

Overview of the synthetic approaches to lysergic acid as a precursor to the psychedelic LSD
Michael J. Nutt A * , Nick Woolf B and Scott G. Stewart A *
A *
A School of Molecular Sciences, The University of Western Australia (M310), 35 Stirling Highway, Crawley, WA 6009, Australia.
B Woke Pharmaceuticals, Suite 301, 10 Bridge Street, Sydney, NSW 2000 Australia.
Australian Journal of Chemistry 76(5) 279-287 https://doi.org/10.1071/CH23055
Submitted: 13 March 2023 Accepted: 24 May 2023 Published: 7 July 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution 4.0 International License (CC BY)
Abstract
In this short primer we will discuss the total synthesis of lysergic acid, an important precursor to both lysergic acid diethylamide (LSD) and its derivatives. Lysergic acid is also noted as a precursor for many drugs targeting the serotonin receptor family of GPCRs, including multiple known hallucinogens. More recently, reinvigorated interest in the therapeutic potential of psychedelics from academic and commercial sectors has placed a renewed importance on practical, scalable means of accessing this complex alkaloid scaffold.
Keywords: amination, cross‐coupling, ergoline, ergot fungus, organic chemical synthesis, total synthesis, lysergic acid, lysergic acid diethylamide, psychedelic.
Introduction
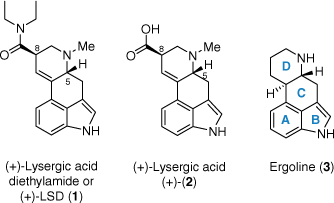
Lysergic acid diethylamide (LSD, 1) is a profoundly powerful psychedelic drug with exquisite potency, capable of inducing dramatic alterations to subjective conscious experience at microgram doses.[1–3] A semi-synthetic compound derived from alkaloids of the ergot fungus, LSD (1) was first synthesised by Swiss chemist Albert Hoffman in 1938 during his time at Sandoz under Professor Arthur Stoll.[4] So named from the German lysergsäurediethylamid, LSD (1) was the 25th entry in a series of amide-substituted ergoline derivates being investigated by Hoffman. The original intent of the program was to improve upon the existing efficacy of several naturally occurring ergot alkaloids as tools in obstetric medicine. Following initial animal testing, LSD (1) returned no notable pharmacological properties and was thus not further investigated by the Sandoz pharmacology team. In a now infamous series of serendipitous events, however, Hoffman felt compelled to return to the compound despite this decision. Upon resynthesising the material several years later on a hunch, his apparent accidental exposure to the compound, on 16 April 1943, lead to the inadvertent discovery of one of the most potent serotonergic psychedelic agents ever reported.
The precursor most commonly used to prepare LSD (and most of its reported derivatives to date) is lysergic acid (2), from which the characteristic tetracyclic ergoline scaffold 3 is derived (Fig. 1).[5] Two stereocentres exist at positions C5 and C8, and the overall shape bestowed on the scaffold by these stereocentres is of critical importance to LSD’s biological activity; only one of the four possible stereoisomers has any appreciable activity as a psychedelic.[5,6] These stereochemical requirements for biological activity place an added challenge on the study and development of LSD analogues from lysergic acid, given its significantly more complex structure compared to the other tryptamine and phenethylamine psychedelics. Owing to the renewed interest in such compounds for their potential in the treatment of various psychiatric disorders,[7] there is a greater desire than ever for convenient, scalable access to lysergic acid as a precursor to LSD and other novel psychedelics.
Herein, we highlight some of the unique approaches used in the existing syntheses[8]A of this pivotal compound, with particular focus on the often-challenging ring forming processes. The aim of our review is to provide readers with a concise and convenient primer of these efforts spanning 1954–2023, categorising works based on which of the four ergoline rings forms the key retrosynthetic disconnection.
Total syntheses with key step D-ring cyclisation
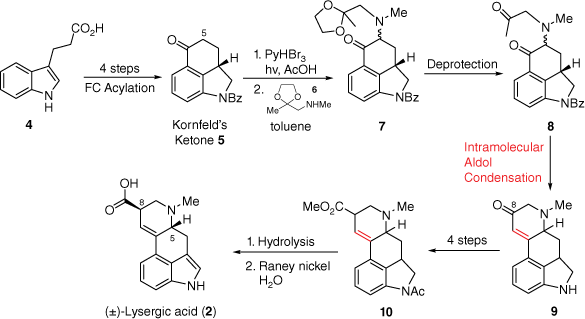
In 1954, the first total synthesis of (±)-lysergic acid was achieved by Ed Kornfeld at Eli Lilly, with R. B. Woodward acting as a consultant on the project.[9]B Initially, a protected version of compound 5, named Kornfeld’s ketone, was prepared through an intramolecular Friedel–Crafts acylation based approach from 3-indolepropionic acid 4 (Scheme 1). From this ketone, α-installation of a bromine at C5 (lysergic acid notation) allowed introduction of nucleophiles such as the amine 6. Deprotection of the acetal within compound 7 set compound 8 up for the second key step, an intramolecular aldol condensation, to form the tetracycle 9 containing the D-ring. Later functional group interconversion reactions at the C8-carbonyl allowed for the production of ester 10. Hydrolysis of 10, along with catalytic dehydrogenation in water using Raney nickel, finalised this landmark synthesis of the target compound (±)-lysergic acid (2).
The synthetic approach from the Rebek Jr research team, followed the pioneering studies of Kornfeld, by also using an α-amino derivative of Uhle’s ketone 11[10]C as a key intermediate. In this example the natural occurring amino acid tryptophan was used as a starting material.[11,12] In the later stages of their synthesis, the D-ring was formed using a lactone ring opening and dehydration sequence. Conversely, in Ninomiya’s approach, the installation of the ergoline D-ring was accomplished by first using reductive photocyclisation using a furan-based tether.[13]
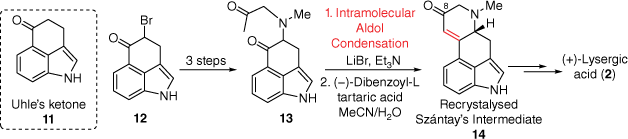
Similar to Kornfeld, the Szántay’s synthetic approach to D-ring formation was through an intramolecular aldol condensation (Scheme 2); however, this route importantly produced enantiomerically pure (+)-lysergic acid.[14] Initially, 3-indolepropionic acid was used to prepare the protected 4-bromo-Uhle’s ketone 12, en route to diketone 13. Following this, the aforementioned aldol condensation, under relatively mild conditions provided the expected enone. Importantly, this racemic mixture containing 14 could be chirally resolved using (−)-dibenzoyl-tartaric acid to give the so-called Szántay’s intermediate 14 which was converted into (+)-lysergic acid. Unique to this synthesis was the initial reaction of the C8 carbonyl within 14 with p-toluenesulfonylmethyl isocyanide to install the necessary additional carbon attachment required at C8. This was the first preparation of (+)-lysergic acid and highlights the strength of chiral resolution in the intermediate stages of the total synthesis.
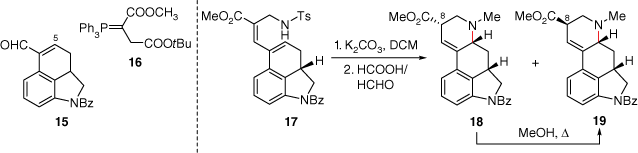
In 1981, Ramage used part of the general synthetic strategy compiled by Kornfeld and Woodward to devise a synthesis of dihydrolysergic acid esters (Scheme 3).[15] In this synthesis α,β-unsaturated aldehyde 15, containing a saturated ABC-ring system, was first used in order to attach a diester tether through a Wittig olefination using 16. Ultimately, the D-ring cyclisation was achieved through nucleophilic cyclisation of the aminodienoic ester 17 to epimers 18 and 19 in a 9:3 ratio respectively (along with an α,β-unsaturated isomer). This significant piece of synthetic chemistry revealed landmark compounds for the syntheses of the ergot ring system. Importantly, this research also highlighted the possibility of epimerisation of the dihydro-isolysergic acid 18 to the epimer 19, containing the correct (+)-lysergic acid (2) stereochemistry at C8.D Later, Kurihara used a modified synthesis based on this work, also incorporating an intermolecular aldol condensation using 15, for the D-ring, in their approach to lysergic acid (2).[16] Likewise, the Ortar group also used the cyclisation of an amino-dienoic ester. In this case the precursor tether was prepared through an intermolecular Mizoroki–Heck reaction using an acrylate bearing the amine nucleophile.[17]
Total syntheses with key step C and D-ring cyclisation
In 1981, the Oppolzer group devised an elegant synthetic approach to lysergic acid (2) using an intramolecular imino-Dies–Alder reaction (Scheme 4) of compound 20, which was first prepared over seven-linear steps from 4-hydroxymethyl-1-tosylindole (18% overall yield).[18] The key to this cycloaddition was an initial retro-Diels–Alder reaction of the methyl bicyclo[2.2.1]hept-5-enyl-2-carboxylate moiety, within 20, to a diene, which under thermolysis, was quenched immediately with the imino-C5 double bond to provide 21 (2:3 mixture of syn- and anti-diastereoisomers, 67% yield). Later, Garner reported an asymmetric cycloaddition-based pathway as the key step in their synthesis of Szántay’s intermediate 14[14] (Scheme 5). This reaction was primed through initial formation of the azomethine ylide (formed through a reaction between the aldehyde at C5 and an amine) bearing a camphorsultam chiral auxiliary. An intramolecular reaction of this ylide, with an alkyne 22, afforded 23 which required a Cossy–Charette ring expansion to furnish the required piperidine D-ring.[19]
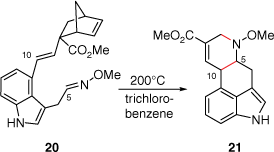
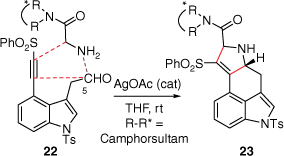
When comparing these two cycloaddition approaches, the Oppolzer group synthesis was performed in 11 steps at 3.9% overall yield while the Garner asymmetric synthesis was carried out in 20 steps from commercially available 4-bromoindole in 0.4% overall yield (if the previously reported Szántay[14] steps are included).
The group of Fujii and Ohno have published several papers involving the synthesis of ergoline alkaloid derivatives such as (±)-lysergic acid (2), (±)-lysergol and (±)-isolysergol. In 2008, this group reported a key synthetic step involving palladium-catalysed domino cyclisation of amino allene to construct the ergoline CD-ring system (Scheme 6).[20] Following oxidative addition, a coordination of the indolylpalladium(II) to the allenic tether was proposed to promote nosylamide anti-attack leading to the palladacycle 26. Overall, this amidopalladation pathway provides 27 as the major product, following reductive elimination. The minor product, the epimer at C5, was proposed to be formed through an alternative carbopalladation and nucleophilic attack of the η3-allylpalladium complex. Unfortunately, although this synthesis is quite elegant, the 13 synthetic steps required to make the precursor 24 make this synthesis less practical.E
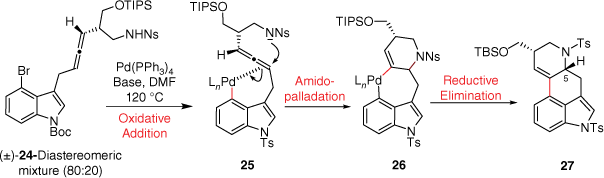
Later, the same group reported two enantioselective total syntheses of (+)-lysergic acid (2) using similar ring closing methodology. Emphasis was placed on producing the enantiopure allenes (derivatives of 39) through a Myers allene formation. Here, the requisite propargyl alcohol precursor was prepared through reductive ring-opening reaction of chiral 2-alkynyl-3-indolyloxirane[21] or through alpine-borane reduction of an ynone.[22]
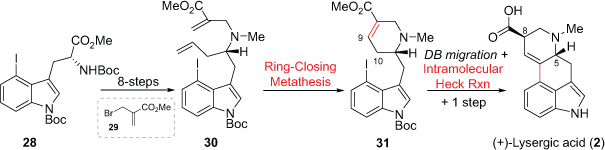
Later, the Jia group reported a stereocontrolled total synthesis of (+)-lysergic acid (2) in 12 steps and 12.7% overall yield (Scheme 7).[23] The synthesis at the time showcased three metal-catalysed and powerful bond-forming strategies for the construction of three rings. The (R)-4-iodotryptophan derivative 28 was obtained from the chiral pool starting material tryptophan. Compound 28 was synthetically converted into the diene 30 over eight steps. In these transformations the key-steps included two Wittig reactions to introduce the mono-substituted olefin moiety and the fragment 29 to introduce the second olefin. Compound 30 readily underwent ring-closing metathesis (RCM) to construct the D-ring of lysergic acid, as represented in 31. It is instructive to note that the C9 to C10 double bond migration was first conducted before a range of conditions for the intramolecular C-ring Mizoroki–Heck cross-coupling reaction were trialled.F The same group later reported a more detailed synthesis along with a modification where a Pd-catalysed indole synthesis was included using 3-chloro-2-iodoaniline.[24]
In 2013, the Fukuyama group expanded their earlier intramolecular Mizoroki–Heck cross-coupling approach by incorporating a key D-ring cyclisation (Scheme 8).[25] This synthesis proceeded firstly through the preparation of two chiral fragments 33 and 34, both synthesised in three steps from the chiral oxazolidinone auxiliary 32 which also created the requisite stereochemistry. The two fragments were connected through a reductive alkylation which was followed by RCM to construct the D-ring. Overall, the total synthesis of (+)-lysergic acid was achieved in 19 steps and 12% calculated from 32. The Fukuyama group also completed another approach using this ring formation strategy.[26] The Bisai group also used a C and D-ring cyclisation synthetic approach to the lysergic acid related compounds (+)-lysergol and (+)-isolysergol. In this report an asymmetric nitronate addition onto an α,β-unsaturated ester aza-Michael addition is used to construct the C-ring while a 6-endo-trig cyclisation is used to install the D-ring.[27]
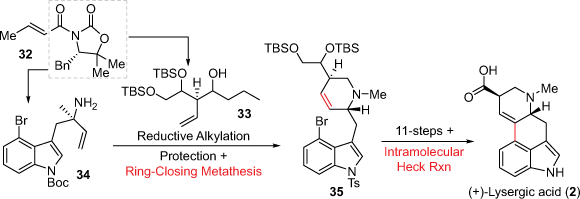
In 2010, the Martin group, during their synthesis of the ergot alkaloids ring system, first applied a reductive Heck coupling cyclisation to generate the key tricyclic compound 37 (Scheme 9).[28] Following this, the introduction of a diene, using bispentadienylzinc and amine 37, was accomplished to afford compound 38 as well as a linear regioisomer. The second ring closure was achieved using the Schrock–Hoveyda molybdenum catalyst (represented as [Mo] in Scheme 9) and microwave to induce a RCM diastereoselectively to 39. This compound was used to later generate the lysergic acid precursor (+)-isolysergol.
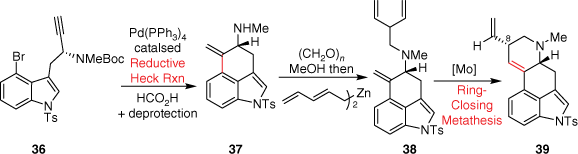
Total syntheses with key step B and C-ring cyclisation
In 1994, the Parsons group reported a tandem radical cyclisation approach to the ergot B and C-rings in a single step, through first radical generation using tin hydride.[29,30] Although this research did not produce lysergic acid (2) itself, several interesting and advanced ABCD-ring analogues were obtained. A synthetically different B and C-ring forming approach to (+)-lysergol was conceived by the Luo group (Scheme 10).[31] In this strategy, it was first conceived that the indole B-ring could be prepared at a later stage in the synthesis. The A–D ring connected piperidine skeleton 40 was prepared through a cyclopropanation ring-opening/nucleophilic substitution cascade reaction from a pyrrole precursor. The key B and C-rings were formed following similar intramolecular dearomatising [3 + 2] annulation methodology using a N-sulfonyl 1,2,3-triazole as developed by Murakami and Miura.[32] The required fully aromatised 3,4-fused indole skeleton 42 was then isolated after a manganese dioxide oxidation. Finally, (+)-lysergol 43, a precursor to lysergic acid, was isolated after sequential deprotections and a reductive amination. Importantly, this sequence was also repeated with a meta-difluorophenyl derivative of 42 to deliver the C13-fluoro equivalent of (+)-lysergol (43). This is especially notable for its potential in the context of medicinal chemistry investigations of LSD, where functionalisation of the ergoline A-ring has historically often been unfeasible.
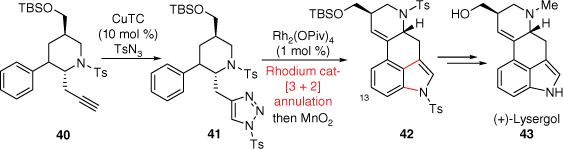
Total syntheses with key step C-ring cyclisation
One of the early lysergic acid total syntheses was through a C-ring cyclisation approach, devised by the Julia group. Firstly 5-bromoisatin (AB-ring) and methyl 6-methylnicotinate (D-ring) were linked through a condensation reaction leading to 44 (Scheme 11). Importantly, for later synthetic approaches to lysergic acid, this work established that the pyridine ring could be methylated and reduced with potassium borohydride to afford the D-ring piperidine α,β-unsaturated ester 45. A reagent consisting of sodium amide in refluxing ammonia then allowed for C-ring formation through reaction with the halogenated indole (AB-ring).[33] This product 46, also an intermediate in the Kornfeld synthesis, was then be converted into (±)-lysergic acid (2).
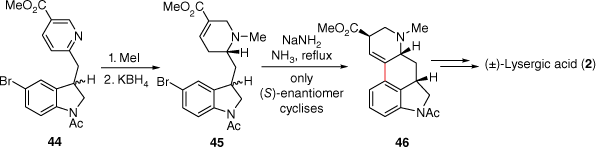
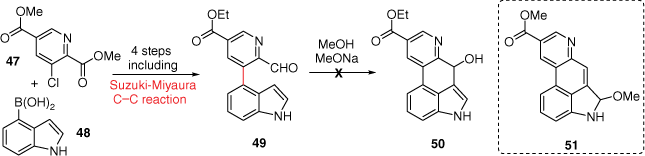
A later approach, through an alternate retrosynthetic disconnection, was attempted by the Hendrickson research group, where indole-4-boronic acid 48 and a chlorinated pyridine derivative 47 underwent coupling using Suzuki–Miyaura protocol to access 49, following additional functional group interconversions.[34] In this original report, cyclisation of aldehyde 49 to the alcohol 50 was purported to occur under strongly basic conditions (Scheme 12). The synthesis was later contested by the Nichols research group, whereupon their attempts to replicate this work instead yielded only B-ring methoxylated product 51.[35]
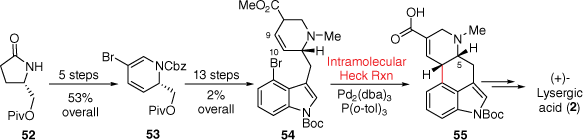
The intramolecular Mizoroki–Heck cross-coupling reaction is now a well-studied and prevalent reaction for creating the six-membered C-ring from precursors resembling the ergoline AB-D-ring. Importantly, in this reaction the retention of the double bond position at C9–C10 is possible through more recent methods. This synthetic approach was first explored by Fukuyama in 2009, outlined in Scheme 13. Initially, the D-ring was prepared starting with an enantiopure pyrrolidinone 52 which was converted through a number of synthetic steps into a piperidine precursor 53, while the AB-ring system was attached using 4-bromoindole.[36] The key palladium-catalysed reaction was ultimately successful providing tetracyclic ring-system 55, following additional double-bond migration. This compound could then be converted into (+)-lysergic acid (2) through the previous method of Oppolzer.[18] The overall yield of this synthesis was quite low; however, this strategy was also used later by Fukuyama in a more effective total synthesis.[37]
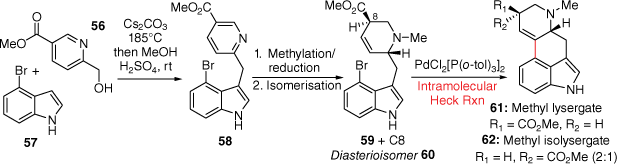
More recently, a Mizoroki–Heck C-ring closing strategy was used by the Wipf group in a short, scalable four-step synthesis of methyl lysergates in a combined 28% yield from a methyl nicotinate precursor (Scheme 14).[38] Initially, a Cs2CO3-mediated hydrogen autotransfer reaction joins the D with the AB-rings using the commercially available 56 and 57 respectively. A combined methylation, reduction and isomerisation reaction sequence afforded 59 and 60 as a 2:1 mixture of diastereosiomers. Importantly, the key Mizoroki–Heck cross-coupling reaction, using PdCl2[P(o-tol)3]2 and Et3N in MeCN, performed well on a > 1 g scale to afford methyl lysergate 61 and methyl isolysergate 62 (2:1 ratio respectively), without migration of the double bond. Finally, reduction of these compounds provided both lysergol and isolysergol.
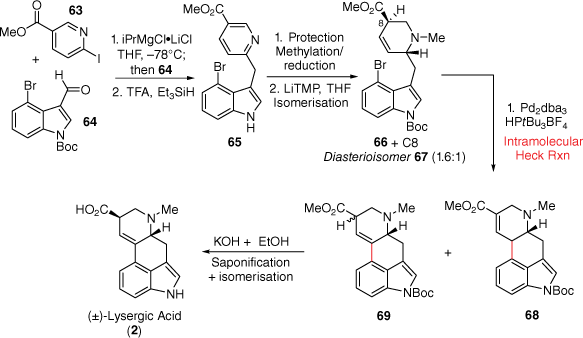
In 2023, the Smith group reported a concise synthesis of (±)-lysergic acid over six synthetic steps (Scheme 15)[39] starting with the two simple commercially available halogenated aromatic precursors 63 and 64. Initially, a magnesium–halogen exchange of iodopyridine 63 and subsequent addition to aldehyde 64 furnishes an intermediate alcohol which underwent simultaneous reduction and N-deprotection to give the AB-D-ring precursor 65. Sequential reprotection, methylation and isomerisation of the enolate with LiTMP gave a 1:1.6 diastereomeric mixture of 66:67. Importantly, re-exposure of the mixture to these conditions further enriched the amount of desired diastereomer 66. The synthesis of (±)-lysergic acid (2) was finalised through the key intramolecular Mizoroki–Heck reaction, leading to three products (the enone 68 and two diastereomeric alkene isomers 69). Conveniently, this mixture of three products could be treated with potassium hydroxide to mediate deprotection, saponification and isomerisation and deliver (±)-lysergic acid (2) diastereoselectively.
Conclusion
Several of the contemporary approaches outlined above have now established efficient, scalable and potentially modular access to lysergic acid (2) and its derivatives, offering more practical opportunities for derivatisation and new structure–activity relationship (SAR) studies. Fortuitously, this progress also comes at a turning point in our understanding of LSD’s pharmacodynamics. Recent landmark reports featuring X-ray crystal structures obtained for LSD bound to two human serotonin receptors (5-HT2A and 5-HT2B) have revealed for the first time key receptor–ligand interactions that may help direct the rational design of new LSD analogues.[40,41] For example, the authors postulate LSD’s uniquely high potency and long duration of action is mediated by conformational changes upon binding that move an extracellular loop of the receptor over the binding pocket containing LSD, acting as a lid to prevent ligand dissociation. From the crystal structure of LSD bound to the 5-HT2AR, it can be seen that hydrophobic residues within this lid motif lay in close proximity to LSD’s A- and D-rings. This discovery places a new importance on routes to accessing LSD analogues functionalised at these sites (such as the method of Yuan et al.[31] described in Scheme 10 above) for their potential to modulate any possible interactions with this region of the receptor. Thus, together with recent advances in our understanding of receptor–ligand interactions, many of the modern syntheses of lysergic acid described herein open the door to an exciting future of furthering the potential held by LSD as a therapeutic tool.
References
1 Savage C. Variations in ego feeling induced by D-lysergic acid diethylamide (LSD-25). Psychoanal Rev 1955; 42: 1-16.
| Google Scholar |
2 Liechti ME. Modern clinical research on LSD. Neuropsychopharmacology 2017; 42: 2114-2127.
| Crossref | Google Scholar |
3 Carhart-Harris RL, Muthukumaraswamy S, Roseman L, Kaelen M, Droog W, Murphy K, Tagliazucchi E, Schenberg EE, Nest T, Orban C, Leech R, Williams LT, Williams TM, Bolstridge M, Sessa B, McGonigle J, Sereno MI, Nichols D, Hellyer PJ, Hobden P, Evans J, Singh KD, Wise RG, Curran HV, Feilding A, Nutt DJ. Neural correlates of the LSD experience revealed by multimodal neuroimaging. Proc Natl Acad Sci USA 2016; 113(17): 4853-4858.
| Crossref | Google Scholar |
7 Nutt D. Psychedelic drugs—a new era in psychiatry? Dialogues Clin Neurosci 2019; 21(2): 139-147.
| Crossref | Google Scholar |
9 (a) Mann J. Turn on and tune in: psychedelics, narcotics and euphoriants, 1st edn. Royal Society of Chemistry; 2009. (b) Kornfeld EC, Fornefeld EJ, Kline GB, Mann MJ, Morrison DE, Jones RG, Woodward RB. The total synthesis of lysergic acid. J Am Chem Soc 1956; 78: 3087–3114. | Crossref | (c) Kornfeld EC, Fornefeld EJ, Kline GB, Mann MJ, Jones RG, Woodward RB. The total synthesis of lysergic acid and engrovine. J Am Chem Soc 1954; 76: 5256–5257. | Crossref |
10 Uhle FC. The synthesis of 5-keto-1,3,4,5-tetrahydrobenz[cd]indole. A synthesis of 4-substituted indoles. J Am Chem Soc 1949; 71: 761-766.
| Crossref | Google Scholar |
11 Rebek J, Tai DF. A new synthesis of lysergic acid. Tetrahedron Lett 1983; 24: 859-860.
| Crossref | Google Scholar |
12 Rebek J, Tai DF, Shue YK. Synthesis of Ergot alkaloids from tryptophan. J Am Chem Soc 1984; 106: 1813-1819.
| Crossref | Google Scholar |
13 Kiguchi T, Hashimoto C, Naito T, Ninomiya I. A new synthesis of (±)-lysergic acid. Heterocycles 1982; 19: 2279-2282.
| Crossref | Google Scholar |
14 Moldvai I, Temesvári-Major E, Incze M, Szentirmay É, Gács-Baitz E, Szántay C. Enantioefficient synthesis of α-ergocryptine: first direct synthesis of (+)-lysergic acid. J Org Chem 2004; 69: 5993-6000.
| Crossref | Google Scholar |
15 Ramage R, Armstrong VW, Coulton S. A new synthetic route to (±)-lysergic acid. Tetrahedron 1981; 37: 157-164.
| Crossref | Google Scholar |
16 Kurihara T, Terada T, Yoneda R. A new synthesis of (±)-lysergic acid. Chem Pharm Bull 1986; 34: 442-443.
| Crossref | Google Scholar |
17 Cacchi S, Giuseppe Ciattini P, Morera E, Ortar G. A concise, palladium-catalyzed approach to (±)-lysergic acid. Tetrahedron Lett 1988; 29: 3117-3120.
| Crossref | Google Scholar |
18 Oppolzer W, Francotte E, Bättig K. Total synthesis of (±)-lysergic acid by an intramolecular imino-Diels–Alder reaction. preliminary communication. Helv Chim Acta 1981; 64: 478-481.
| Crossref | Google Scholar |
19 Rathnayake U, Garner P. Asymmetric synthesis of lysergic acid via an intramolecular (3+2) dipolar cycloaddition/ring-expansion sequence. Org Lett 2021; 23: 6756-6759.
| Crossref | Google Scholar |
20 Inuki S, Oishi S, Fujii N, Ohno H. Total synthesis of (±)-lysergic acid, lysergol, and isolysergol by palladium-catalyzed domino cyclization of amino allenes bearing a bromoindolyl group. Org Lett 2008; 10: 5239-5242.
| Crossref | Google Scholar |
21 Iwata A, Inuki S, Oishi S, Fujii N, Ohno H. Formal total synthesis of (+)-lysergic acid via zinc(II)-mediated regioselective ring-opening reduction of 2-alkynyl-3-indolyloxirane. J Org Chem 2011; 76: 5506-5512.
| Crossref | Google Scholar |
22 Inuki S, Iwata A, Oishi S, Fujii N, Ohno H. Enantioselective total synthesis of (+)-lysergic acid, (+)-lysergol, and (+)-isolysergol by palladium-catalyzed domino cyclization of allenes bearing amino and bromoindolyl groups. J Org Chem 2011; 76: 2072-2083.
| Crossref | Google Scholar |
23 Liu Q, Jia Y. Total synthesis of (+)-lysergic acid. Org Lett 2011; 13: 4810-4813.
| Crossref | Google Scholar |
24 Liu Q, Zhang YA, Xu P, Jia Y. Total synthesis of (+)-lysergic acid. J Org Chem 2013; 78: 10885-10893.
| Crossref | Google Scholar |
25 Umezaki S, Yokoshima S, Fukuyama T. Total synthesis of lysergic acid. Org Lett 2013; 15: 4230-4233.
| Crossref | Google Scholar |
26 Kanno R, Yokoshima S, Kanai M, Fukuyama T. Total synthesis of (+)-lysergic acid. J Antibiot 2018; 71: 240-247.
| Crossref | Google Scholar |
27 Bhunia S, Chaudhuri S, Bisai A. Total syntheses of pyroclavine, festuclavine, lysergol, and isolysergol via a catalytic asymmetric nitro-Michael reaction. Chem Eur J 2017; 23: 11234-11238.
| Crossref | Google Scholar |
28 Deck JA, Martin SF. Enantioselective synthesis of (+)-isolysergol via ring-closing metathesis. Org Lett 2010; 12: 2610-2613.
| Crossref | Google Scholar |
29 Cladingboel DE, Parsons PJ. Synthesis of lysergic acid derivatives by triple radical cyclisation. J Chem Soc Chem Commun 1990; 1990: 1543-1544.
| Crossref | Google Scholar |
30 Özlü Y, Cladingboel DE, Parsons PJ. Tandem radical cyclisations: synthesis of lysergic acid derivatives. Tetrahedron 1994; 50: 2183-2206.
| Crossref | Google Scholar |
31 Yuan H, Guo Z, Luo T. Synthesis of (+)-lysergol and its analogues to assess serotonin receptor activity. Org Lett 2017; 19: 624-627.
| Crossref | Google Scholar |
32 Miura T, Funakoshi Y, Murakami M. Intramolecular dearomatizing [3 + 2] annulation of α-imino carbenoids with aryl rings furnishing 3,4-fused indole skeletons. J Am Chem Soc 2014; 136: 2272-2275.
| Crossref | Google Scholar |
33 Julia M, Le Goffic F, Igolen J, Baillarge M. Une nouvelle synthese de l’acide lysergique. Tetrahedron Lett 1969; 10: 1569-1571 [In French].
| Crossref | Google Scholar |
34 Hendrickson JB, Wang J. A new synthesis of lysergic acid. Org Lett 2004; 6: 3-5.
| Crossref | Google Scholar |
35 Bekkam M, Mo H, Nichols DE. A reported “new synthesis of lysergic acid” yields only the derailment product: methyl 5-methoxy-4,5-dihydroindolo[4,3-f,g]quinoline-9-carboxylate. Org Lett 2012; 14: 296-298.
| Crossref | Google Scholar |
36 Inoue T, Yokoshima S, Fukuyama T. Synthetic studies toward (+)-lysergic acid: construction of the tetracyclic ergoline skeleton. Heterocycles 2009; 79: 373-378.
| Crossref | Google Scholar |
37 Kurokawa T, Isomura M, Tokuyama H, Fukuyama T. Synthesis of lysergic acid methyl ester via the double cyclization strategy. Synlett 2009; 2009: 775-778.
| Crossref | Google Scholar |
38 Tasker NR, Wipf P. Short synthesis of Ergot alkaloids and evaluation of the 5-HT1/2 receptor selectivity of lysergols and isolysergols. Org Lett 2022; 24: 7255-7259.
| Crossref | Google Scholar |
39 Knight BJ, Harbit RC, Smith JM. Six-Step Synthesis of (±)-Lysergic Acid. J Org Chem 2023; 88: 2158-2165.
| Crossref | Google Scholar |
40 Kim K, Che T, Panova O, DiBerto JF, Lyu J, Krumm BE, Wacker D, Robertson MJ, Seven AB, Nichols DE, Shoichet BK, Skiniotis G, Roth BL. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 2020; 182: 1574-1588.e19.
| Crossref | Google Scholar |
41 Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, Shoichet BK, Dror RO, Roth BL. Crystal structure of an LSD-bound human serotonin receptor. Cell 2017; 168: 377-389.e12.
| Crossref | Google Scholar |
Footnotes
A Individual steps of the total syntheses of lysergic acid have been discussed in detail in there references cited here.
B The first synthesis of lysergic acid was achieved in the Eli Lilly laboratories in Indianapolis by Ed Kornfeld and his co-workers. It is reported Kornfeld conceived and carried out the synthesis while Woodward was an Eli Lilly consultant. Please see the references cited here. For the synthesis, please see Kornfeld et al.
C Uhle’s ketone, shown in Scheme 2, was first prepared in 1949.