL3 = 9) (Chart 6).[80] An X-ray structure (Fig. 7) determination showed that this cationic species has an overall distorted rhombododecahedral geometry, with six palladium(ii) centres defining a truncated octahedron while eight ruthenium(ii) metalloligands cap the faces. Once again, each individual cage is homochiral, displaying either ΛΛΛΛΛΛΛΛ- or ΔΔΔΔΔΔΔΔ-configurations around the respective ruthenium centres; the product crystallises as a racemic mixture of each form. This cage system was demonstrated to readily encapsulate aromatic guest molecules that include phenanthrene, pyrene, and anthracene.
|
Following this initial study, it was demonstrated that when individual optically resolved isomers of metalloligand 9 (M = RuII) were reacted with palladium(ii), then enantiopure Λ- or Δ-cage products were in turn isolated.[81] The synthetic procedure involved the use of racemic Λ/Δ-[Ru(1,10-phenanthroline)3] as a precursor. In an initial step, this racemate was first classically resolved with the aid of K2[Sb2{(+)-tartrate}2]·3H2O and the individual stereoisomers were then chemically modified to ultimately yield the separated Λ- and Δ-forms of metalloligand 9 (M = RuII). In turn, these were then used to form the corresponding Λ- and Δ-forms of the [Pd6(RuL3)8]28+ (where RuL3 = 9) cage. The absolute configurations of these cage products were successfully established by X-ray diffraction following the growth of single crystals of the individual isomers from acetonitrile in the presence of S-binol and R-binol (binol = 1,1′-bi-2-naphthol) respectively. Individual isomers of the latter cocrystallise with the respective cages and serve as absolute structure reference compounds. The X-ray structures also revealed that the palladium(ii) centres adopt fixed chiral configurations. Thus, the Pd(pyridyl)4 fragments in each stereoisomer each have a ‘fan-like’ arrangement giving rise to either all clockwise or all anticlockwise stereoconfigurations about the palladium centres in the respective structures; the ‘handedness’ of the palladium centres is opposite to that of the ruthenium centres in each case.
In aqueous media, although the individual homochiral cages showed rather poor stereoselectivity towards chiral compounds containing asymmetric carbon stereocentres, they did show significant enantio-differentiation between (atropisomeric) S-binol and R-binol molecules. These enter the respective cage cavities driven by the hydrophobic effect, with the process attributed to be moderated by differences in the dynamic exchange behaviour of the respective host–guest diastereomers.
In further studies, the photoredox-active nature of the metalloligand 9 (M = RuII) was also exploited.[82] Both the racemic and enantiopure [Pd6(RuL3)8]28+ (RuL3 = 9) cages discussed above were employed to encapsulate naphthol guests. These were then shown to undergo regiospecific 1,4-coupling on radiation (λ 453 nm) to yield 4-(2-hydroxy-1-naphthyl)-1,2-napthoquinones. This coupling differs from the normal 1,1-coupling observed in the absence of such a cage. Moderate stereochemical control was also achieved when the homochiral cage was employed. Thus, dual functionality was observed for this system, with both photoinduced redox reactivity and with a measure of stereo-selectivity being achieved.
Related studies involving the use of the configurationally more labile metalloligand [FeL3]2+ of type 9 have also been reported.[83] The preparation of [FeL3](BF4)2 was achieved via reaction of the presynthesised ligand[84] with Fe(BF4)2·6H2O in DMSO in a 3 : 1 molar ratio. Addition of a solution of the resulting product in DMSO with [Pd(CH3CN)4](BF4)2 in a molar ratio of 4 : 3 yielded the deep red heteronuclear cage [Pd6(FeL3)8](BF4)28 (where FeL3 = 9), as a racemic mixture of individual homochiral cage molecules. In this study, the relative stereolability of ‘free’ 9 (with M = FeII) towards both dissociation and racemisation was demonstrated to be greatly reduced when incorporated as part of the superstructure of the [Pd6(FeL3)8]28+ cage product. This resulting enhanced configurational stability clearly reflects that the eight [FeL3]2+ units are ‘buttressed’ by their connections to the six adjacent palladium(ii) centres. Indeed, the resolved cage molecules were found to retain their chirality in D2O at room temperature for more than 5 months.
The first example of a discrete 3d/4f cage, [Cu6Dy8L8(MeOH)8(H2O)6](NO3)12·(solvent)n (where H3L = tripodal tris{[2-{(imidazole)methylidene}amino]ethyl}amine, 3), was reported by Li et al.[85] The two-step synthesis of this purple crystalline product initially involved formation of the robust non-centrosymmetric metalloligand [DyH3L(NO3)]2+, where H3L is 3.[86] Reaction of copper(ii) nitrate with this metalloligand in a 1 : 1.4 molar ratio in the presence of base led to assembly of the above cage, which crystallised as a racemic mixture with either ΛΛΛΛΛΛΛΛ- or ΔΔΔΔΔΔΔΔ-configurations at the Dy3+ centres. The X-ray structure of this product (Fig. 8) confirmed the presence of a slightly distorted square-prismatic coordination structure in which the Dy3+ ions are positioned at the corners of the prism while the Cu2+ ions are located on the centres of its faces; four deprotonated imidazole groups from different metalloligands coordinate to each copper centre, which are also associated with coordinated waters. Each Dy3+ centre has a non-symmetric, eight-coordinate coordination geometry that contributes to the distortion from cubic symmetry observed for the cage. This heterometallic cage displays weak molecular magnetic behaviour.

|
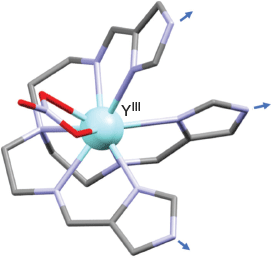
Subsequently, the related non-centrosymmetric metalloligand [Y(H3L)(NO3)]2+ (where H3L = 3) was also reported and its rigid, distorted capped square-antiprism structure was confirmed by X-ray diffraction (Fig. 9).[86] Tripodal H3L acts as a heptadentate ligand towards the yttrium(iii) ion, which is also bound to a bidentate nitrato ligand to yield a nine-coordinate, non-symmetric coordination shell.
|
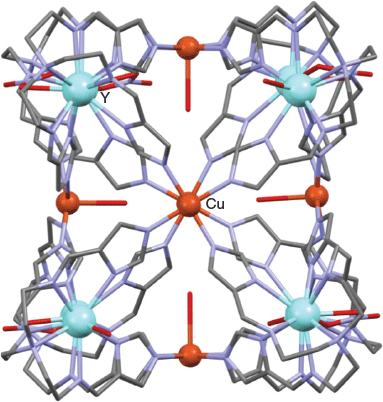
Reaction of the above non-centrosymmetric metalloligand with copper(ii) nitrate in methanol under basic conditions, followed by filtration of the reaction solution, then slow diffusion of THF into the filtrate, yielded purple crystals of [Cu6Y8L8(NO3)5(H2O)3](NO3)7·21H2O (where H3L = 3).[86] The X-ray structure confirmed the distorted cubic arrangement of the resulting cage (Fig. 10). As expected, the eight corners of the cube are defined by the eight Y3+ ions (each displaying non-centrosymmetric metal coordination geometries). Five of these are nine-coordinate with NO3− anions as coligands while three are eight-coordinate with H2O coligands. The six Cu2+ ions are located on the six faces of the cube, with four of these copper sites being coordinated to water molecules, while the remaining two (opposite) copper(ii) sites are bound to nitrato ligands. Individual cage molecules are chiral, but once again the product crystallised as a racemic mixture of both enantiomeric forms. This study of a Y8Cu6L8 cage along with that of the related Dy8Cu6L8 cage discussed above illustrate the way that substitution of different metal centres in an otherwise similar metalloligand can result in variation of the corresponding metal coordination spheres and, in turn, in modification of the resulting cage’s topology.
Miscellaneous Other Cage Systems
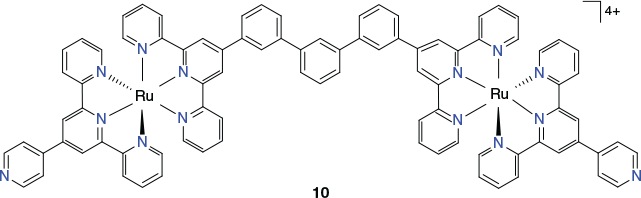
Beves et al. have reported the use of metalloligands incorporating [Ru(terpyridine)2]2+ chromophores for the assembly of extended cage molecules incorporating large internal cavities.[87,88] As an example, this group reported the synthesis of the 4 nm long metalloligand 10 (Chart 7), which they reacted with [Pd(CH3CN)4](BF4)2 in a 3 : 5 molar ratio in acetonitrile.[88] Rapid assembly in solution occurred to form a [Pd2(metalloligand 10)4]20+ species, which was characterised using high-resolution electrospray mass spectrometry and NMR spectroscopy (including diffusion measurements). Although no X-ray structure could be obtained, the results from the physical measurements combined with molecular mechanics modelling were in accord with the resulting structure being a symmetric discrete (elongated) cage in which the two palladium(ii) centres are spanned by four metalloligands of type 10; thus, each palladium(ii) centre is bound to four terminal pyridyl groups arising respectively from the four metalloligands.
|
Nitschke et al. have developed an innovative one-pot strategy for generating [M8(metalloligand)6]16+ cubic cages in which C4-symmetric metalloligands of type 12 (Fig. 11), each incorporating a dimolybdenum(ii) paddle-wheel core connected to four potentially bidentate α-diimine donor functions, are formed in situ in acetonitrile.[89,90] Four cages of type [M8(metalloligand 12)6]16+ (M = FeII or ZnII and X = H or F) were synthesised. These were obtained by Schiff base condensation of 2-formylpyridine (24 equiv.) with tetrakis(p-aminobenzoato)dimolybdenum(ii) (11, X = H or F) (6 equiv.) in the presence of the required iron(ii) or zinc(ii) triflate (8 equiv.). The respective product cages were isolated as their triflate salts.[90]

|
The X-ray structure (Fig. 12) of the [Zn8(metalloligand 12)6]16+ (X = H) cage is representative of this cage series. The structure confirmed that each of the six faces of the cube is occupied by a dimolybdenum(ii) paddlewheel while the eight vertices are defined by eight zinc(ii) centres, each of which is six-coordinate with a tris(α-diimine) coordination shell. The coordinatively unsaturated molybdenum centres were expected to act as binding sites for coordinating species and this was shown to be the case towards halide ions. Although the presence of fluoro substituents (that is, the cages with X = F) resulted in only minor perturbations of the corresponding cubic structures, their introduction was nevertheless sufficient to significantly influence the relative binding affinities of individual cages towards particular halide ions.[90]
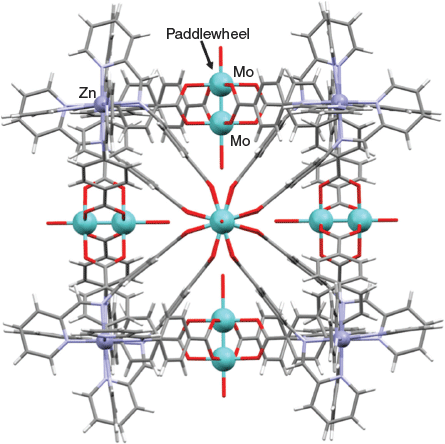
|
Metalloligands of type ML (where M = CoIII, FeIII and H3L = tris{[2-{(imidazole)methylidene}amino]ethyl}amine, 3) incorporating the triply deprotonated form of 3[66] each react with manganese(ii) perchlorate in methanol to yield, as expected, the corresponding face-centred cubic cages of type [(MIII)8(MnII)6L8]12+ (where M = FeIII or CoIII, H3L = 3) of the general type discussed in the preceding section.[66] However, in marked contrast, when zinc(ii) nitrate was substituted for manganese(ii) perchlorate in the general synthetic procedure, then two open-sided cubic structures of type [MIII4ZnII4L8]4+ (M = Fe, Co, H3L = 3) were obtained in which the heterometal sites alternate around the eight corners of the cube; both structures were confirmed by X-ray diffraction (and were shown to include a perchlorate anion in their respective cavities). The structure of [FeIII4ZnII4L8]4+ is given in Fig. 13.
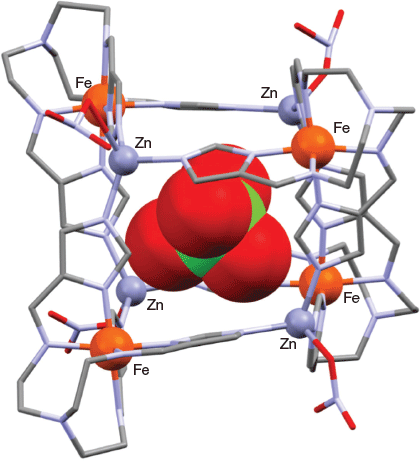
|
Metherell and Ward have reported a related [M′4M″4L12]12+ cubic cage that also displays alternating M′ and M″ metal sites as well as, in this case, alternating chiralities around the eight metal centres defining the cube.[91] The metalloligand [RuII(L)3](PF6)2 (L = 13) was prepared directly by reacting 13 (Chart 8) with [Ru(dmso)4Cl2] in ethylene glycol at low temperature; the product was isolated as a non-statistical 7 : 1 mer-/fac-isomer mixture, undoubtedly reflecting the non-equilibrium conditions present during the crystallisation process. This stereoisomeric mixture was then reacted with Co(BF4)2 in dichloromethane/methanol at room temperature and the product formed was recrystallised from nitromethane over several months (aided by THF vapour diffusion) to yield a yellow crystalline product of type [Ru4Co4(L)12{Na(BF4)4}](PF6)6(BF4)7 (L = 13). An X-ray structure determination showed that the cage cation consists of an approximately cubic Ru4Co4 framework in which alternating cobalt(ii) and ruthenium(ii) ions are positioned around each of the eight metal sites occupying the corners of the cube. These metal sites also show alternating fac- (for the cobalt(ii) sites) and mer- (for the ruthenium(ii) sites) stereogenic configurations.
|
The internal cavity of the cationic [Ru4Co4(L)12{Na(BF4)4}]13+ (L = 13) cube is occupied by a tetrahedral array of tetrafluoroborate anions that are in turn coordinated to a central sodium ion to provide an unusual example of an [Na(BF4)4]3− anionic guest residing inside a metal–organic cage. The X-ray structure of this host–guest cage product is shown in Fig. 14.
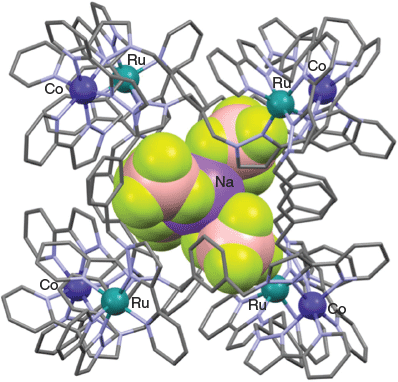
|
Finally, it is noted that in a previous study the same authors also prepared a related [Ru4Cd4L12]16+ cubic structure in which the ruthenium(ii) and cadmium(ii) centres again alternate around adjacent corners of the cube.[92]
Concluding Remarks
Metalloligand strategies for the assembly of a variety of metal–organic cage types have now been well delineated and clearly point the way for the further rational design and synthesis of new homo- and heteronuclear cages exhibiting predefined topologies in the future. The stepwise nature of most cage syntheses of this type, involving both primary and secondary metal binding sites, tends to facilitate the synthesis of new cage systems incorporating heterometal centres that, for example, may vary widely in their nature and give rise to less common metal ion combinations. This, along with the prospect that the metal sites in such assemblies often communicate with each other, yields the prospect of generating heterometallic systems displaying unusual (and potentially useful) properties – in particular, optical, electronic (including redox), and magnetic properties. Although some success along this path has been achieved so far, clearly this aspect is ripe for further exploitation. Overall, there still remains much scope for the creative design of new metalloligand systems with defined electronic and structural natures leading to systems displaying desired functionality. Greater emphasis on this aspect will undoubtedly be rewarded by significant advances in the future.
Conflicts of Interests
The authors declare no conflicts of interest.
Acknowledgements
No specific funding was received for this review.
References
[1] N. B. Debata, D. Tripathy, H. S. Sahoo, Coord. Chem. Rev. 2019, 387, 273.| Crossref | GoogleScholarGoogle Scholar |
[2] M. Pan, K. Wu, J. H. Zhang, C. Y. Su, Coord. Chem. Rev. 2019, 378, 333.
| Crossref | GoogleScholarGoogle Scholar |
[3] H. S. Scott, R. W. Staniland, P. E. Kruger, Coord. Chem. Rev. 2018, 362, 24.
| Crossref | GoogleScholarGoogle Scholar |
[4] M. D. Ward, C. A. Hunter, N. H. Williams, Acc. Chem. Res. 2018, 51, 2073.
| Crossref | GoogleScholarGoogle Scholar | 30085644PubMed |
[5] P. M. Bogie, T. F. Miller, R. J. Hooley, Isr. J. Chem. 2019, 59, 130.
| Crossref | GoogleScholarGoogle Scholar |
[6] R. W. Hogue, S. Singh, S. Brooker, Chem. Soc. Rev. 2018, 47, 7303.
| Crossref | GoogleScholarGoogle Scholar | 30124687PubMed |
[7] D. Zhang, T. K. Ronson, J. R. Nitschke, Acc. Chem. Res. 2018, 51, 2423.
| Crossref | GoogleScholarGoogle Scholar | 30207688PubMed |
[8] A. J. Brock, J. K. Clegg, F. Li, L. F. Lindoy, Coord. Chem. Rev. 2018, 375, 106.
| Crossref | GoogleScholarGoogle Scholar |
[9] S. Pullen, G. H. Clever, Acc. Chem. Res. 2018, 51, 3052.
| Crossref | GoogleScholarGoogle Scholar | 30379523PubMed |
[10] L. F. Lindoy, J. Incl. Phenom. Macrocycl. Chem. 2019, 94, 121.
| Crossref | GoogleScholarGoogle Scholar |
[11] Z. X. Wu, K. Zhou, A. V. Ivanov, M. Yusobov, F. Verpoort, Coord. Chem. Rev. 2017, 353, 180.
| Crossref | GoogleScholarGoogle Scholar |
[12] L. J. Chen, H. B. Yang, M. Shionoya, Chem. Soc. Rev. 2017, 46, 2555.
| Crossref | GoogleScholarGoogle Scholar | 28452389PubMed |
[13] A. J. McConnell, Supramol. Chem. 2018, 30, 858.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. Brock, H. Al-Fayaad, M. C. Pfrunder, J. K. Clegg, in Solid-State Supramolecular Materials (Ed. F. R. Banerjee) 2017, Ch. 10, pp. 325–387 (RSC: London).
[15] R. A. S. Vasdev, D. Preston, J. D. Crowley, Chem. Asian J. 2017, 12, 2513.
| Crossref | GoogleScholarGoogle Scholar |
[16] T. R. Cook, P. J. Stang, Chem. Rev. 2015, 115, 7001.
| Crossref | GoogleScholarGoogle Scholar | 25813093PubMed |
[17] L. Chen, Q. Chen, M. Wu, F. Jiang, M. Hong, Acc. Chem. Res. 2015, 48, 201.
| Crossref | GoogleScholarGoogle Scholar | 25517043PubMed |
[18] L. Avram, Y. Cohen, Chem. Soc. Rev. 2015, 44, 586.
| Crossref | GoogleScholarGoogle Scholar | 25110858PubMed |
[19] A. M. Castilla, W. J. Ramsay, J. R. Nitschke, Acc. Chem. Res. 2014, 47, 2063.
| Crossref | GoogleScholarGoogle Scholar | 24793652PubMed |
[20] F. J. Rizzuto, L. K. S. von Krbek, J. R. Nitschke, Nat. Rev. Chem. 2019, 3, 204.
| Crossref | GoogleScholarGoogle Scholar |
[21] X. Jing, C. He, L. Zhao, C. Duan, Acc. Chem. Res. 2019, 52, 100.
| Crossref | GoogleScholarGoogle Scholar | 30586276PubMed |
[22] R. Custelcean, Chem. Soc. Rev. 2014, 43, 1813.
| Crossref | GoogleScholarGoogle Scholar | 24384869PubMed |
[23] M. D. Ward, P. R. Raithby, Chem. Soc. Rev. 2013, 42, 1619.
| Crossref | GoogleScholarGoogle Scholar | 22797247PubMed |
[24] M. D. Pluth, K. N. Raymond, Chem. Soc. Rev. 2007, 36, 161.
| Crossref | GoogleScholarGoogle Scholar | 17264920PubMed |
[25] H. Amouri, C. Desmarets, J. Moussa, Chem. Rev. 2012, 112, 2015.
| Crossref | GoogleScholarGoogle Scholar | 22251425PubMed |
[26] W. Q. Xu, Y. Z. Fan, H. P. Wang, J. Teng, Y. H. Li, C. X. Chen, D. Fenske, J. J. Jiang, C. Y. Su, Chem. – Eur. J. 2017, 23, 3542.
| Crossref | GoogleScholarGoogle Scholar | 28094459PubMed |
[27] W. Cullen, S. Turega, C. A. Hunter, M. D. Ward, Chem. Sci. 2015, 6, 625.
| Crossref | GoogleScholarGoogle Scholar | 28936311PubMed |
[28] N. Ahmad, H. A. Younus, A. H. Chughtai, F. Verpoort, Chem. Soc. Rev. 2015, 44, 9.
| Crossref | GoogleScholarGoogle Scholar | 25319756PubMed |
[29] A. Galan, P. Ballester, Chem. Soc. Rev. 2016, 45, 1720.
| Crossref | GoogleScholarGoogle Scholar | 26797259PubMed |
[30] H. Vardhan, F. Verpoort, Adv. Synth. Catal. 2015, 357, 1351.
| Crossref | GoogleScholarGoogle Scholar |
[31] W.-X. Gao, H.-N. Zhang, G.-X. Jin, Coord. Chem. Rev. 2019, 386, 69.
| Crossref | GoogleScholarGoogle Scholar |
[32] V. Marti-Centelles, A. L. Lawrence, P. J. Lusby, J. Am. Chem. Soc. 2018, 140, 2862.
| Crossref | GoogleScholarGoogle Scholar | 29406705PubMed |
[33] W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams, M. D. Ward, Nat. Chem. 2016, 8, 231.
| Crossref | GoogleScholarGoogle Scholar | 26892554PubMed |
[34] C. Tan, D. Chu, X. Tang, Y. Liu, W. Xuan, Y. Cui, Chem. – Eur. J. 2019, 25, 662.
| Crossref | GoogleScholarGoogle Scholar | 30076749PubMed |
[35] C. M. Hong, R. G. Bergman, K. N. Raymond, F. D. Toste, Acc. Chem. Res. 2018, 51, 2447.
| Crossref | GoogleScholarGoogle Scholar | 30272943PubMed |
[36] P. S. Bols, H. L. Anderson, Acc. Chem. Res. 2018, 51, 2083.
| Crossref | GoogleScholarGoogle Scholar | 30156831PubMed |
[37] L. J. Jongkind, X. Caumes, A. P. T. Hartendorp, J. N. H. Reek, Acc. Chem. Res. 2018, 51, 2115.
| Crossref | GoogleScholarGoogle Scholar | 30137959PubMed |
[38] I. Sinha, P. S. Mukherjee, Inorg. Chem. 2018, 57, 4205.
| Crossref | GoogleScholarGoogle Scholar | 29578701PubMed |
[39] C. Deraedt, D. Astruc, Coord. Chem. Rev. 2016, 324, 106.
| Crossref | GoogleScholarGoogle Scholar |
[40] M. Otte, ACS Catal. 2016, 6, 6491.
| Crossref | GoogleScholarGoogle Scholar |
[41] M. D. Ward, C. A. Hunter, N. H. Williams, Chem. Lett. 2017, 46, 2.
| Crossref | GoogleScholarGoogle Scholar |
[42] C. J. Brown, F. D. Toste, R. G. Bergman, K. N. Raymond, Chem. Rev. 2015, 115, 3012.
| Crossref | GoogleScholarGoogle Scholar | 25898212PubMed |
[43] M. D. Pluth, R. G. Bergman, K. N. Raymond, Acc. Chem. Res. 2009, 42, 1650.
| Crossref | GoogleScholarGoogle Scholar | 19591461PubMed |
[44] D. Fiedler, D. H. Leung, R. G. Bergman, K. N. Raymond, Acc. Chem. Res. 2005, 38, 349.
| Crossref | GoogleScholarGoogle Scholar | 15835881PubMed |
[45] J. S. Train, A. B. Wragg, A. J. Auty, A. J. Metherell, D. Chekulaev, C. G. P. Taylor, S. P. Argent, J. A. Weinstein, M. D. Ward, Inorg. Chem. 2019, 58, 2386.
| 30688057PubMed |
[46] Y. Y. Zhang, W. X. Gao, L. Lin, G. X. Jin, Coord. Chem. Rev. 2017, 344, 323.
| Crossref | GoogleScholarGoogle Scholar |
[47] M. D. Wise, K. Severin, Chimia 2015, 69, 191.
| Crossref | GoogleScholarGoogle Scholar | 26668936PubMed |
[48] H. Li, Z. J. Yao, D. Liu, G. X. Jin, Coord. Chem. Rev. 2015, 293–294, 139.
| Crossref | GoogleScholarGoogle Scholar |
[49] G. Kumar, R. Gupta, Chem. Soc. Rev. 2013, 42, 9403.
| Crossref | GoogleScholarGoogle Scholar | 24081027PubMed |
[50] D. Rota Martir, D. Escudero, D. Jacquemin, D. B. Cordes, A. M. Z. Slawin, H. A. Fruchtl, S. L. Warriner, E. Zysman-Colman, Chem. – Eur. J. 2017, 23, 14358.
| Crossref | GoogleScholarGoogle Scholar | 28783869PubMed |
[51] L. Li, D. J. Fanna, N. D. Shepherd, L. F. Lindoy, F. Li, J. Incl. Phenom. Macrocycl. Chem. 2015, 82, 3.
| Crossref | GoogleScholarGoogle Scholar |
[52] S. S. Nurttila, W. Brenner, J. Mosquera, K. M. van Vliet, J. R. Nitschke, J. N. H. Reek, Chem. – Eur. J. 2019, 25, 609.
| 30351486PubMed |
[53] T. Y. Cen, S. P. Wang, Z. B. Zhang, J. Wu, S. J. Li, J. Porphyr. Phthalocyanines 2018, 22, 726.
| Crossref | GoogleScholarGoogle Scholar |
[54] (a) See, for example: E. T. Luis, H. Iranmanesh, K. S. A. Arachchige, W. A. Donald, G. Quach, E. G. Moore, J. E. Beves, Inorg. Chem. 2018, 57, 8476.
| Crossref | GoogleScholarGoogle Scholar | 29969245PubMed |
(b) G. L. Liu, M. Zeller, K. Z. Su, J. D. Pang, Z. F. Ju, D. Q. Yuan, M. C. Hong, Chem. – Eur. J. 2016, 22, 1734.
[55] Y. Y. Zhang, L. Zhang, Y. J. Lin, G. X. Jin, Chem. – Eur. J. 2015, 21, 14983.
[56] L. Li, A. R. Craze, D. J. Fanna, A. J. Brock, J. K. Clegg, L. F. Lindoy, J. R. Aldrich-Wright, J. K. Reynolds, F. Li, Polyhedron 2017, 125, 44.
| Crossref | GoogleScholarGoogle Scholar |
[57] Y. J. Zhang, M. Bhadbhade, L. G. Kong, I. Karatchevtseva, R. K. Zheng, Polyhedron 2017, 138, 82.
| Crossref | GoogleScholarGoogle Scholar |
[58] S. M. Jansze, K. Severin, Acc. Chem. Res. 2018, 51, 2139.
| Crossref | GoogleScholarGoogle Scholar | 30156828PubMed |
[59] S. Komine, T. Tateishi, T. Kojima, H. Nakagawa, Y. Hayashi, S. Takahashi, S. Hiraoka, Dalton Trans. 2019, 4139.
| Crossref | GoogleScholarGoogle Scholar | 30785436PubMed |
[60] S. M. Jansze, D. Ortiz, F. Fadaei Tirani, R. Scopelliti, L. Menin, K. Severin, Chem. Commun. 2018, 9529.
| Crossref | GoogleScholarGoogle Scholar |
[61] G. Cecot, M. Marmier, S. Geremia, R. De Zorzi, A. V. Vologzhanina, P. Pattison, E. Solari, F. F. Tirani, R. Scopelliti, K. Severin, J. Am. Chem. Soc. 2017, 139, 8371.
| Crossref | GoogleScholarGoogle Scholar | 28603972PubMed |
[62] G. Cecot, B. Alameddine, S. Prior, R. D. Zorzi, S. Geremia, R. Scopelliti, F. T. Fadaei, E. Solari, K. Severin, Chem. Commun. 2016, 11243.
| Crossref | GoogleScholarGoogle Scholar |
[63] D. Rota Martir, E. Zysman-Colman, Chem. Commun. 2019, 139.
| Crossref | GoogleScholarGoogle Scholar |
[64] S. Sanz, H. M. O’Connor, V. Marti-Centelles, P. Comar, M. B. Pitak, S. J. Coles, G. Lorusso, E. Palacios, M. Evangelisti, A. Baldansuren, N. F. Chilton, H. Weihe, E. J. L. McInnes, P. J. Lusby, S. Piligkos, E. K. Brechin, Chem. Sci. 2017, 8, 5526.
| Crossref | GoogleScholarGoogle Scholar | 28970932PubMed |
[65] M. Hardy, N. Struch, F. Topić, G. Schnakenburg, K. Rissanen, A. Lützen, Inorg. Chem. 2018, 57, 3507.
| Crossref | GoogleScholarGoogle Scholar | 29185725PubMed |
[66] R. Saha, D. Samanta, A. J. Bhattacharyya, P. S. Mukherjee, Chem. – Eur. J. 2017, 23, 8980.
| Crossref | GoogleScholarGoogle Scholar | 28471006PubMed |
[67] X.-R. Wu, S.-Y. Yao, L.-Q. Wei, L.-P. Li, B.-H. Ye, Inorg. Chim. Acta 2018, 482, 605.
| Crossref | GoogleScholarGoogle Scholar |
[68] S. Wang, J. L. Zuo, H. C. Zhou, H. J. Choi, Y. X. Ke, J. R. Long, X. Z. You, Angew. Chem. Int. Ed. 2004, 43, 5940.
| Crossref | GoogleScholarGoogle Scholar |
[69] H. B. Wu, Q. M. Wang, Angew. Chem. Int. Ed. 2009, 48, 7343.
| Crossref | GoogleScholarGoogle Scholar |
[70] M. B. Duriska, S. M. Neville, B. Moubaraki, J. A. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J. F. Letard, K. S. Murray, C. J. Kepert, S. R. Batten, Angew. Chem. Int. Ed. 2009, 48, 2549.
| Crossref | GoogleScholarGoogle Scholar |
[71] M. B. Duriska, S. M. Neville, J. Z. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert, S. R. Batten, Angew. Chem. Int. Ed. 2009, 48, 8919.
| Crossref | GoogleScholarGoogle Scholar |
[72] M. M. J. Smulders, A. Jimenez, J. R. Nitschke, Angew. Chem. Int. Ed. 2012, 51, 6681.
| Crossref | GoogleScholarGoogle Scholar |
[73] F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C. G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H. C. Yao, F. Li, Inorg. Chem. 2014, 53, 688.
| Crossref | GoogleScholarGoogle Scholar | 24393071PubMed |
[74] D. Simond, S. E. Clifford, A. F. Vieira, C. Besnard, A. F. Williams, RSC Adv. 2014, 4, 16686.
| Crossref | GoogleScholarGoogle Scholar |
[75] H. M. O’Connor, S. Sanz, M. B. Pitak, S. J. Coles, G. S. Nichol, S. Piligkos, P. J. Lusby, E. K. Brechin, CrystEngComm 2016, 18, 4914.
| Crossref | GoogleScholarGoogle Scholar |
[76] S. Sanz, H. M. O’Connor, E. M. Pineda, K. S. Pedersen, G. S. Nichol, O. Mønsted, H. Weihe, S. Piligkos, E. J. L. McInnes, P. J. Lusby, E. K. Brechin, Angew. Chem. Int. Ed. 2015, 54, 6761.
| Crossref | GoogleScholarGoogle Scholar |
[77] S. Sanz, H. M. O’Connor, P. Comar, A. Baldansuren, M. B. Pitak, S. J. Coles, H. Weihe, N. F. Chilton, E. J. L. McInnes, P. J. Lusby, S. Piligkos, E. K. Brechin, Inorg. Chem. 2018, 57, 3500.
| Crossref | GoogleScholarGoogle Scholar | 29323893PubMed |
[78] Y. Yang, J. H. Jia, X. L. Pei, H. Zheng, Z. A. Nan, Q. M. Wang, Chem. Commun. 2015, 3804.
| Crossref | GoogleScholarGoogle Scholar |
[79] Y. Yang, Y. Wu, J. H. Jia, X. Y. Zheng, Q. Zhang, K. C. Xiong, Z. M. Zhang, Q. M. Wang, Cryst. Growth Des. 2018, 18, 4555.
| Crossref | GoogleScholarGoogle Scholar |
[80] K. Li, L. Y. Zhang, C. Yan, S. C. Wei, M. Pan, L. Zhang, C. Y. Su, J. Am. Chem. Soc. 2014, 136, 4456.
| Crossref | GoogleScholarGoogle Scholar | 24611560PubMed |
[81] K. Wu, K. Li, Y. J. Hou, M. Pan, L. Y. Zhang, L. Chen, C. Y. Su, Nat. Commun. 2016, 7, 10487.
| Crossref | GoogleScholarGoogle Scholar | 26839048PubMed |
[82] J. Guo, Y. W. Xu, K. Li, L. M. Xiao, S. Chen, K. Wu, X. D. Chen, Y. Z. Fan, J. M. Liu, C. Y. Su, Angew. Chem. Int. Ed. 2017, 56, 3852.
| Crossref | GoogleScholarGoogle Scholar |
[83] Y.-J. Hou, K. Wu, Z.-W. Wei, K. Li, Y.-L. Lu, C.-Y. Zhu, J.-S. Wang, M. Pan, J.-J. Jiang, G.-Q. Li, C.-Y. Su, J. Am. Chem. Soc. 2018, 140, 18183.
| Crossref | GoogleScholarGoogle Scholar | 30512934PubMed |
[84] X.-L. Chen, Z.-X. Han, H.-M. Hu, J.-J. Wang, S.-H. Chen, F. Fu, N. Li, M.-L. Yang, G.-L. Xue, Inorg. Chim. Acta 2009, 362, 3963.
| Crossref | GoogleScholarGoogle Scholar |
[85] L. Li, Y. J. Zhang, M. Avdeev, L. F. Lindoy, D. G. Harman, R. K. Zheng, Z. X. Cheng, J. R. Aldrich-Wright, F. Li, Dalton Trans. 2016, 9407.
| Crossref | GoogleScholarGoogle Scholar | 27227419PubMed |
[86] Y. Zhang, D. G. Harman, M. Avdeev, I. Karatchevtseva, Inorg. Chim. Acta 2019, 484, 521.
| Crossref | GoogleScholarGoogle Scholar |
[87] J. J. Yang, M. Bhadbhade, W. A. Donald, H. Iranmanesh, E. G. Moore, H. Yan, J. E. Beves, Chem. Commun. 2015, 4465.
| Crossref | GoogleScholarGoogle Scholar |
[88] C. Shen, A. D. W. Kennedy, W. A. Donald, A. M. Torres, W. S. Price, J. E. Beves, Inorg. Chim. Acta 2017, 458, 122.
| Crossref | GoogleScholarGoogle Scholar |
[89] W. J. Ramsay, T. K. Ronson, J. K. Clegg, J. R. Nitschke, Angew. Chem. Int. Ed. 2013, 52, 13439.
| Crossref | GoogleScholarGoogle Scholar |
[90] W. J. Ramsay, F. J. Rizzuto, T. K. Ronson, K. Caprice, J. R. Nitschke, J. Am. Chem. Soc. 2016, 138, 7264.
| Crossref | GoogleScholarGoogle Scholar | 27213555PubMed |
[91] A. J. Metherell, M. D. Ward, RSC Adv. 2016, 6, 10750.
| Crossref | GoogleScholarGoogle Scholar |
[92] A. J. Metherell, M. D. Ward, Chem. Commun. 2014, 6330.
| Crossref | GoogleScholarGoogle Scholar |
* This manuscript is dedicated to Professor Richard Robson, an extraordinary chemist and a pioneer in the development of both macrocyclic chemistry and metal–organic framework (MOF) chemistry.