

Probing the properties of molecules and complex materials using machine learning
David A. Winkler A B C *
A B C *
A Biochemistry and Chemistry, School of Agriculture, Biology and Engineering and La Trobe Institute for Molecular Science, La Trobe University, Bundoora, 3046, Australia.
B School of Medicinal Chemistry, Monash Institute of Pharmaceutical Science, Monash University, Parkville, 3154, Australia.
C School of Pharmacy, University of Nottingham, Nottingham, NG7 2QL, UK.
Australian Journal of Chemistry 75(11) 906-922 https://doi.org/10.1071/CH22138
Submitted: 16 June 2022 Accepted: 29 July 2022 Published: 13 September 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The application of machine learning to predicting the properties of small and large discrete (single) molecules and complex materials (polymeric, extended or mixtures of molecules) has been increasing exponentially over the past few decades. Unlike physics-based and rule-based computational systems, machine learning algorithms can learn complex relationships between physicochemical and process parameters and their useful properties for an extremely diverse range of molecular entities. Both the breadth of machine learning methods and the range of physical, chemical, materials, biological, medical and many other application areas have increased markedly in the past decade. This Account summarises three decades of research into improved cheminformatics and machine learning methods and their application to drug design, regenerative medicine, biomaterials, porous and 2D materials, catalysts, biomarkers, surface science, physicochemical and phase properties, nanomaterials, electrical and optical properties, corrosion and battery research.
Keywords: artificial intelligence, batteries, Bayesian methods, biomaterials, catalysts, complex systems, computational molecular design, drug design, machine learning, nanomaterials, organic photovoltaic (OPV) devices, porous materials, quantitative structure-activity relationships (QSAR), regenerative medicine, science, 2D materials.
Introduction
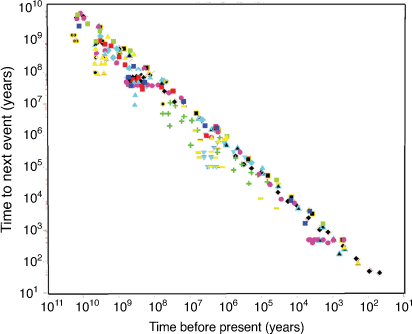
Science has always been fascinated by change, uncovering new aspects of Nature and finding useful ways to exploit them to meet global challenges. The rate of change is accelerating, with average time between innovations decreasing exponentially (Fig. 1).
|
Computational molecular design prior to ~1990 was focused on the use of computationally expensive physics-based methods like molecular modelling, molecular mechanics, molecular dynamics and quantum chemistry. The quantitative structure–activity relationship (QSAR) methods, developed by Hansch and Fujita in the 1960s, were based on the observation that changes in the constitution of small organic molecules generated a corresponding change in their biological activities. Regression methods were used to find relationships between structure, encoded by mathematical entities called descriptors or features, and biological properties of small organic molecules, also numerically encoded. QSAR use was limited to modelling of small data sets of molecules with similar scaffolds, with the primary aim of understanding the molecular basis for drug (or agrochemical) action. As they were not mechanism- or physics-based, their empirical nature created doubt as to their efficacy, the question of when correlation means causation (still an important issue), and lack of data were major barriers to their wider adoption.
After that time, technological developments involving automation, computational power, algorithms, synthesis and informatics have maintained this exponential acceleration. As all scientists can attest, there have been massive increases in the number of small molecules and materials that can be synthesised and characterised, triggered by the invention of combinatorial and other high throughput synthesis methods for drugs and agrochemicals, and by genomics (and subsequently other ‘omics’) technologies in the late 20th century. In the past two decades many of these technological developments have been adopted by materials, nanomaterials and biomaterials researchers, triggering an explosion of materials research.
As the molecular and biological systems that can be studied have become more complex, and analytical methods have become much more sensitive and selective, the amount of data generated has also increased exponentially. These massive data sets defy human interpretation. The key responses to this system complexity and overwhelming data and information are machine learning (ML) and complex systems science. Inspired by the success of QSAR methods using largely statistical regression and classification methods, ML differs from previous hard coded expert systems and rule-based algorithms in being able to autonomously learn complex relationships and patterns in data. Being data-driven, it is ideally matched to modelling complexity and extracting information and meaning from very large, complex, multidimensional data sets. ML is revolutionising many areas of science, technology, medicine and business. The application of ML to extracting patterns, rules and relationships from these rich, complex data sets has seen a broadening of the QSAR concept from mainly mechanistic understanding to accurate, robust and broadly applicable prediction of molecular properties and biological activity. This divergence was reviewed recently in Fujita’s final published work.[1]
Complex systems
Complex systems science studies deep connections between diverse areas of science, technology, medicine, business, sociology etc. and the emergent properties generated by very complicated systems that contains many simpler interacting elements. Key complexity concepts are the interconnectedness of components into a network with different properties to those of the components, chaotic behaviour, phase changes, self-organisation and self-assembly, similar power law behaviour in seemly unconnected phenomena and non-equilibrium systems.[2] Components of complex systems most easily seen to be relevant to chemistry are self-organisation and self-assembly (e.g. porous materials, DNA origami),[3] non-equilibrium systems (e.g. Belousov–Zhabotinsky or BZ reaction) and emergent properties of complex systems[4] (exemplified by the success of ML in modelling overt properties of complicated, multidimensional phenomena). A study of complex systems provides a new way of thinking about and analysing complex molecular and biological systems.[5] The use of ML and other AI methods to find deep connections between parameters and complicated patterns in data can be thought of modelling emergent properties of molecular systems using information about their basic building blocks.
Machine learning
The first QSAR models were generated using basic regression and classification methods. The recognition that many relationships are non-linear and the need to remove human bias from the definition of QSAR models (e.g. assuming a parabolic dependency for certain descriptors) opened the door for adoption of ML methods from ~1990 onwards. A popular ML method, the neural network, is a universal approximator able to model any continuous function given sufficient data. Neural networks and other ML methods are trained on descriptors and response variables and can automatically determine the degree of non-linearity and interactions between descriptors, without the need for subjective decisions that were a feature of the early QSAR models. When statistical models such as regression and classification are used for prediction rather than inference, they are considered ML methods.
Being a pattern recognition method, ML essentially models emergent properties of complex systems, without needing to know all mechanistic details (e.g. between administration and response for acute toxicities of molecules towards mice) or between structure and physicochemical properties and useful materials properties. The use of ML models to predict biological or physical properties of molecules and materials has expanded markedly from an initial QSAR focus on the activities of drugs and agrochemicals, and prediction of logP (octanol/water) and aqueous solubilities.[6,7]
The QSAR method was developed at a time when computational resources and data were very limited. The key steps in QSAR modelling are descriptor generation, feature selection, structure–activity/structure–property mapping, model validation and model interpretation. An important element of my research, working with long-time collaborator, Frank Burden, has involved deconstructing and improving these steps using modern mathematical and computational methods.
Descriptors
To generate ML models, it is essential to convert molecules or materials into mathematical entities (descriptors) that are relevant to the property being modelled. Descriptors make the largest contribution to model quality, robustness and predictivity, much greater than the choice of ML method. We conducted early research on generating ‘universal’ descriptors that can be used to model most biological and materials properties. We generated descriptors and fingerprints representing the connectivity of atoms in molecules and the partial charge distributions in molecules and used these to model biological properties.[8–10] More recently, we tackled the problem of generating descriptors for micron-scale topographical features on the surfaces of biomaterials (discussed below), a conceptually different problem to generating molecular descriptors.
Feature selection
Many thousands of descriptors can be generated for molecules, but the most relevant ones for modelling a given property are context dependent. It is important to remove the least informative descriptors before building models, as including them can lead to overfitting of the model, difficulties with model interpretation, and degradation of model quality. While there are a wide range of statistical methods to do this, we have adopted sparse feature selection methods such as LASSO (least absolute shrinkage and selection operator),[11] MLREM (multiple linear regression with expectation maximisation),[12] automatic relevance determination[13] and Bayesian regularised neural networks with a sparsity inducing Laplacian Bayesian prior[14] to provide the most relevant subset of descriptors for a given modelled property. These methods remove the less relevant descriptors and generate parsimonious models that have excellent predictive power and are easier to interpret because they have fewer features.
Structure–activity and structure–property mapping
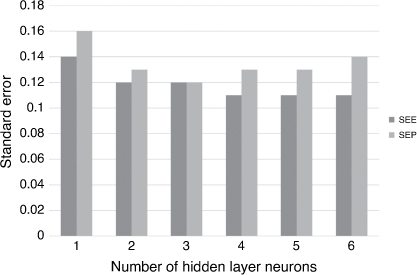
Once a sparse set of relevant features has been generated, a wide range of ML methods can be used to generate the model. The main difference in performance occurs between linear models (e.g. multiple linear regression (MLR) and MLREM), and non-linear models. For a given set of features and dependent properties, most non-linear ML methods will generate models of similar quality. The main issue with generating robust models from given training data is ensuring that the model has optimal complexity. If the model is too simple (bias, e.g. using a linear model for a non-linear relationship) or too complex (variance, model fits noise as well as the underlying relationship), its predictive power will be compromised. We solved this problem by employing Bayesian regularisation of a neural network (BRANN) to automatically control the complexity of models and generate optimum predictive power.[15,16] We employ Gaussian priors (BRANNGP) and sparsity-inducing Laplacian priors (BRANNLP), both automatically pruning the number of effective parameters (complexity) in the model with the latter also performing non-linear pruning of less relevant descriptors. As Fig. 2 shows, the performance of neural network models employing Bayesian regularisation is almost independent of the number of units (commonly called neurons, neurodes, nodes, processing elements or units) or in the hidden layer beyond a minimum number.[17]
|
Another common ML method used for classification (and regression) is the support vector machine (SVM), which can be prone to overfitting. We reported that by using the sparse Bayesian form of this algorithm, the relevant vector machine (RVM), sparser models of similar performance or less sparse models with superior performance could be generated from the same data sets.[18]
Deep learning methods such as deep neural networks have emerged very recently as novel ways of modelling a wide range of chemical, physical, medical and business phenomena. Unlike shallow neural networks that contain a single hidden layer with few neurons, deep neural networks have multiple hidden layers with large numbers of neurons in each. Overfitting is minimised by use of regularising methods such as weight drop out and the problem of vanishing gradients in deep neural networks is addressed by linear rectifier transfer functions in the neurons. Two of the main advantages of DNNs over shallow NNs are their abilities to generate effective latent features from very simple representations of molecules or other objects, and their ability to decode latent features back into new synthesisable molecules that potentially have better properties (e.g. encoder–decoder networks or generative–adversarial networks, GANs, Fig. 3).[19]

|
Given the same descriptors and properties, the performance of DNNs is similar to that of shallow NNs, consistent with the universal approximation theorem.[21] As the availability of data is sometimes still an issue for data-driven ML methods, meta models (an ensemble method in which strong models are generated from a consensus of weak models) and active learning (adaptive experimental design) approaches can greatly improve the efficiency of model generation by identifying the most important training data required to improve the generalisation ability of models.[22,23]
Model validation
It is important to validate how predictive models are, that is, how well they can predict the properties of molecules or materials not used to train them. A range of statistical methods such as cross validation and bootstrapping are commonly employed to assess the predictivity of ML models. However, the use of an independent test set, partitioned from the training set and never used in model generation, provides a more realistic estimate of predictive power. Analysis of test set predictions to generate estimates of model predictivity is intrinsically simple,[24] but many variations have appeared in the literature, somewhat confusing the issue, and our research in this area has created much needed clarity. Measures of statistical dispersion such as standard errors of estimation (SEE, for training sets), standard errors of prediction (SEP, for test sets), root-mean-square error (RMSE) and mean average error (MAE, better when outliers occur) are preferred over squared correlation coefficients (square of the correlation between observed and predicted y values, r2) as they are independent of the size of the training data set and number of parameters in the model. In some regressions, r2 can depend on the number of parameters in the model unless it is adjusted for these degrees of freedom.
Model interpretation
Interpretability of models is a function of the types of descriptors used and the type of model generated. If arcane but efficient descriptors are used, interpreting these features in terms of chemical structure is extremely hard. There has been a significant move away from arcane descriptors to those that can be more easily visualised, a trend initiated by the development of molecular field descriptors by Cramer et al.[25] Common interpretable descriptors involve fragments (molecular fingerprints, molecular signatures), or smooth overlap of atomic positions (SOAP) that can be mapped back onto exemplar molecules (Fig. 4) in the training set to provide guidance to chemists on how to improve their lead molecules or materials.[26–28]

|
Feature importance is also important for interpreting models. While MLR models are easily interpretable from the regression coefficients, in non-linear models feature importance is a local property that is context dependent. Working with colleagues from the University of Nottingham, we found that fuzzy fusion methods can help clarify the importance of molecular features and elucidate the types of molecular changes needed to improve target properties (D Rengasamy, JM Mase, M Torres Torres, B Rothwell, DA Winkler, GP Figueredo, unpubl. data).
Diverse examples of the use of ML models
Machine learning methods are platform technologies that are applicable to modelling and prediction of a very wide range of molecular and biological properties of molecules and materials. The following are examples from my various teams’ research of the breadth of applications in which ML methods have made substantial impact.
Stem cells, adhesion, media and bioreactors
We employed Bayesian regularised neural networks to model several types of stem cell experiments. The performance of haematopoietic stem cell (HSC, blood stem cell) bioreactors was modelled to identify the key process variables and factors that control proliferation and differentiation of stem and progenitor cells. The literature was scanned to identify 262 experiments with 21 process variables that yielded expansion of 7 types of cell populations: nucleated cells, CD34 positive cells, colony forming units, proerythroblasts, myelomonocytic progenitors, erythrocyte burst-forming units and long-term culture-initiating cells.[29] The non-linear models were more accurate than linear models and had useful levels of predictivity for new data not previously seen by the models (able to predict fold expansion to within a factor of between 1.5 (BFU-E) and 4.0 (NC)) and identified the most important factors driving expansion of each cell type.
Polymers have been used to control the attachment, proliferation and differentiation of stem cells.[30,31] In a recent study with colleagues from Monash University we showed how data from experiments on a polymer library could be used to identify the most relevant physicochemical properties driving the fate of human dental pulp-derived stem cells (hDPSC).[32] An array of 141 homopolymers was assessed for hDPSC attachment, proliferation and osteogenic (bone forming) differentiation. The best homopolymers were used to derive a second-generation library of copolymers. Linear regression models could not accurately predict the attachment, proliferation and differentiation of hDPSCs on changes to polymer surface chemistry so non-linear MLR methods, SVM and BRANN were employed. The biological data were bimodal and binary classification models of the three cell properties using a BRANN had accuracies of 85, 85 and 95% respectively, with those for the SVM models being slightly worse. In complementary studies with the University of Nottingham and the Langer group at MIT, the attachment of human embryoid bodies (hEB, a cluster of embryonic stem cells) to a library of 496 polymers was also successfully modelled using neural networks.[33] An MLREM model successfully predicted the hEB adhesion on polymers in the test set with an r2 value of 0.66, and a standard error of prediction (SEP) of 0.15 log EB. The sparse non-linear BRANNLP model predicted hEB adhesion of test set polymers with an r2 of 0.82, and an SEP of 0.10 log EB (predicted EB binding within a factor of 1.3), suggesting significant non-linearity in the relationship between the polymer surface chemistry and hEB attachment.
Bioglasses (BG) containing strontium have been shown to increase bone growth or reduce bone loss but the mechanism by which this is achieved has remained elusive. With our collaborators from Imperial College London, we conducted experiments with mesenchymal stem cells (MSC) exposed to varying levels of strontium and other bioglass components. We performed a genome wide expression analysis of the effects.[34] Using an unbiased sparse Bayesian feature selection method for the MSC gene expression fold ratios, we surprisingly discovered a group of key genes related to fatty acid and steroid biosynthesis that were highly relevant. Fig. 5 shows changes in hMSC global mRNA expression mediated by treatment with BG- and SrBG-conditioned media, and the most relevant sparse genes from the MLREM model. Subsequent experimental qPCR and lipid raft experiments validated the predictions of the sparse feature selection and identified a novel mechanism by which strontium drives MSC down the osteogenic pathway.[34]

|
Biomarkers
Sparse Bayesian feature selection and ML modelling have proven useful for identifying biomarkers for the symmetry of ESC division, an important problem in stem cell biology. Working with colleagues from Korea and University of Massachusetts Medical School, ESCs were induced to divide symmetrically (producing two stem cells) or asymmetrically (producing one stem cell and one progenitor cell) using several types of physical and chemical stimuli, and the gene expression profiles of the ESC and daughter cells measured. Again, sparse feature selection identified two markers, H2A.Z and BTG1, that were specific for symmetric versus asymmetric ESC division, providing IP for the Boston start up biotechnology company, Asymmetrex.[35] Fig. 6 shows how the marker only binds to SCs, not progenitor cells.
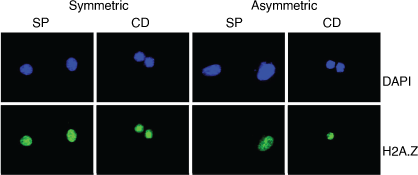
|
Small molecule physicochemical properties
The physicochemical properties of small molecules are very useful for designing drugs and agrochemicals and for aerospace applications, for example.[36] Aqueous solubility is a critical property of small organic molecules, both for synthesis and for useful pharmacokinetics. We probed the relationship between crystal lattice interactions, enthalpy of sublimation and aqueous solubility, an important unresolved issue in understanding the dissolution of organic crystals.[37] We trained an MLR model on the enthalpy of sublimation of 1302 small organic molecules and found a four-parameter equation that fitted that data with an r2 value of 0.96 and an average absolute error of 7.9 ± 0.3 kJ mol–1. A melting point model could predict this property with a standard error of 45 ± 1 K and r2 value of 0.79.
Using the enthalpy of sublimation as a surrogate for crystal lattice interactions, we generated ML models of aqueous solubility using a large and highly diverse data set of 4558 organic compounds.[38] MLR-EM and BRANNLP methods were used to derive optimal predictive models of aqueous solubility. The BRANNLP model had the best statistics, with a test set prediction r2 of 0.90 and a standard error of 0.67 log(S). Surprisingly, including descriptors that captured crystal lattice interactions did not significantly improve the quality of these aqueous solubility models. The model was applicable over more than 10 orders of magnitude of aqueous solubility and had a very broad domain of applicability, making it useful for prediction of this property for a wide range of unsynthesised small molecule drug candidates.
Drug transport and action
Given the history of the early application of statistical and ML models to the modelling and design of pharmaceutical and agrochemical properties of small organic molecules, ML continues to contribute strongly to these fields. More complex models based on neural networks and other ML methods have a strong and increasing literature base. The introduction of robust and sparse Bayesian regularised neural networks and, more recently, deep learning methods to bioactive small molecules has seen a renaissance in the use of these very useful methods.[39] ML has been applied at CSIRO to very complex problems of the structure of liquid crystal and self-assembling nanoparticle drug delivery systems in which multiple coexisting phases can occur, only one of which may be useful for drug delivery.[40–42] There are three main phases, the gyroid (space group Ia3d), diamond (space group Pn3m) and primitive (space group Im3m) bicontinuous cubic phases, plus the inverse hexagonal phase (HII) consisting of cylindrical inverse micellar-like structures packed in a hexagonal configuration. We used the BRANNGP ML methods to model each individual phase for a range of drugs, loadings and temperatures. As Fig. 7 exemplifies, we could model the different, coexisting phases in two lipids with accuracies > 99% for the training drugs, and 82% for a new set of drugs predicted by the model and tested subsequently.
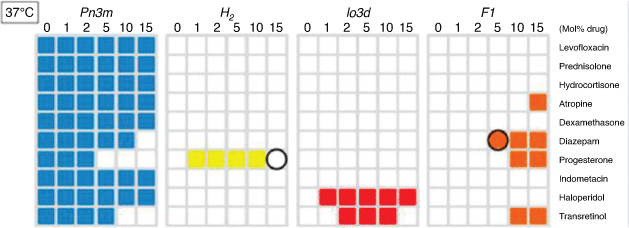
|
We have also applied ML to report some of the first robust, predictive models of intestinal absorption of drugs[43] and penetration of drugs across the blood–brain barrier (BBB).[44] For intestinal absorption, we trained a BRANNGP model on absorption values for 169 diverse small molecules. Using descriptors encoding physicochemical properties of the drugs, the test set absorptions could be predicted with r2 of 0.86–0.89 with a standard error of < 10%. The BBB work used BBB partition coefficients for 106 compounds to develop ML models of this property. BRANNGP models of BBB partition could predict the property with an r2 of 0.65 and a standard error of 0.54 logBBB. Analysis of the feature importance identified logP (octanol/water), molecular flexibility (conformational entropy) and polar surface area as being the most relevant.
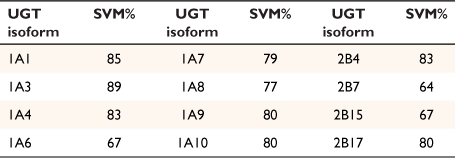
With colleagues from Flinders University, we also applied the SVM ML method and quantum chemical descriptors to predicting the phase 2 metabolism (glucuronidation) of small molecule drugs. Twelve isoform-specific data sets of substrates and non-substrates for each UGT isoform, ranging in size from 50 to 250 chemicals, were collated from the literature. We successfully assigned the appropriate phase 2 metabolism pathway of the drugs to the 12 isoforms of the key metabolic enzyme, UDP-glucuronosyltransferase (Table 1).[45–47]
|
We published one the first studies that showed how to simultaneously model both efficacy and selectivity of anticancer drug candidates inhibiting farnesyl transferase in a single ML model.[48] Farnesyl transferase inhibition for compounds in the test set was predicted with r2 of 0.76 and SEP of 0.16 and for geranylgeranyl transferase with r2 = 0.78 and SEP = 0.38. The selectivity index that denoted molecules with high FTase inhibition active and highly selectivity (low GGTase inhibition) was predicted with an r2 = 0.77 (Fig. 8).

|
ML models have been very useful in modelling and predicting toxic effects of small molecules, as well as their useful biological effects.[49–52] The current pandemic has shone a bright light on the importance of developing better drugs for ‘neglected’ tropical diseases. Although the impact of ML methods on this field is relatively small, large increases in data from screening campaigns has stimulated substantial effort on the use of ML methods to discover drugs for these diseases, which have a disproportionately massive impact on the lives of people in developing countries.[19,53]
Biomaterials
With the ageing population, extended lifespans and rapid expansion of medical device technologies, there is a greatly increased need for materials that are biocompatible and bioactive, that can be used to modulate biology and improve the performance of implantable and indwelling medical devices and cell therapies. These materials must undergo rigorous testing prior to registration for medical use, resulting in a relatively small number of approved materials (mainly polymers and metals/alloys) being available for a very wide range of medical needs. Research into much superior polymeric materials has expanded greatly over the past two decades, and the recognition of the almost infinite number of materials that could be synthesised is driving development of high throughput synthesis and characterisation methods, and the use of ML methods to extract knowledge and information from the resulting large data sets.
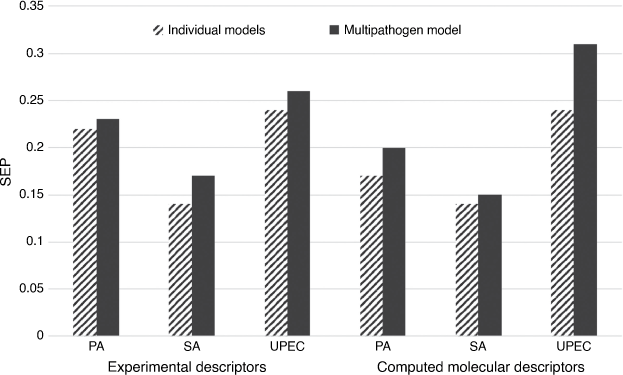
Working with researchers from MIT and Nottingham, we have been developing descriptors and models describing the interactions of different types of polymer surface chemistries with a range of cell types and soluble proteins. We were one of the first to successfully model the attachment of bacterial pathogens to a polymer library, work aimed at generating very low attachment polymers for biomedical coatings. We could successfully model and predict attachment of three important nosocomial pathogens, Staphylococcus aureus, Pseudomonas aeruginosa and uropathogenic E coli whose attachment was assessed using microbes transformed with green fluorescent protein (fluorescence was proportional to number of bacteria). Initially we developed robust models of the attachment of each pathogen alone[54] but subsequently found we could generate a model that could simultaneously predict the attachment of all three pathogens to the polymers.[55] Non-linear models were clearly better at predicting the attachment of multiple pathogens to the polymers in a test set than the linear model (SEP of 0.19 logF versus 0.28 logF), and the multipathogen model had a very similar accuracy to the average of the test set predictions for the three individual pathogen models (Fig. 9). Computed descriptors generated more accurate MLR model predictions of multipathogen attachment than those derived from experimental time-of-flight secondary ion mass spectrometry (ToF-SIMS) ion peaks (SEP of 0.28 logF versus 0.33 logF), but the non-linear BRANN models had similar predictive power.
|
We also used ML methods to generate design rules for low protein fouling polymers for biomedical applications in a collaboration with RMIT. By appropriate choice of efficient and interpretable descriptors for the polymers in the study, we could not only quantitatively predict the attachment of proteins to different polymers, but also improve the reliability of earlier antifouling polymer design rules reported by Whiteside.[56] Using a set of 48 molecules forming self-assembled monolayers, we assessed the adsorption of lysozyme and fibrinogen at 3 and 30 min exposure times. These prototype proteins were used because they have different properties such as size, shape and pI. The combined data set of 176 points was used to train the ML models using descriptors from the Whitesides rules, and those from our augmented rule set. The prediction of the protein adsorption on the monolayers improved markedly from r2 = 0.35, SEP = 24% for the original Whitesides rules to r2 = 0.82, SEP = 12% for the augmented rules.
Recently, we have used microtopographies on the surface of chemically diverse polymers to add an additional control over cells (Fig. 10) (M Vassey, L Ma, L Kämmerling, C Mbadugha, GF Trindade, GP Figueredo, F Pappalardo, R Markus, S Rajani, Q Hu, DA Winkler, D Irvine, R Hague, AM Ghaemmaghami, R Wildman, MR Alexander, unpubl. data). ML methods could determine the relative importance of deliberately introduced surface topographies and surface chemistries for modulating the behaviour of a diverse range of cell types, notably macrophages and other immune cells.[57,58] We found that surface microtopographies alone could polarise macrophages into pro- and anti-inflammatory phenotypes, although a combination of surface chemistry and topography is more powerful. For surface chemistries alone we studied a library of 400 polymers encoded using molecular descriptors to train two class (M2 and M1 polarisation) RF, SVM and neural network models of macrophage polarisation with 80% accuracies. We used a LASSO to eliminate less informative descriptors. For the surface topography studies we generated topographical features from primitive features (circle, triangle and rectangle; sized 3–23 μm in diameter and 10 μm in height). 2176 designs were arranged periodically to form 290 × 290 μm TopoUnits. The TopoUnit topographies were used to construct the features in addition to parameters from Cell Profiler that describe characteristics of surface feature area and shape. 246 descriptors were investigated. Pearson correlation analysis was applied to remove overlapping and non-intuitive descriptors (≥ 0.85). A regression model for polarisation had r2 of 0.84 and 0.56 for the macrophage phenotype training and test sets respectively.

|
Subsequently we reported that microtopographies alone could affect the attachment of GFP-transformed representative Gram negative (Ps. aeruginosa) and Gram positive (S. aureus) bacterial pathogens to a polymer surface (M Romero, J Luckett, GP Figueredo, AM Carabelli, A Carlier, A Vasilevich, S Vermeulen, D Scurr, AL Hook, J‐F Dubern, AC da Silva, DA Winkler, A Ghaemmaghami, J de Boer, P Williams, MR Alexander, unpubl. data). We experimentally surveyed 2176 combinatorially generated shapes using an unbiased high throughput micro-topographical polystyrene polymer chip. Bacterial surface attachment was sensitive to surface topography, reducing colonisation in vitro by up to 15-fold compared with a flat surface for both motile and non-motile bacterial pathogens. Using similar topographical descriptors to those in the prior study, we elucidated how the topographies drive phenotypes. A RF model predicted the observed attachment values for topographies in the test sets for both bacterial species models with high efficacy: r2 = 0.85 for P. aeruginosa and r2 = 0.81 for S. aureus average fluorescence.
Working with colleagues from Eindhoven University of Technology, Maastricht University, and the Broad Institute of MIT and Harvard, we extended the work on microtopographies by using evolutionary methods (genetic algorithm) to ‘evolve’ topographies towards those generating desired cell phenotypes.[59] We converted the information about design topography from a set of design parameters into a ‘topography genes’. We selected 81 parent topographies, based on their induction of ALP expression in MSCs (an osteogenic marker), from a pool of 2176 TopoChip topographies. These ‘parents’ were used to generate millions of diverse topographies using genetic mutation methods. Breeding and mutation were performed over multiple cycles in which groups of 10 parents were selected from an initial pool of 81 parent surfaces. These generated 10 × 10 parent pairs, plus the 10 best original parents (elitism operator), a total of 110 topographies to be assessed for fitness. We showed that a few cycles of evolutions could identify topographies that could induce markedly better cell responses that the initial pool.
Corrosion and batteries
Corrosion control is a > US$1Tn impost on industry and conventional methods of control are being phased out or banned due to carcinogenicity. In the flip side, control of metal dissolution is very important for many new, existing and potential battery technologies. Small organic molecules are excellent candidates for corrosion control agents and dissolution modulators for batteries and potentially provide improved performance. Given the vastness of small organic molecules space, high throughput assessment methods have been developed, providing data for training ML models. Although the application of ML to these applications is embryonic, it shows great potential for exploring large areas of chemical space to find very effective dissolution modulators. Colleagues at the CSIRO and Helmholtz-Zentrum Geesthacht and I have reported several seminal ML studies in this area, for both batteries and corrosion inhibitors[28,60–62] (T Würger, L Wang, D Snihirova, SV Lamaka, DA Winkler, D Höche, ML Zheludkevich, RH Meißner, C Feiler, unpubl. data). Using high throughput experiments to assess corrosion inhibition of aerospace aluminium alloys AA2024 and AA7075 by 100 small organic molecules, we generated robust, predictive, quantitative computational models of inhibitor efficiency at pH 4 and 10 using these data. BRANNGP models could predict corrosion inhibition with standard errors of ≤ 10% for test set compounds except for AA7075 at pH 10, which exhibited a standard error of 16%. ML studies of 71 organic compounds at a concentration of 50 mM that modulate the dissolution of two Mg alloys could predict the acceleration or inhibition of a blind test set not used in training with a useful r2 of 0.82.
2D materials properties
2D layered materials are attracting much research attention currently because of their wide range of applications, their superlubricant, superconductivity, magnetism, and photelectric properties and their almost endless potential to be tuned to specific applications. These properties can usually be predicted using expensive and resource intensive high level quantum chemical methods that are intractable for very large numbers of multilayer hybrid 2D materials especially. Researchers at the University of Melbourne, University of Queensland and University of Technology Sydney and I have shown how ML can effective leverage DFT calculations on a relatively small number of carefully chosen materials to predict properties of a much larger set not yet synthesised. This allows prioritising of the difficult syntheses of these materials toward those most likely to be useful. We recently successfully applied ML methods to a data set of DFT bandgap predictions, allowing the ML model to estimate the likely bandgaps and optical properties of a wide range of new materials and to focus on those with optimal bandgaps for different applications (Fig. 11).[63,64] 109 quantum chemical bandgap calculations were used to build an initial Bayesian neural network (BNN) model. Given the cost of DFT calculations and synthesis of complex hybrid 2D materials, we adopted an active learning approach to maximise the predictive range of ML models while minimising the number of DFT calculations and experiments required. Active learning involves generating an ML model from an existing modest data set then predicting beyond the domain of the model. The relevant properties of materials with the largest prediction uncertainty are then predicted by DFT calculations and the results added to the data set. This process continues until all materials in the desired prediction domain can be estimated with acceptable error.[23] Using this active learning approach, a final training set of 473 structures generated models in which bandgaps were predicted with an r2 of 0.81 and mean absolute percentage error of 0.16, and the test set was predicted with an r2 of 0.92 and mean absolute percentage error of 0.11 (Fig. 11).

|
We adopted a similar approach with colleagues from University of Technology Sydney, Univeristy of Queensland and the University of North Carolina Chapel Hill to predict the super lubricant properties of layered 2D materials, identifying several materials with significant commercial potential as advanced lubricants.[65] Bayesian neural network models of bilayer interlayer energy or the elastic constant (C33), trained on DFT values for 282 and 226 structures respectively and graph-based Voronoi tessellation down-selected by LASSO, predicted the test sets with r2 of 0.80 and MAE of 0.035 eV Å–2 for interlayer energy and r2 of 0.80 and MAE of 16.0 GPa for C33. These models were used to screen a virtual library of 18 million bilayer 2D materials to identify those with promising super lubricant properties.
Nanomaterials
Nanomaterials have unique and useful properties relative to their bulk forms, due to greatly increased surface to volume ratios. The number of commercial products containing nanomaterials has been rising rapidly, raising concerns about their human and environmental safety, and the ability of regulatory agencies to manage their safe and responsible use. Nanosafety concerns are driving substantial research investment, with a CSIRO nanosafety project then a cluster of EU Horizon 2020 projects addressing various aspects of nanosafety, including computational nanotoxicology. The aim of computational nanosafety research is to use ML models to predict useful and potentially deleterious properties of nanomaterials and use these to create a ‘safe-by-design’ paradigm for industrial applications of nanomaterials.[66,67] With colleagues from CSIRO, we have reported some of the first successful ML models of nanomaterials properties,[68] and showed how to generate accurate and interpretable models of their properties using different ML approaches.[66] With colleagues from the Izmir Institute of Technology, we have also reviewed the application of ML methods to modelling properties and nanomaterials and the protein corona that modulates their interactions with biology.[69,70]
ML methods again have been very successful in modelling and predicting the properties of these complex materials, the complexity being increased by their interactions with biological macromolecules to generate a surface coating or corona, their size and poorly defined structures and their tendency to agglomerate. We used Bayesian NNs to model the biological effects of a library of 45 types of ZnO nanoparticles with varying particle sizes, aspect ratios, doping types, doping concentrations and surface coatings.[66] Biological assays measuring cell viability, membrane integrity (LDH release) and oxidative stress were used to study the responses of human umbilical endothelial cells (HUVECs) or human hepatocellular liver carcinoma cells (HepG2) to the nanoparticles. Bayesian neural network models could predict the test set of nanoparticles with r2 values of 0.89 for cell viability, 0.86 for LDH release and 0.67 for oxidative stress (Fig. 12).

|
Surface science
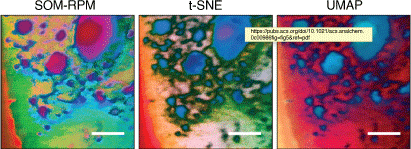
The surfaces of materials control many of their important properties such as corrosion, catalysis, and biological responses. Surface analysis instrumentation has undergone a spectacular increase in capabilities over the past decade, and methods such as ToF SIMS now generate very large and information-rich datasets for a wide variety of engineered and biological samples. Modelling and analysis of these large data sets has fallen behind the instrumental development, creating an opportunity for ML researchers to significantly increase the utility of these and other surface analysis methods. Our team at La Trobe University applied information theory and a particular type of neural network, the self-organising map (SOM), to the analysis of complex ToF SIMS data sets. For example, by binning mass spectra we could investigate the information content of different resolutions, finding ~1 m/z being the point at which information is optimum. This process avoids subjective manual peak picking commonly used by ToF-SIMS researchers for analysis of their data. Applying a SOM (Fig. 13) to mass spectrometric data provided enhanced information and performance compared to traditional data analysis methods such as PCA. Subsequent use of a deep learning algorithm, a convolutional neural network, provided spectacular spatial and mass resolution enhancement of hyperspectral 2D and 3D mass spectrometric images of both non-biological and biological samples such as tumour sections. This resulted in greatly improved understanding of surface characteristics of materials and biological samples such as breast cancer tissue samples.[71–75]
|
Porous materials and catalysts for energy and environment
Porous materials such as metal–organic frameworks (MOFs), zeolitic imidazoline frameworks (ZIFs) and covalent organic frameworks (COFs) have become an important class of materials due to their large surface areas and materials spaces, and their ability to be tuned for specific applications. They are particularly important for energy and environmental applications such as hydrogen storage, CO2 capture and, with integrated catalysts, CO2 reduction to useful fuels. Electrocatalysts and photocatalysts are also important technologies for a sustainable energy and environmental future. Working with colleagues from RMIT, we recently reviewed the application of ML methods to the modelling and design of these types of industrially important catalysts.[76]
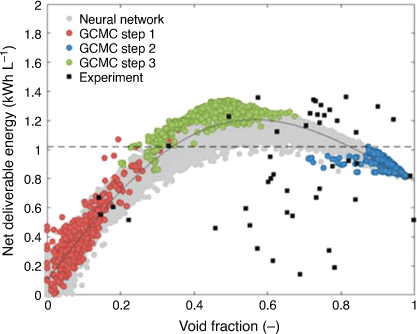
ML methods have been shown to be useful for leveraging a relatively small number of accurate but computationally expensive Grand Canonical Monte Carlo (GCMC) calculations into a much larger number of porous materials. The GCMC calculations can reliably predict the loading of gases, and using these data to train ML models provides a rapid method of estimating loading capacities of large porous materials datasets. With other collaborators from CSIRO, we initially used ML methods to generate a model for CO2 storage with a view to identifying the best materials for storage and catalytic reduction of CO2.[77] We modelled 167 ideal silica zeolites, 164 hypothetical silica zeolites plus an additional ‘smart’ set of 60 zeolites chosen by the ML model. The BRANN model predictions for both CO2 and H2 uptake were excellent, with r2 values of 0.93 and 0.97 and standard errors of 9.5 cm3 STP cm–3 (CO2), and 1.3 cm3 STP cm–3 (H2). We recently applied the same approach to modelling the storage limits of porous materials for hydrogen storage (e.g. for hydrogen powered vehicles). We also adopted an evolutionary approach, where GCMC results trained ML models that predicted a new limited set of materials with improved performance to be subjected to GCMC calculations. This cycle continued until the performance limits for hydrogen storage inherent in these materials were identified (Fig. 14).[78]
|
OLEDs, OPV and optical polymers
Large organic molecules and polymers are also playing a leading role in the development of next generation organic photovoltaic (OPV) devices, organic LEDs for display applications and for sensor applications. Collaborators at RMIT and I employed ML methods to model a universal polymer platform for charge transfer-dependent full-colour emission.[79] A chemically diverse library of 71 naphthalene diimide polymers was synthesised and their photoluminescence (PL) properties, λem, quantum yields (Φ) and CIE 1931 chromaticity coordinates (CIE x and CIE y) in aggregate or solid state, were measured. These were used to train MLREM and BRANNLP ML models to predict these four properties. For λem, the test set prediction SEP for the neural network was significantly smaller than that for the MLREM model (0.072 versus 0.096). For the prediction of Φ, BRANNLP and MLREM gave very similar results for the prediction of the test set (with SEP of 0.037 and 0.039) while the non-linear BRANNLP models for the CIE coordinates of the polymers were superior to those generated by the MLREM algorithm. The BRANNLP test set prediction SEP values of CIE x and CIE y (0.059 and 0.085) were about half those of those for the MLREM model. Thus, a full-colour tuneable polymer platform was achieved, guided by ML algorithms (Fig. 15).

|
We also curated a large set of experimental studies on organic photovoltaic devices for solar energy conversion and used these to identify the key materials and device characteristics controlling four important device parameters, conversion efficiency (PCE), open circuit voltage (Voc), short circuit current (Jsc) and frontier orbital energies. We generated ML models trained on this large data set and could predict these properties for new materials with good accuracy.[80] We generated models for PCE, Voc, Jsc, HOMO energy, LUMO energy and the HOMO–LUMO gap for the 344 compounds in the dataset. These donor–acceptors pairs, with donors encoded by signature descriptors and acceptors captured by 1-hot binary vectors were used to train sparse MLREM and BRANNLP models. The models predicted the test set properties with the following fit statistics: PCE% r2 = 0.78 and SEP = 0.48%; Voc r2 = 0.58 and SEP = 0.16 V; Jsc r2 = 0.60 and SEP = 22 mA cm–2; EHOMO r2 = 0.49 and SEP = 0.007 eV; ELUMO r2 = 0.67 and SEP = 0.008 eV. The model was also useful for subsequent de novo prediction of OPV properties of materials from the literature not used in the modelling study.
Summary and perspective
My research on ML methods and applications at CSIRO and several universities has expanded greatly over the last three decades, demonstrating the great utility of ML methods for molecular science. Clearly, ML methods will continue to be widely and increasingly applied to a myriad of applications across diverse domains of science, technology, medicine, business and beyond. This trend will continue and accelerate as larger data sets become available, new and more effective algorithms are proposed and new applications and unmet needs are addressed. In chemistry, several important innovations have occurred recently. It has now become possible for ML methods to design chemical syntheses, freeing organic chemists for more creative aspects of the task.[81]
The ability to use trained ML models to predict synthesisable molecules and materials is also now possible, and an increasing number of examples are appearing in the literature. Deep learning methods can now be trained on large numbers of high-level quantum chemical calculations, allowing them to make accurate predictions of molecular properties millions of times faster.[82] The application of other AI methods such as evolutionary algorithms is likely to be the next innovative computational paradigm adopted broadly. Evolutionary algorithms can search very large chemical spaces more efficiently than other methods and are starting to be used for the discovery and optimisation of drugs[83,84] and materials.[59,85] Ultimately, the fusion of synthesis design, synthesis robots, evolutionary methods and ML will make possible autonomous chemists[86] and materials scientists,[87] greatly expanding the range and reliability of drugs and useful materials in the short to medium term future (Fig. 16).
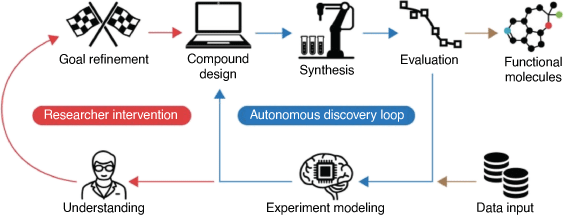
|
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of interest
The author declares no conflicts of interest.
Declaration of funding
The work described in this manuscript was supported by ARC Discovery grants, a DAAD grant, CSIRO internal and postdoctoral fellow funding sources, an Australian Stem Cell Centre postdoctoral fellowship, an EPSRC Next Generation Biomaterials grant and Boeing.
Acknowledgements
I’m extremely grateful to my long-term collaborator, Frank Burden, and graduate students, research scientists and postdoctoral fellows who conducted much of the work described in this Account: Mitchell Polley, Julianne Halley, Anna Tarasova, Tu Le, Monika Szabo, Nicholas Welch, Wil Gardner, Marco Fronzi, Vidana Epa, Paulius Mikulskis, Grazziela Figueredo. I have had the privilege to work with many gifted senior scientists at CSIRO, some of whose work has been showcased here. I’m also very grateful to the computational and experimental scientists with whom I have collaborated, providing fascinating problems to solve using machine learning and other computational methods and giving me a unique window into leading edge research I would otherwise not be involved with: Paul Pigram and colleagues at La Trobe, Ben Muir and Aaron Thornton and colleagues at CSIRO, Morgan Alexander and colleagues at Nottingham, Sviatlana Lamaka and colleagues at HZG, Nico Voelcker and colleagues at Monash, Toshio Fujita at Kyoto University, Alex Tropsha and colleagues at UNC Chapel Hill, Andrew Christofferson and colleagues at RMIT, Rachel Caruso and colleagues at RMIT, Molly Stevens and colleagues at Imperial College London, John Miners and colleagues at Flinders University, Igor Tetko and colleagues at the Herzberg Institute Munich, Joe Shapter, Mike Ford, Amanda Ellis and colleagues at UQ, UTS, and Melbourne, Ceyda Oksel at Imperial College and Izmir Institute of Technology, Maryam Salahinejad and colleagues at the Nuclear Science and Technology Research Institute (NSTRI) in Tehran, Ira Katz and Géraldine Farjot at Air Liquide Santé in Paris, Roger Martin and colleagues at Peter Mac Cancer Institute and James Sherley and colleagues at Asymmetrex in Boston. Apologies and thanks to those I may have inadvertently omitted.
References
[1] T Fujita, DA Winkler, Understanding the Roles of the “Two QSARs”. J Chem Inf Model 2016, 56, 269.| Understanding the Roles of the “Two QSARs”.Crossref | GoogleScholarGoogle Scholar |
[2] Mitchell M. Complexity: a guided tour. New York: Oxford University Press; 2011.
[3] JD Halley, DA Winkler, Consistent concepts of self‐organization and self‐assembly. Complexity 2008, 14, 10.
| Consistent concepts of self‐organization and self‐assembly.Crossref | GoogleScholarGoogle Scholar |
[4] JD Halley, DA Winkler, Classification of emergence and its relation to self‐organization. Complexity 2008, 13, 10.
| Classification of emergence and its relation to self‐organization.Crossref | GoogleScholarGoogle Scholar |
[5] JD Halley, DA Winkler, Classification of self-organization and emergence in chemical and biological systems. Aust J Chem 2006, 59, 849.
| Classification of self-organization and emergence in chemical and biological systems.Crossref | GoogleScholarGoogle Scholar |
[6] T Le, VC Epa, FR Burden, DA Winkler, Quantitative structure-property relationship modeling of diverse materials properties. Chem Rev 2012, 112, 2889.
| Quantitative structure-property relationship modeling of diverse materials properties.Crossref | GoogleScholarGoogle Scholar |
[7] EN Muratov, J Bajorath, RP Sheridan, IV Tetko, D Filimonov, V Poroikov, et al. QSAR without borders. Chem Soc Rev 2020, 49, 3525.
| QSAR without borders.Crossref | GoogleScholarGoogle Scholar |
[8] FR Burden, MJ Polley, DA Winkler, Toward novel universal descriptors: charge fingerprints. J Chem Inf Model 2009, 49, 710.
| Toward novel universal descriptors: charge fingerprints.Crossref | GoogleScholarGoogle Scholar |
[9] FR Burden, A Chemically Intuitive Molecular Index Based on the Eigenvalues of a Modified Adjacency Matrix. Quant Struct-Act Relat 1997, 16, 309.
| A Chemically Intuitive Molecular Index Based on the Eigenvalues of a Modified Adjacency Matrix.Crossref | GoogleScholarGoogle Scholar |
[10] DA Winkler, FR Burden, AJR Watkins, Atomistic topological indices applied to benzodiazepines using various regression methods. Quant Struct-Act Relat 1998, 17, 14.
| Atomistic topological indices applied to benzodiazepines using various regression methods.Crossref | GoogleScholarGoogle Scholar |
[11] R Tibshirani, Regression Shrinkage and Selection via the Lasso. J R Stat Soc Series B Stat Methodol 1996, 58, 267.
| Regression Shrinkage and Selection via the Lasso.Crossref | GoogleScholarGoogle Scholar |
[12] FR Burden, DA Winkler, Optimal sparse descriptor selection for QSAR using Bayesian methods. QSAR Comb Sci 2009, 28, 645.
| Optimal sparse descriptor selection for QSAR using Bayesian methods.Crossref | GoogleScholarGoogle Scholar |
[13] FR Burden, MG Ford, DC Whitley, DA Winkler, Use of automatic relevance determination in QSAR studies using Bayesian neural networks. J Chem Inf Comput Sci 2000, 40, 1423.
| Use of automatic relevance determination in QSAR studies using Bayesian neural networks.Crossref | GoogleScholarGoogle Scholar |
[14] FR Burden, DA Winkler, An optimal self‐pruning neural network and nonlinear descriptor selection in QSAR. QSAR Comb Sci 2009, 28, 1092.
| An optimal self‐pruning neural network and nonlinear descriptor selection in QSAR.Crossref | GoogleScholarGoogle Scholar |
[15] Burden FR, Winkler DA. Bayesian Regularization of Neural Networks, in Artificial Neural Networks: Methods and Applications, In Livingston D, editor. Methods in Molecular Biology, Vol. 458. Totowa, NJ 07512 USA: Humana Press; 2009. pp 25‐44. ISBN: 978‐1‐58829‐718‐1
[16] DA Winkler, Sparse QSAR modelling methods for therapeutic and regenerative medicine. J Comput Aided Mol Des 2018, 32, 497.
| Sparse QSAR modelling methods for therapeutic and regenerative medicine.Crossref | GoogleScholarGoogle Scholar |
[17] FR Burden, DA Winkler, Robust QSAR models using Bayesian regularized neural networks. J Med Chem 1999, 42, 3183.
| Robust QSAR models using Bayesian regularized neural networks.Crossref | GoogleScholarGoogle Scholar |
[18] FR Burden, DA Winkler, Relevance Vector Machines: Sparse Classification Methods for QSAR. J Chem Inf Model 2015, 55, 1529.
| Relevance Vector Machines: Sparse Classification Methods for QSAR.Crossref | GoogleScholarGoogle Scholar |
[19] DA Winkler, Potent antimalarial drugs with validated activities. Nat Mach Intell 2022, 4, 102.
| Potent antimalarial drugs with validated activities.Crossref | GoogleScholarGoogle Scholar |
[20] X Tong, X Liu, X Tan, X Li, J Jiang, Z Xiong, et al. Generative Models for De Novo Drug Design. J Med Chem 2021, 64, 14011.
| Generative Models for De Novo Drug Design.Crossref | GoogleScholarGoogle Scholar |
[21] DA Winkler, TC Le, Performance of Deep and Shallow Neural Networks, the Universal Approximation Theorem, Activity Cliffs, and QSAR. Mol Inform 2017, 36, 1600118.
| Performance of Deep and Shallow Neural Networks, the Universal Approximation Theorem, Activity Cliffs, and QSAR.Crossref | GoogleScholarGoogle Scholar |
[22] H Mai, TC Le, T Hisatomi, D Chen, K Domen, DA Winkler, et al. Use of Meta Models for Rapid Discovery of Narrow Bandgap Oxide Photocatalysts. iScience 2021, 24, 103068.
| Use of Meta Models for Rapid Discovery of Narrow Bandgap Oxide Photocatalysts.Crossref | GoogleScholarGoogle Scholar |
[23] M Fronzi, O Isayev, DA Winkler, JG Shapter, AV Ellis, PC Sherrell, et al. Active learning in Bayesian neural networks for bandgap predictions of novel Van der Waals heterostructures. Adv Intell Syst 2021, 3, 2100080.
| Active learning in Bayesian neural networks for bandgap predictions of novel Van der Waals heterostructures.Crossref | GoogleScholarGoogle Scholar |
[24] DLJ Alexander, A Tropsha, DA Winkler, Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J Chem Inf Model 2015, 55, 1316.
| Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models.Crossref | GoogleScholarGoogle Scholar |
[25] RD Cramer, DE Patterson, JD Bunce, Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc 1988, 110, 5959.
| Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins.Crossref | GoogleScholarGoogle Scholar |
[26] FR Burden, BS Rosewarne, DA Winkler, Predicting maximum bioactivity by effective inversion of neural networks using genetic algorithms. Chemometr Intell Lab Syst 1997, 38, 127.
| Predicting maximum bioactivity by effective inversion of neural networks using genetic algorithms.Crossref | GoogleScholarGoogle Scholar |
[27] P Mikulskis, MR Alexander, DA Winkler, Toward interpretable machine learning models for materials discovery. Adv Intell Syst 2019, 1, 1900045.
| Toward interpretable machine learning models for materials discovery.Crossref | GoogleScholarGoogle Scholar |
[28] T Würger, D Mei, B Vaghefinazari, DA Winkler, SV Lamaka, ML Zheludkevich, et al. Exploring structure-property relationships in magnesium dissolution modulators. npj Mater Degrad 2021, 5, 2.
| Exploring structure-property relationships in magnesium dissolution modulators.Crossref | GoogleScholarGoogle Scholar |
[29] DA Winkler, FR Burden, Robust, quantitative tools for modelling ex-vivo expansion of haematopoietic stem cells and progenitors. Mol Biosyst 2012, 8, 913.
| Robust, quantitative tools for modelling ex-vivo expansion of haematopoietic stem cells and progenitors.Crossref | GoogleScholarGoogle Scholar |
[30] AD Celiz, JGW Smith, AK Patel, AL Hook, D Rajamohan, VT George, et al. Discovery of a Novel Polymer for Human Pluripotent Stem Cell Expansion and Multilineage Differentiation. Adv Mater 2015, 27, 4006.
| Discovery of a Novel Polymer for Human Pluripotent Stem Cell Expansion and Multilineage Differentiation.Crossref | GoogleScholarGoogle Scholar |
[31] AD Celiz, JGW Smith, R Langer, DG Anderson, DA Winkler, DA Barrett, et al. Materials for stem cell factories of the future. Nat Mater 2014, 13, 570.
| Materials for stem cell factories of the future.Crossref | GoogleScholarGoogle Scholar |
[32] S Rasi Ghaemi, B Delalat, S Gronthos, MR Alexander, DA Winkler, AL Hook, et al. High-Throughput Assessment and Modeling of a Polymer Library Regulating Human Dental Pulp-Derived Stem Cell Behavior. ACS Appl Mater Interfaces 2018, 10, 38739.
| High-Throughput Assessment and Modeling of a Polymer Library Regulating Human Dental Pulp-Derived Stem Cell Behavior.Crossref | GoogleScholarGoogle Scholar |
[33] VC Epa, J Yang, Y Mei, AL Hook, R Langer, DG Anderson, et al. Modelling human embryoid body cell adhesion to a combinatorial library of polymer surfaces. J Mater Chem 2012, 22, 20902.
| Modelling human embryoid body cell adhesion to a combinatorial library of polymer surfaces.Crossref | GoogleScholarGoogle Scholar |
[34] H Autefage, E Gentleman, E Littmann, MAB Hedegaard, T Von Erlach, M O’Donnell, et al. Sparse feature selection methods identify unexpected global cellular response to strontium-containing materials. Proc Natl Acad Sci U S A 2015, 112, 4280.
| Sparse feature selection methods identify unexpected global cellular response to strontium-containing materials.Crossref | GoogleScholarGoogle Scholar |
[35] YH Huh, M Noh, FR Burden, JC Chen, DA Winkler, JL Sherley, Sparse feature selection identifies H2A.Z as a novel, pattern-specific biomarker for asymmetrically self-renewing distributed stem cells. Stem Cell Res 2015, 14, 144.
| Sparse feature selection identifies H2A.Z as a novel, pattern-specific biomarker for asymmetrically self-renewing distributed stem cells.Crossref | GoogleScholarGoogle Scholar |
[36] TC Le, M Ballard, P Casey, MS Liu, DA Winkler, Illuminating Flash Point: Comprehensive Prediction Models. Mol Inform 2015, 34, 18.
| Illuminating Flash Point: Comprehensive Prediction Models.Crossref | GoogleScholarGoogle Scholar |
[37] M Salahinejad, TC Le, DA Winkler, Capturing the crystal: prediction of enthalpy of sublimation, crystal lattice energy, and melting points of organic compounds. J Chem Inf Model 2013, 53, 223.
| Capturing the crystal: prediction of enthalpy of sublimation, crystal lattice energy, and melting points of organic compounds.Crossref | GoogleScholarGoogle Scholar |
[38] M Salahinejad, TC Le, DA Winkler, Aqueous solubility prediction: do crystal lattice interactions help? Mol Pharm 2013, 10, 2757.
| Aqueous solubility prediction: do crystal lattice interactions help?Crossref | GoogleScholarGoogle Scholar |
[39] II Baskin, D Winkler, IV Tetko, A renaissance of neural networks in drug discovery. Expert Opin Drug Discov 2016, 11, 785.
| A renaissance of neural networks in drug discovery.Crossref | GoogleScholarGoogle Scholar |
[40] TC Le, X Mulet, FR Burden, DA Winkler, Predicting the complex phase behavior of self-assembling drug delivery nanoparticles. Mol Pharm 2013, 10, 1368.
| Predicting the complex phase behavior of self-assembling drug delivery nanoparticles.Crossref | GoogleScholarGoogle Scholar |
[41] BTC Le, N Tran, X Mulet, DA Winkler, Modeling the Influence of Fatty Acid Incorporation on Mesophase Formation in Amphiphilic Therapeutic Delivery Systems. Mol Pharm 2016, 13, 996.
| Modeling the Influence of Fatty Acid Incorporation on Mesophase Formation in Amphiphilic Therapeutic Delivery Systems.Crossref | GoogleScholarGoogle Scholar |
[42] TC Le, CE Conn, FR Burden, DA Winkler, Computational modeling and prediction of the complex time-dependent phase behavior of lyotropic liquid crystals under in meso crystallization conditions. Crystal Growth Des 2013, 13, 1267.
| Computational modeling and prediction of the complex time-dependent phase behavior of lyotropic liquid crystals under in meso crystallization conditions.Crossref | GoogleScholarGoogle Scholar |
[43] MJ Polley, FR Burden, DA Winkler, Predictive human intestinal absorption QSAR models using Bayesian regularized neural networks. Aust J Chem 2005, 58, 859.
| Predictive human intestinal absorption QSAR models using Bayesian regularized neural networks.Crossref | GoogleScholarGoogle Scholar |
[44] DA Winkler, FR Burden, Modelling blood-brain barrier partitioning using Bayesian neural nets. J Mol Graph Model 2004, 22, 499.
| Modelling blood-brain barrier partitioning using Bayesian neural nets.Crossref | GoogleScholarGoogle Scholar |
[45] MJ Sorich, P Smith, D Winkler, FR Burden, R McKinnon, J Miners, In Silico Prediction of Chemical Metabolism by Human Udp-Glucuronosyltransferase Isoforms: Evaluation of Classification Algorithms. Drug Metab Rev 2003, 35, 167.
[46] MJ Sorich, JO Miners, RA McKinnon, DA Winkler, FR Burden, PA Smith, Comparison of linear and nonlinear classification algorithms for the prediction of drug and chemical metabolism by human UDP-glucuronosyltransferase isoforms. J Chem Inf Comput Sci 2003, 43, 2019.
| Comparison of linear and nonlinear classification algorithms for the prediction of drug and chemical metabolism by human UDP-glucuronosyltransferase isoforms.Crossref | GoogleScholarGoogle Scholar |
[47] MJ Sorich, RA McKinnon, JO Miners, DA Winkler, PA Smith, Rapid prediction of chemical metabolism by human UDP-glucuronosyltransferase isoforms using quantum chemical descriptors derived with the electronegativity equalization method. J Med Chem 2004, 47, 5311.
| Rapid prediction of chemical metabolism by human UDP-glucuronosyltransferase isoforms using quantum chemical descriptors derived with the electronegativity equalization method.Crossref | GoogleScholarGoogle Scholar |
[48] MJ Polley, DA Winkler, FR Burden, Broad-based quantitative structure-activity relationship modeling of potency and selectivity of farnesyltransferase inhibitors using a Bayesian regularized neural network. J Med Chem 2004, 47, 6230.
| Broad-based quantitative structure-activity relationship modeling of potency and selectivity of farnesyltransferase inhibitors using a Bayesian regularized neural network.Crossref | GoogleScholarGoogle Scholar |
[49] FR Burden, DA Winkler, A quantitative structure–activity relationships model for the acute toxicity of substituted benzenes to Tetrahymena pyriformis using Bayesian-regularized neural networks. Chem Res Toxicol 2000, 13, 436.
| A quantitative structure–activity relationships model for the acute toxicity of substituted benzenes to Tetrahymena pyriformis using Bayesian-regularized neural networks.Crossref | GoogleScholarGoogle Scholar |
[50] DA Winkler, FR Burden, Bayesian neural nets for modeling in drug discovery. Drug Discov Today: BIOSILICO 2004, 2, 104.
| Bayesian neural nets for modeling in drug discovery.Crossref | GoogleScholarGoogle Scholar |
[51] DA Winkler, Neural networks as robust tools in drug lead discovery and development. Mol Biotechnol 2004, 27, 139.
| Neural networks as robust tools in drug lead discovery and development.Crossref | GoogleScholarGoogle Scholar |
[52] DA Winkler, Neural networks in ADME and toxicity prediction. Drugs Future 2004, 29, 1043.
| Neural networks in ADME and toxicity prediction.Crossref | GoogleScholarGoogle Scholar |
[53] DA Winkler, Use of Artificial Intelligence and Machine Learning for Discovery of Drugs for Neglected Tropical Diseases. Front Chem 2021, 9, 614073.
| Use of Artificial Intelligence and Machine Learning for Discovery of Drugs for Neglected Tropical Diseases.Crossref | GoogleScholarGoogle Scholar |
[54] VC Epa, AL Hook, C Chang, J Yang, R Langer, DG Anderson, et al. Modelling and prediction of bacterial attachment to polymers. Adv Funct Mater 2014, 24, 2085.
| Modelling and prediction of bacterial attachment to polymers.Crossref | GoogleScholarGoogle Scholar |
[55] P Mikulskis, A Hook, AA Dundas, D Irvine, O Sanni, D Anderson, et al. Prediction of Broad-Spectrum Pathogen Attachment to Coating Materials for Biomedical Devices. ACS Appl Mater Interfaces 2018, 10, 139.
| Prediction of Broad-Spectrum Pathogen Attachment to Coating Materials for Biomedical Devices.Crossref | GoogleScholarGoogle Scholar |
[56] TC Le, M Penna, DA Winkler, I Yarovsky, Quantitative design rules for protein-resistant surface coatings using machine learning. Sci Rep 2019, 9, 265.
| Quantitative design rules for protein-resistant surface coatings using machine learning.Crossref | GoogleScholarGoogle Scholar |
[57] L Burroughs, MH Amer, M Vassey, B Koch, GP Figueredo, B Mukonoweshuro, et al. Discovery of synergistic material-topography combinations to achieve immunomodulatory osteoinductive biomaterials using a novel in vitro screening method: The ChemoTopoChip. Biomaterials 2021, 271, 120740.
| Discovery of synergistic material-topography combinations to achieve immunomodulatory osteoinductive biomaterials using a novel in vitro screening method: The ChemoTopoChip.Crossref | GoogleScholarGoogle Scholar |
[58] MJ Vassey, GP Figueredo, DJ Scurr, AS Vasilevich, S Vermeulen, A Carlier, et al. Immune Modulation by Design: Using Topography to Control Human Monocyte Attachment and Macrophage Differentiation. Adv Sci (Weinh) 2020, 7, 1903392.
| Immune Modulation by Design: Using Topography to Control Human Monocyte Attachment and Macrophage Differentiation.Crossref | GoogleScholarGoogle Scholar |
[59] A Vasilevich, A Carlier, DA Winkler, S Singh, J de Boer, Evolutionary design of optimal surface topographies for biomaterials. Sci Rep 2020, 10, 22160.
| Evolutionary design of optimal surface topographies for biomaterials.Crossref | GoogleScholarGoogle Scholar |
[60] C Feiler, D Mei, SV Lamaka, T Würger, RH Meißner, DA Winkler, BJC Luthringer-Feyerabend, et al. In silico Screening of Modulators of Magnesium Dissolution. Corros Sci 2019, 163, 108245.
| In silico Screening of Modulators of Magnesium Dissolution.Crossref | GoogleScholarGoogle Scholar |
[61] DA Winkler, M Breedon, AE Hughes, FR Burden, AS Barnard, TG Harvey, et al. Towards chromate-free corrosion inhibitors: structure–property models for organic alternatives. Green Chem 2014, 16, 3349.
| Towards chromate-free corrosion inhibitors: structure–property models for organic alternatives.Crossref | GoogleScholarGoogle Scholar |
[62] DA Winkler, M Breedon, P White, AE Hughes, ED Sapper, I Cole, Using high throughput experimental data and in silico models to discover alternatives to toxic chromate corrosion inhibitors. Corros Sci 2016, 106, 229.
| Using high throughput experimental data and in silico models to discover alternatives to toxic chromate corrosion inhibitors.Crossref | GoogleScholarGoogle Scholar |
[63] SA Tawfik, O Isayev, C Stampfl, J Shapter, DA Winkler, MJ Ford, Efficient Prediction of Structural and Electronic Properties of Hybrid 2D Materials Using Complementary DFT and Machine Learning Approaches. Adv Theor Simul 2019, 2, 1800128.
| Efficient Prediction of Structural and Electronic Properties of Hybrid 2D Materials Using Complementary DFT and Machine Learning Approaches.Crossref | GoogleScholarGoogle Scholar |
[64] SA Tawfik, O Isayev, DA Winkler, M Spencer, Predicting thermal properties of crystals using machine learning. Adv Theor Simul 2019, 3, 1900208.
| Predicting thermal properties of crystals using machine learning.Crossref | GoogleScholarGoogle Scholar |
[65] M Fronzi, SA Tawfik, MA Ghazaleh, O Isayev, DA Winkler, J Shapter, MJ Ford, High Throughput Screening of Millions of van der Waals Heterostructures for Superlubricant Applications. Adv Theor Simul 2020, 3, 2000029.
| High Throughput Screening of Millions of van der Waals Heterostructures for Superlubricant Applications.Crossref | GoogleScholarGoogle Scholar |
[66] TC Le, H Yin, R Chen, Y Chen, L Zhao, PS Casey, et al. An Experimental and Computational Approach to the Development of ZnO Nanoparticles that are Safe by Design. Small 2016, 12, 3568.
| An Experimental and Computational Approach to the Development of ZnO Nanoparticles that are Safe by Design.Crossref | GoogleScholarGoogle Scholar |
[67] DA Winkler, Role of Artificial Intelligence and Machine Learning in Nanosafety. Small 2020, 16, e2001883.
| Role of Artificial Intelligence and Machine Learning in Nanosafety.Crossref | GoogleScholarGoogle Scholar |
[68] VC Epa, FR Burden, C Tassa, R Weissleder, S Shaw, DA Winkler, Modeling biological activities of nanoparticles. Nano Lett 2012, 12, 5808.
| Modeling biological activities of nanoparticles.Crossref | GoogleScholarGoogle Scholar |
[69] DA Winkler, Recent advances, and unresolved issues, in the application of computational modelling to the prediction of the biological effects of nanomaterials. Toxicol Appl Pharmacol 2016, 299, 96.
| Recent advances, and unresolved issues, in the application of computational modelling to the prediction of the biological effects of nanomaterials.Crossref | GoogleScholarGoogle Scholar |
[70] DA Winkler, E Mombelli, A Pietroiusti, L Tran, A Worth, B Fadeel, et al. Applying quantitative structure-activity relationship approaches to nanotoxicology: current status and future potential. Toxicology 2013, 313, 15.
| Applying quantitative structure-activity relationship approaches to nanotoxicology: current status and future potential.Crossref | GoogleScholarGoogle Scholar |
[71] W Gardner, R Maliki, SM Cutts, BW Muir, D Ballabio, DA Winkler, et al. Self-Organizing Map and Relational Perspective Mapping for the accurate visualization of high-dimensional hyperspectral data. Anal Chem 2020, 92, 10450.
| Self-Organizing Map and Relational Perspective Mapping for the accurate visualization of high-dimensional hyperspectral data.Crossref | GoogleScholarGoogle Scholar |
[72] W Gardner, DA Winkler, SM Cutts, SA Torney, GA Pietersz, BW Muir, PJ Pigram, Two-Dimensional and Three-Dimensional Time-of-Flight Secondary Ion Mass Spectrometry Image Feature Extraction Using a Spatially Aware Convolutional Autoencoder. Anal Chem 2022, 94, 7804.
| Two-Dimensional and Three-Dimensional Time-of-Flight Secondary Ion Mass Spectrometry Image Feature Extraction Using a Spatially Aware Convolutional Autoencoder.Crossref | GoogleScholarGoogle Scholar |
[73] W Gardner, DA Winkler, D Ballabio, BW Muir, PJ Pigram, Analyzing 3D hyperspectral TOF-SIMS depth profile data using self-organizing map-relational perspective mapping. Biointerphases 2020, 15, 061004.
| Analyzing 3D hyperspectral TOF-SIMS depth profile data using self-organizing map-relational perspective mapping.Crossref | GoogleScholarGoogle Scholar |
[74] W Gardner, DA Winkler, SM Cutts, SA Torney, GA Pietersz, BW Muir, et al. Two-Dimensional and Three-Dimensional Time-of-Flight Secondary Ion Mass Spectrometry Image Feature Extraction Using a Spatially Aware Convolutional Autoencoder. Anal Chem 2022, 94, 7804.
| Two-Dimensional and Three-Dimensional Time-of-Flight Secondary Ion Mass Spectrometry Image Feature Extraction Using a Spatially Aware Convolutional Autoencoder.Crossref | GoogleScholarGoogle Scholar |
[75] W Gardner, DA Winkler, BW Muir, PJ Pigram, Applications of multivariate analysis and unsupervised machine learning to ToF-SIMS images of organic, bioorganic, and biological systems. Biointerphases 2022, 17, 020802.
| Applications of multivariate analysis and unsupervised machine learning to ToF-SIMS images of organic, bioorganic, and biological systems.Crossref | GoogleScholarGoogle Scholar |
[76] H Mai, TC Le, D Chen, DA Winkler, RA Caruso, Machine Learning for Electrocatalyst and Photocatalyst Design and Discovery. Chem Rev 2022, 16, 13478.
| Machine Learning for Electrocatalyst and Photocatalyst Design and Discovery.Crossref | GoogleScholarGoogle Scholar |
[77] AW Thornton, DA Winkler, MS Liu, M Haranczyk, DF Kennedy, Towards computational design of zeolite catalysts for CO2 reduction. RSC Adv 2015, 5, 44361.
| Towards computational design of zeolite catalysts for CO2 reduction.Crossref | GoogleScholarGoogle Scholar |
[78] AW Thornton, CM Simon, J Kim, O Kwon, KS Deeg, K Konstas, et al. Materials Genome in Action: Identifying the Performance Limits of Physical Hydrogen Storage. Chem Mater 2017, 29, 2844.
| Materials Genome in Action: Identifying the Performance Limits of Physical Hydrogen Storage.Crossref | GoogleScholarGoogle Scholar |
[79] Y Bao, Y Ye, N Meftahi, I Lyskov, T Tian, S Kumar, et al. Machine Learning-assisted Exploration of a Universal Polymer Platform with Charge Transfer-dependent Full-color Emission. 2022,
| Machine Learning-assisted Exploration of a Universal Polymer Platform with Charge Transfer-dependent Full-color Emission.Crossref | GoogleScholarGoogle Scholar |
[80] N Meftahi, M Klymenko, AJ Christofferson, U Bach, DA Winkler, SP Russo, Machine Learning Property Prediction for Organic Photovoltaic Devices. npj Comput Mater 2020, 6, 166.
| Machine Learning Property Prediction for Organic Photovoltaic Devices.Crossref | GoogleScholarGoogle Scholar |
[81] Y Shen, JE Borowski, MA Hardy, R Sarpong, AG Doyle, T Cernak, Automation and computer-assisted planning for chemical synthesis. Nat Rev Methods Primers 2021, 1, 23.
| Automation and computer-assisted planning for chemical synthesis.Crossref | GoogleScholarGoogle Scholar |
[82] JS Smith, BT Nebgen, R Zubatyuk, N Lubbers, C Devereux, K Barros, et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat Commun 2019, 10, 2903.
| Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning.Crossref | GoogleScholarGoogle Scholar |
[83] TC Le, DA Winkler, A Bright Future for Evolutionary Methods in Drug Design. ChemMedChem 2015, 10, 1296.
| A Bright Future for Evolutionary Methods in Drug Design.Crossref | GoogleScholarGoogle Scholar |
[84] DA Winkler, Biomimetic molecular design tools that learn, evolve, and adapt. Beilstein J Org Chem 2017, 13, 1288.
| Biomimetic molecular design tools that learn, evolve, and adapt.Crossref | GoogleScholarGoogle Scholar |
[85] TC Le, DA Winkler, Discovery and Optimization of Materials Using Evolutionary Approaches. Chem Rev 2016, 116, 6107.
| Discovery and Optimization of Materials Using Evolutionary Approaches.Crossref | GoogleScholarGoogle Scholar |
[86] V Dragone, V Sans, AB Henson, JM Granda, L Cronin, An autonomous organic reaction search engine for chemical reactivity. Nat Commun 2017, 8, 15733.
| An autonomous organic reaction search engine for chemical reactivity.Crossref | GoogleScholarGoogle Scholar |
[87] G Pilania, Machine learning in materials science: From explainable predictions to autonomous design. Comput Mater Sci 2021, 193, 110360.
| Machine learning in materials science: From explainable predictions to autonomous design.Crossref | GoogleScholarGoogle Scholar |