

Temperature Replica Exchange Molecular Dynamics Simulations of Cyclic Peptide Conformation
Karen M. Corbett A , Colin W. Pouton A and David K. Chalmers A B
A , Colin W. Pouton A and David K. Chalmers A B
A Monash Institute of Pharmaceutical Sciences, Monash University, 381 Royal Parade, Parkville, Vic. 3052, Australia.
B Corresponding author. Email: david.chalmers@monash.edu

Karen Corbett is a post-doctoral Research Fellow at the Monash Institute of Pharmaceutical Sciences. She obtained her Ph.D. in molecular biophysics at Florida State University. Karen’s interests are in the development and application of enhanced sampling molecular dynamics methods to study functional motions of biomolecules, especially for conformational determinants of peptide permeability. |
Australian Journal of Chemistry 74(9) 684-685 https://doi.org/10.1071/CH21120
Submitted: 22 May 2021 Accepted: 8 July 2021 Published: 30 July 2021
Journal Compilation © CSIRO 2021 Open Access CC BY-NC
Keywords: molecular dynamics, replica exchange, parallel tempering, temperature replica exchange molecular dynamics, cyclic peptide structure, cyclic peptide conformation, structure-function relationships, enhanced sampling.
Background
Cyclic peptides (CPs) have applications as pharmaceuticals, agricultural chemicals, and nanomaterials.[1–4] CPs often take specific, interchanging 3D structures (conformations). The activity of CPs depends on their atomic-level structures and conformational dynamics. Characterising these properties is important to a fundamental understanding of CP function but has historically proven difficult.
Molecular dynamics simulations (MDS) represent each atom of the system of interest explicitly and propagate each atom with Newtonian dynamics. MDS allow characterisation of 3D structures of molecules like CPs with atomic-level and femtosecond-level detail. However, systems simulated using conventional MDS can become stuck in free energy minima (Fig. 1), meaning that they are challenged in capturing long-timescale (microsecond and above) CP conformational changes.
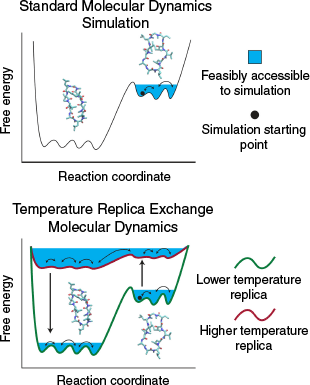
|
A special kind of MDS, temperature replica exchange MDS (TREMDS),[5] have increasingly been used to study CPs. TREMDS have the advantage of maintaining the atomic-level detail of MDS and capturing long-timescale peptide conformational changes. TREMDS involve concurrently running a series of MDS at various temperatures and swapping between them every so often, allowing enhanced conformational sampling at the lower temperature of interest compared with MDS (Fig. 1).[5] TREMDS are publicly available within several MDS engines, including GROMACS,[6] GENESIS,[7] CHARMM,[8] and NAMD.[9] We note that other kinds of replica exchange MDS also exist; for example, see refs [10–12].
Several studies have reported the remarkable accuracy of TREMDS in the structure prediction of CPs. Geng et al. conducted TREMDS of 20 CPs with known crystal structures.[13] Depending on the simulation parameters (i.e. ‘force fields’), approximately half to 15 of the CP crystal structures were predicted by TREMDS with sub-angstrom accuracy.[13] A separate TREMDS study by Hu et al. found that the most populated conformation observed in the TREMDS of Ac–(cyclo-1,5)-[CAAIS5(2-Me)(R)]–NH2 (S5, (S)-pentenylglycine) was ‘almost identical to its solved structure.’[14] Zhao et al. calculated the 3JNHCHα couplings and NOEs from TREMDS simulations of Ac–cyclo(Asp–Ala–Ala–Dap)–Ala–Ala–NH2 and Ac–cyclo(iso-Asp–Ala–Ala–Dap)–Ala–Ala]–NH2 (Dap, 2,3-diaminopropionic acid); these agreed with NMR measurements.[15]
Applications
TREMDS have recently supplemented experimental studies. Berger et al. used TREMDS to understand different solvent-dependent and temperature-dependent conformational effects of two CPs observed by NMR and FT-IR spectroscopy.[16] Hou et al. used TREMDS in conjunction with CD spectroscopy on two CP epimers to determine the relationship between chirality and helicity of the two peptides.[17] Hyung et al. studied several cyclosporin CPs using TREMDS and ion mobility mass spectrometry (IM-MS); the TREMDS simulations enabled conformational interpretation of the IM-MS collision cross-section profiles.[18]
TREMDS have been used to predict conformational determinants of CP activity, allowing the development of new, active compounds. Macaluso et al. studied four CP analogues of a vasoactive peptide. Using TREMDS and binding affinity experiments, they determined a conformational motif important for the target binding affinities of these CPs.[19] Kirschner et al. used TREMDS to characterise the conformations of active, antioestrogenic peptides (both cyclic and linear).[20] TREMDS then identified analogues with similar conformational motifs to the active peptides. These new TREMDS-identified analogues were synthesised and found to be active.[20]
Conclusion
TREMDS have shown success in studying of the conformation of CPs and their structure–function relationships. With increasing computational power and improved methodologies, we believe that TREMDS will be increasingly important for CP development.
Data Availability Statement
The data that support this study are available in the article and accompanying references.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
K. M. C. was supported by an American Australian Association Sir Keith Murdoch Scholarship.
References
[1] X. Wang, M. Lin, D. Xu, D. Lai, L. Zhou, Molecules 2017, 22, 2069.| Crossref | GoogleScholarGoogle Scholar |
[2] R. J. Brea, C. Reiriz, J. R. Granja, Chem. Soc. Rev. 2010, 39, 1448.
| Crossref | GoogleScholarGoogle Scholar | 20419200PubMed |
[3] A. Zorzi, K. Deyle, C. Heinis, Curr. Opin. Chem. Biol. 2017, 38, 24.
| Crossref | GoogleScholarGoogle Scholar | 28249193PubMed |
[4] R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe, S. Perrier, Chem. Soc. Rev. 2012, 41, 6023.
| Crossref | GoogleScholarGoogle Scholar | 22875035PubMed |
[5] Y. Sugita, Y. Okamoto, Chem. Phys. Lett. 1999, 314, 141.
| Crossref | GoogleScholarGoogle Scholar |
[6] M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess, E. Lindah, SoftwareX 2015, 1–2, 19.
| Crossref | GoogleScholarGoogle Scholar |
[7] C. Kobayashi, J. Jung, Y. Matsunaga, T. Mori, T. Ando, K. Tamura, M. Kamiya, Y. Sugita, J. Comput. Chem. 2017, 38, 2193.
| Crossref | GoogleScholarGoogle Scholar | 28718930PubMed |
[8] B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, et al. J. Comput. Chem. 2009, 30, 1545.
| Crossref | GoogleScholarGoogle Scholar | 19444816PubMed |
[9] J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, et al. J. Chem. Phys. 2020, 153, 044130.
| Crossref | GoogleScholarGoogle Scholar | 32752662PubMed |
[10] H. Fukunishi, O. Watanabe, S. Takada, J. Chem. Phys. 2002, 116, 9058.
| Crossref | GoogleScholarGoogle Scholar |
[11] Y. Sugita, A. Kitao, Y. Okamoto, J. Chem. Phys. 2000, 113, 6042.
| Crossref | GoogleScholarGoogle Scholar |
[12] L. Wang, R. A. Friesner, B. J. Berne, J. Phys. Chem. B 2011, 115, 9431.
| Crossref | GoogleScholarGoogle Scholar | 21714551PubMed |
[13] H. Geng, F. Jiang, Y.-D. Wu, J. Phys. Chem. Lett. 2016, 7, 1805.
| Crossref | GoogleScholarGoogle Scholar | 27128113PubMed |
[14] K. Hu, H. Geng, Q. Zhang, Q. Liu, M. Xie, C. Sun, W. Li, H. Lin, et al. Angew. Chem. 2016, 128, 8145.
| Crossref | GoogleScholarGoogle Scholar |
[15] H. Zhao, Q.-S. Liu, H. Geng, Y. Tian, M. Cheng, Y.-H. Jiang, M.-S. Xie, X.-G. Niu, et al. Angew. Chem. 2016, 128, 12267.
| Crossref | GoogleScholarGoogle Scholar |
[16] N. Berger, L. J. B. Wollny, P. Sokkar, S. Mittal, J. Mieres-Perez, R. Stoll, W. Sander, E. Sanchez-Garcia, ChemPhysChem 2019, 20, 1664.
| Crossref | GoogleScholarGoogle Scholar | 31045298PubMed |
[17] Z. Hou, C. Sun, H. Geng, K. Hu, M. Xie, Y. Ma, F. Jiang, F. Yin, Z. Li, Bioconjug. Chem. 2018, 29, 2904.
| Crossref | GoogleScholarGoogle Scholar | 30193458PubMed |
[18] S.-J. Hyung, X. Feng, Y. Che, J. G. Stroh, M. Shapiro, Anal. Bioanal. Chem. 2014, 406, 5785.
| Crossref | GoogleScholarGoogle Scholar | 25064599PubMed |
[19] N. J. M. Macaluso, R. C. Glen, ChemMedChem 2010, 5, 1247.
| Crossref | GoogleScholarGoogle Scholar |
[20] K. N. Kirschner, K. W. Lexa, A. M. Salisburg, K. A. Alser, L. Joseph, T. T. Andersen, J. A. Bennett, H. I. Jacobson, G. C. Shields, J. Am. Chem. Soc. 2007, 129, 6263.
| Crossref | GoogleScholarGoogle Scholar | 17441722PubMed |