

Controlling emission energy in metal–organic frameworks featuring cyclometalated iridium(III) linkers
Carol Hua A * and Timothy U. Connell B *
A * and Timothy U. Connell B *
A
B
Handling Editor: Curt Wentrup
Australian Journal of Chemistry 76(10) 686-695 https://doi.org/10.1071/CH23127
Submitted: 30 June 2023 Accepted: 16 August 2023 Published: 20 September 2023
Abstract
Efficient solid-state luminescent materials are critical components in varied optoelectronic devices. Here, we report three metal–organic frameworks combining calcium(II) with heteroleptic iridium(III) metalloligands containing the same 1,2-diimine ancillary ligand but different cyclometalating ligands. The synthesised frameworks exhibit similar supramolecular structure but different emission properties depending on the cyclometalating ligand. Binding calcium(II) to the metalloligands further affects framework emission depending on the relative contribution of triplet charge transfer (3MLLCT) or ligand-centred (3LC) transitions to the emissive excited state.
Keywords: calcium, coordination chemistry: structure, iridium, luminescence, metal–organic framework, photochemistry, solid state, transition metals.
Introduction
Luminescent materials are critical components in numerous technologies important to modern society, including lighting, sensing and other optoelectronic devices. Metal–organic frameworks (MOFs) are an exciting design platform for solid-state luminescent materials as their highly ordered crystalline nature provides well-defined luminophore environments that may increase emission lifetime and quantum efficiency.1 Modular synthetic routes enable the ready incorporation of luminophores as either a component of the rigid framework or as an encapsulated guest within accessible pores, which provides a plethora of options to modify and optimise the optical properties.2,3 Combining a luminescent linking ligand with a closed-shell metal ion (e.g. zinc(II), cadmium(II)) is a common approach to synthesising luminescent MOFs. Although luminescence of the linking ligand often translates to the synthesised MOF, metal ion coordination may instead result in modifying or even quenching the linker’s emission.1
Most luminescent linkers are organic molecules4 that are limited by short emission lifetimes, small Stokes shifts (causing inner-filter effect and self-quenching) and a propensity to photobleach. Luminescent cyclometalated iridium(III) complexes also find wide application in lighting,5 sensing,6 solar fuels,7 organic synthesis8,9 and bioimaging.10,11 Although they exhibit attractive photophysical properties similar to other luminescent d6 transition metal complexes, including good photostability and long-lived triplet excited states, common heteroleptic [Ir(C^N)2(N^N)]+ complexes (where C^N is a cyclometalating and N^N a 1,2-diimine ancillary ligand) additionally boast frontier orbitals (the highest occupied and lowest unoccupied molecular orbital, HOMO and LUMO respectively) spatially partitioned within the molecule. The HOMO is generally localised across the metal and cyclometalated arene ring, while the LUMO is exclusively localised on the ancillary ligand.12,13 This separation also allows unprecedented tuning of emission energy by controlling the ligand sphere using robust synthetic pathways.14,15 In general, orbital energies can be modified with functional groups on the corresponding ligand (HOMO and cyclometalating ligand, LUMO and ancillary ligand, Fig. 1); electron-withdrawing groups stabilise whereas donating groups destabilise.16 The resulting lowest-energy triplet state is typically dominated by one of two possible conformations: the dπ → π* metal–ligand-to-ligand charge transfer excited state (3MLLCT) and π → π* ligand-centred excited states (3LC).17 The 3MLLCT state is readily accessed by efficient intersystem crossing from the analogous low-energy 1MLLCT state, facilitated by the large spin-orbit coupling constant of the heavy iridium atom. Highly stabilised 3LC states typically occur through strong exchange interactions,18 and are accessed following internal conversion from the corresponding 3MLLCT state.
The frontier orbitals of heteroleptic cyclometalated iridium(III) complexes can be selectively controlled by manipulating the ligand sphere.
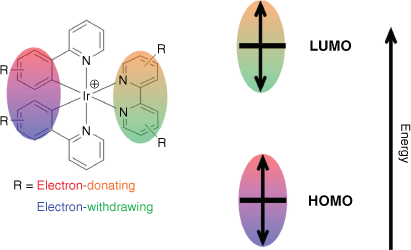
The robust photophysical properties of cyclometalated iridium(III) complexes make them attractive building blocks in self-assembled frameworks. Both homoleptic [Ir(C^N)3] and heteroleptic [Ir(C^N)2(N^N)]+ complexes can act as linking metalloligands, binding a range of metal salts (alkaline and transition metals, as well as lanthanoids) through pyridine or carboxylate donor groups to construct supramolecular frameworks.19 Heteroleptic iridium(III) metalloligands most commonly employ regioisomers of 2,2′-bipyridinedicarboxylic acid as the ancillary ligand, functionalised at either the 4,4′- or 5,5′-positions.20,21 Carboxylate substitution at the 4,4′-position yields densely packed 1-D chains featuring a looping motif.22–25 Conversely, 5,5′-substitution yields linear linkers that are predominately employed to synthesise zirconium(IV) UiO-type MOFs, where the metalloligand is incorporated through either direct synthesis or post-synthetic modification.26–28 It is surprising that despite the ready synthetic control of emission energy in heteroleptic iridium(III) complexes, their prior use as MOF metalloligands has focussed on the archetypal 2-phenylpyridine (Hppy) cyclometalating ligand. Select examples incorporate fluorinated derivatives,29,30 a common strategy for stabilising the HOMO and hypsochromically shifting emission energy. Using carboxylated cyclometalating ligands to prepare either homoleptic Ir(C^N)3 or heteroleptic complexes in combination with 2,2′-bipyridine-4,4′-dicarboxylic acid (H2dcbpy) afforded an octahedral metal binding metalloligand, resulting in porous cubic frameworks.21 In one recent report, linear iridium(III) metalloligands with similar carbazole functionalised cyclometalating ligands (one also containing a trifluoromethyl group) were incorporated into zirconium(IV) UiO-type frameworks and compared for photocatalytic activity.27 Although the trifluoromethylated derivative performed higher for all substrate combinations, comparing the emission showed no difference in spectral profile.
Here, we compare three MOFs combining calcium(II) cations with cyclometalated iridium(III) metalloligands that contain the same 2,2′-bipyridine-4,4′-dicarboxylic acid ancillary ligand (H2dcbpy) but different cyclometalating ligands. Two complexes gave isostructural frameworks of empirical formula {Ca[Ir(C^N)2(dcbpy)]2(DMF)2}·2H2O (DMF = N,N′-dimethylformamide) with different higher-order packing. The third complex, [Ir(bt)2(Hdcbpy)]2 (where Hbt corresponds to 2-phenylbenzothiazole), slowly dissociates in solution and an ordered framework could only be isolated after co-crystallisation with an equivalent of the free H2dcbpy ligand. Modulating the frontier orbitals through ligand design resulted in MOFs with tunable solid-state emission and we found that triplet excited state character (i.e. either 3MLLCT or 3LC) exhibited different sensitivity to calcium(II) binding on MOF incorporation.
Results and discussion
The iridium(III) metalloligands were prepared and isolated as amphiphilic neutral molecules using a modified reported procedure.22 Briefly, precursor iridium(III) dimers and H2dcbpy were combined in a basic methanolic solution and the neutral products (where only one carboxylate is protonated) selectively precipitated from water through pH control during workup (Scheme 1). Three different cyclometalating ligands were selected: Hppy, 1-phenylisoquinoline (Hpiq) and Hbt. We favoured extended conjugation in our choice of cyclometalating ligands, in contrast to prior reports functionalising Hppy with electron-withdrawing substituents,28–30 to elicit a bathochromic shift in emission.
Synthesis of iridium(III) metalloligands used in this study along with the high-resolution mass spectrum of [Ir(ppy)2(H2dcbpy)]+ (inset).
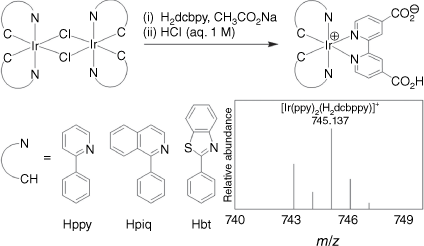
Although 1H NMR spectra were readily collected, poor solubility of amphiphilic iridium(III) complexes in common deuterated solvents (e.g. CDCl3, [D6]DMSO, CD3CN, CD3OD) prevented 13C{1H} NMR spectroscopy. All 1H spectra exhibited C2v symmetry, which we attribute to rapid intermolecular exchange of the protonated carboxylate with residual water in the NMR solvent ([D6]DMSO, see Supporting information). Proton spectra of both [Ir(ppy)2(Hdcbpy)] and [Ir(piq)2(Hdcbpy)] displayed the correct number and integration of hydrogen environments; however, residual H2dcbpy ligand peaks were visible in the 1H NMR spectra of [Ir(bt)2(Hdcbpy)]. These peaks persisted even after repeated recrystallisation from ethanol and an additional spectrum (collected 7 days later) showed a greater distribution of species in solution (Supplementary Fig. S1), suggesting the complex dissociates over time. High-resolution mass spectrometry (HRMS) of the three complexes exhibited the expected mass and isotope distribution (Scheme 1, Supplementary Fig. S2).
The two MOFs {Ca[Ir(ppy)2(dcbpy)]2(DMF)2}·2H2O (1), {Ca[Ir(piq)2(dcbpy)]2(DMF)2}·2H2O (2) were synthesised through the reaction of [Ir(ppy)2(Hdcbpy)] and [Ir(piq)2(Hdcbpy)] respectively, with Ca(NO3)2·4H2O at 100°C for 48 h in a 3 : 1 mixture of N,N′-dimethylformamide (DMF) and water to yield either yellow block (1) or orange needle (2) crystals. Dark yellow block crystals of {Ca[Ir(bt)2(dcbpy)]2(dcbpy)(H2O)2]}2DMF (3) were prepared using a similar method with one equivalent of H2dcbpy added to the solution. Phase purity of the bulk samples was demonstrated using powder X-ray diffraction (XRD) for all compounds (Supplementary Fig. S3–S5).
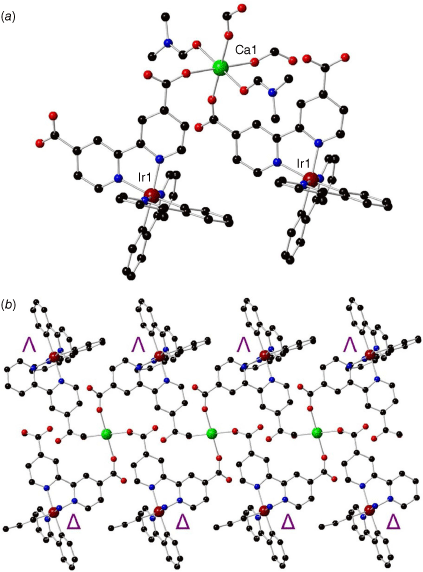
The crystal structure of 1 was solved and refined in the monoclinic space group P21/c with unit cell parameters of a = 8.8330(18), b = 36.811(7), c = 10.533(2) Å and β = 100.29(3)° (full crystallographic parameters in Supplementary Table S1). The calcium(II) ion exhibits an octahedral coordination sphere with four O-donors from the dcbpy ancillary ligand of the iridium(III) metalloligand and two O-donors from axial DMF molecules containing typical Ca–O bond distances of 2.283–2.405 Å (Fig. 2a). Monodentate coordination of the dcbpy carboxylates can likely be attributed to the steric bulk of the iridium(III) metalloligand. Hydrogen bonding interactions are formed with one molecule of water between uncoordinated carboxylate O-donors of two different dcbpy ligands with distances of d(O3–H2O) = 2.754(4) Å and d(O1–H2O) = 2.737(5) Å such that the water molecule sits within a ‘pocket’ within the framework structure (Supplementary Fig. S6). The linking metalloligand in 1 retains all the structural features expected of a cyclometalated iridium(III) complex, with bond lengths of Ir–N (dcbpy) = 2.132(3), 2.135(3) Å, Ir–N(ppy) = 2.037(3) Å and Ir–C(ppy) = 2.009(4), 2.015(3) Å.
Structure of {Ca[Ir(ppy)2(dcbpy)]2(DMF)2}·2H2O (1) showing (a) the coordination environment around the calcium(II) ion (black, carbon; red, oxygen; blue, nitrogen; maroon, iridium; green, calcium), (b) the 1-D chain containing Δ- and Λ-iridium(III) metalloligands on the top and bottom (coordinated DMF molecules were removed for clarity).
The crystal structure of {Ca[Ir(piq)2(dcbpy)]2(DMF)2}·2H2O (2) was solved and refined in the triclinic space group P̄1 with unit cell parameters of a = 8.8580(18), b = 16.029(3), c = 19.758(4) Å and α = 106.75(3), β = 96.19(3), γ = 105.00(3)°. Changing the cyclometalating ligand from ppy (1) to piq (2) results in an isostructural compound with a similar octahedral coordination sphere around the calcium(II) ion (Supplementary Fig. S7a): bond lengths Ca–O(dcbpy) = 2.282–2.361 Å and Ca–O(DMF) = 2.361 Å. The iridium(III) metalloligand also contains typical bond distances Ir–N(dcbpy) = 2.134(3), 2.155(4) Å, Ir–N(piq) = 2.034(5), 2.035(5) Å and Ir–C(piq) = 2.002(4), 2.014(4) Å.
Two molecules of either iridium(III) metalloligand ([Ir(ppy)2(dcbpy)]− or [Ir(piq)2(dcbpy)]−) bridge each calcium(II) ion, resulting in a continuous 1-D chain, with calcium(II) located along the centre of the chain and iridium(III) metalloligands above and below (Fig. 2b, Supplementary Fig. S7b). A racemic mixture of both Δ- and Λ-isomers of each iridium(III) complex is present in the respective structure, alternating along the 1-D chain. The cyclometalating ligands are orientated along the same direction because of offset π–π stacking interactions (4.006 Å) between the phenyl ring and the pyridyl ring of a ppy ligand in an adjacent 1-D chain (Fig. 3a). Similar offset π–π stacking interactions (4.208 Å) exist in structure 2 between the phenyl ring and an isoquinoline in the adjacent 1-D chain. The 1-D chain features a distorted looping motif with the carboxylates of the dcbpy ligand only coordinated in a monodentate manner. The structures of compounds 1 and 2 are isostructural with the reported [Mg[Ir(ppy)2(dcbpy)])2(DMF)2]∙3.5H2O framework, which features an octahedral magnesium(II) ion (chemically very similar to calcium(II)) with four monodentate O-donors from the dcbpy carboxylates and DMF molecules in the two axial positions.23 The distorted looping motif in these frameworks contrasts with previously reported 1-D chains combining [Ir(ppy)2(dcbpy)]− with zinc(II), cadmium(II), cobalt(II) and nickel(II).20 These, instead, exhibit a regular looping 1-D chain with bidentate metal coordination of the [Ir(ppy)2(dcbpy)]− carboxylate groups.
Packing of the 1-D chains along the a axis for (a) {Ca[Ir(ppy)2(dcbpy)]2(DMF)2}·2H2O (1), and (b) {Ca[Ir(piq)2(dcbpy)]2(DMF)2}·2H2O (2).
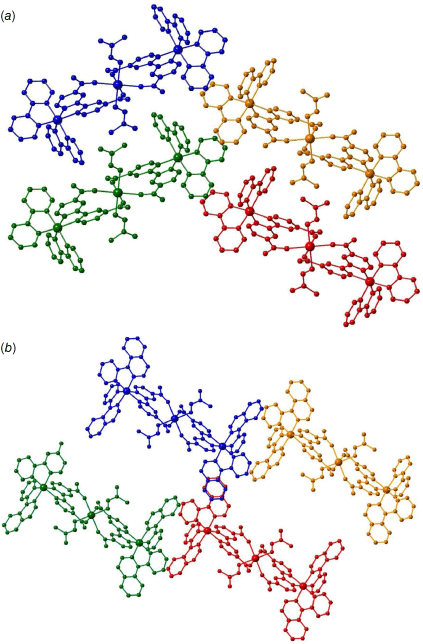
The higher-order structure of 1 is densely packed, with a calculated accessible void volume of only 1.1% (Fig. 3a). Thermo gravimetric analysis (TGA) of 1 reflects the lack of accessible voids; an initial steep mass loss corresponds to evaporation of surface water and DMF molecules before a plateau between 60 and 230°C (Supplementary Fig. S8). A subsequent 6.5% mass loss between 230 and 290°C may correspond to the liberation of the DMF molecules coordinated to the calcium(II) centres. The higher temperature required to remove the DMF molecules (230–290°C vs the boiling of DMF at 153°C) reflects the additional energy required to break the coordination bond with calcium(II). Decomposition of the framework occurs above 350°C. The extra conjugation of the isoquinoline ring in the cyclometalating ligands of 2 compared with 1 increases the steric bulk and prevents adjacent 1-D chains from packing tightly together (Fig. 3b). This results in a much larger accessible void space of 33.4%, also reflected in TGA of 2 with a 15% mass loss below 150°C corresponding to evaporation of water and DMF solvent molecules within the MOF voids (Supplementary Fig. S9). Decomposition of 2 occurs above 300°C.
In contrast to 1 and 2, the framework {Ca[Ir(bt)2(dcbpy)]2(dcbpy)(H2O)2]}·2DMF (3) contains an additional dcbpy linker in its structure. The crystal structure was solved and refined in the triclinic space group P̄1 with unit cell parameters of a = 9.6380(19), b = 18.941(4), c = 22.687(5) Å, α = 75.51(3), β = 88.64(3), γ = 88.30(3)°. The asymmetric unit of compound 3 comprises of two distinct calcium(II) ions, both with octahedral coordination geometries of O-donors (Fig. 4a). For both Ca1 and Ca2, and similarly to the structures of 1 and 2, four O-donors are coordinated from carboxylate groups of [Ir(bt)2(dcbpy)]−; only one carboxylate is bound in a monodentate fashion – the other donors feature a bridging coordination mode between Ca1 and Ca2. One of the axial O-donors is from a coordinated water molecule, while the final axial position is from the free dcbpy ligand, which also bridges between Ca1 and Ca2. The nitrogen atoms in dcbpy remain uncoordinated, with the pyridyl rings orientated 180° relative to each other such that the N-donors are pointing in opposite directions. The uncoordinated N–N binding site in dcbpy may offer a possible catalytic site or be amenable to post-synthetic modification if rotation around the central C–C bond can be achieved.
Structure of {Ca[Ir(bt)2(dcbpy)]2(dcbpy)(H2O)2]}·2DMF (3) showing (a) the coordination environment around the calcium(II) ion (black, carbon; red, oxygen; blue, nitrogen; maroon, iridium; green, calcium); (b) the 1-D chain containing iridium(III) metalloligands on the top and bottom; (c) the free dcbpy ligand (yellow) linking adjacent 1-D chains (blue) to form a 2-D sheet; and (d) the packing of the 2-D sheets along the a axis.
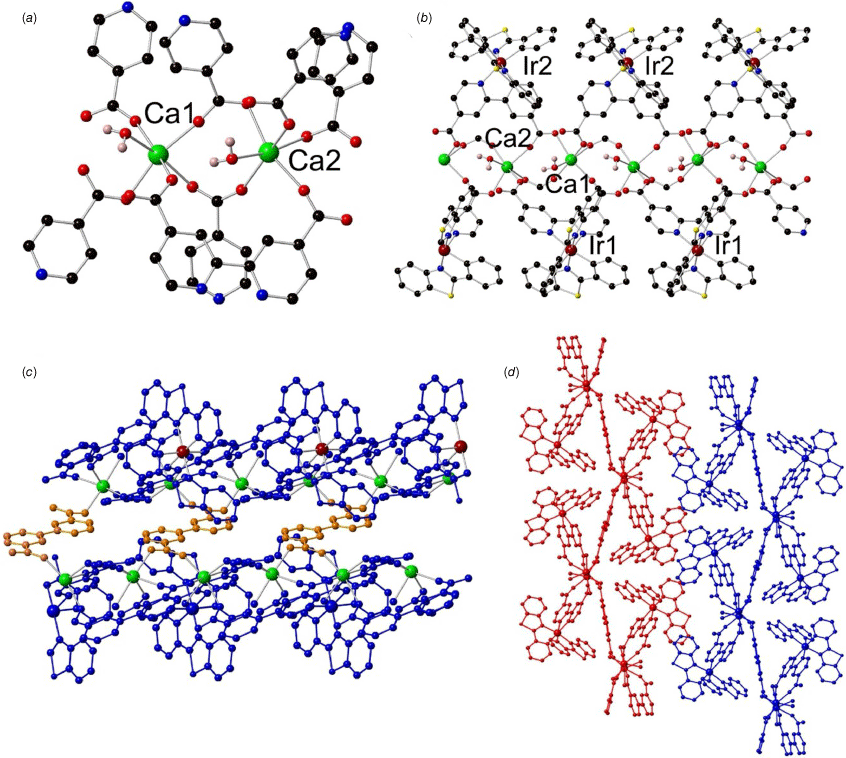
The [Ir(bt)2(dcbpy)]− metalloligand is present as a racemic mixture, resulting in the centrosymmetric space group of P̄1, and contains standard bond distances: Ir–N(dcbpy) = 2.141(3), 2.168(2) Å, Ir–N(bt) = 2.040(2), 2.060(2) Å and Ir–C(bt) = 2.009(3), 2.018(3) Å. In analogous fashion to frameworks 1 and 2, the calcium(II) ions are in a 1-D chain between iridium(III) metalloligands above and below (Fig. 4b). The 1-D chains are additionally bridged by free dcbpy to form 2-D sheets (Fig. 4c). One DMF molecule is associated with the structure, located within a ‘pocket’ formed by two iridium(III) metalloligands. Hydrogen bonding interactions occur between the DMF molecule and the uncoordinated O-donor from the carboxylate group of [Ir(bt)2(dcbpy)]−, d(O–CH) = 3.624(5) Å, and with the water molecules coordinated to the calcium(II) ions, d(O–CH) = 3.456(5), 3.552(5) Å. The 2-D sheets are densely packed in an AAA fashion relative to each other with only 0.5% accessible void space (Fig. 4d). The dense nature of the structure is reflected in TGA, which showed ~6% mass loss between 25 and 300°C corresponding to the loss of the DMF molecules intercalated within the structure (Supplementary Fig. S10). Decomposition of the framework occurs above 300°C where a large mass loss is observed. Further close-contact interactions support the closely packed 2-D sheets of 3; the sulfur atom in the benzothiazole heterocycle enables sulfur–sulfur interactions (4.052 Å) between benzothiazole groups from iridium(III) metalloligands in adjacent 1-D chains. Additional π–sulfur interactions between the benzothiazole sulfur atom and the phenyl ring of an adjacent cyclometalating ligand are also present, with bond distance 3.861 Å.
The 1-D chain [Ca(CO2)2]∞ in compound 3 bears similarities to reported MOFs synthesised with [Ir(ppy)2(dcbpy)]− and various lanthanoid ions or yttrium(III).22,25 The metal ions in all cases comprise five O-donors from the iridium(III) metalloligand along with coordinated solvent molecules to complete the coordination sphere (seven-coordinate for the lanthanoid ions with two solvent molecules, and six-coordinate for calcium(II) with one solvent molecule). A ‘pocket’ is formed between two iridium(III) metalloligands in the lanthanoid 1-D chains, enabling the encapsulation of a water molecule through hydrogen bonding interactions in an analogous manner to the encapsulated DMF molecule in compound 3 (Supplementary Fig. S6). The inclusion of the larger DMF molecule (instead of the smaller water molecule) may be attributed to the larger void space induced by the sterically bulky 2-phenylbenzothiazole cyclometalating ligand.
The synthesised iridium(III) metalloligands all exhibit similar absorption spectra measured in dichloromethane (Supplementary Fig. S11). Consistent with related heteroleptic iridium(III) complexes,14 spectra were assigned to intense π–π* ligand centred (LC) transitions at higher energies (>350 nm) followed by charge transfer bands (metal-to-ligand and ligand-to-ligand, MLLCT) at longer wavelengths (350–550 nm).
Luminescence spectroscopy of iridium(III) metalloligands was investigated both in solution and the solid state (Table 1, Fig. 5). All three complexes exhibited strong orange–red emission as dichloromethane solutions with emission maxima (λsol) at 613, 630 and 573 nm for [Ir(ppy)2(Hdcbpy)], [Ir(piq)2(Hdcbpy)] and [Ir(bt)2(Hdcbpy)] respectively. Spectral shape analysis provided some insight into the nature of each complex’s triplet excited state.17 The broad featureless emission of [Ir(ppy)2(Hdcbpy)] (Fig. 5a, dotted line) is consistent with a dominant 3MLLCT excited state, whereas the resolved vibronic peaks in [Ir(piq)2(Hdcbpy)] (Fig. 5b, dotted line) instead support an excited state mostly 3LC in character. Between these two extremes, the broad but asymmetric emission profile of the [Ir(bt)2(Hdcbpy)] complex (Fig. 5c, dotted line) suggests a mixed excited state, where the 3LC and 3MLLCT transitions are close in energy. In the solid state, all iridium(III) complexes exhibit a bathochromic shift in their emission maxima, which is common in related complexes and likely a result of packing interactions in the crystal lattice.31 Although the change in emission maxima was small for [Ir(ppy)2(Hdcbpy)] (λss 622 nm, Δem 9 nm), the [Ir(piq)2(Hdcbpy)] exhibits a large bathochromic shift (λss 699 nm, Δem 69 nm). Furthermore, [Ir(piq)2(Hdcbpy)] emission is heavily quenched in the solid state and at the instrument’s limit of detection (Supplementary Fig. S12). Both observations suggest a dominant non-radiative relaxation pathway. We also observed a larger bathochromic shift for [Ir(bt)2(Hdcbpy)] (λss 604 nm, Δem 41 nm) as well as a more symmetrical spectral profile; such pronounced spectral changes between states further support similar energy levels of both 3LC and 3MLLCT.
Normalised emission spectra of (a) [Ir(ppy)2(Hdcbpy)], (b) [Ir(piq)2(Hdcbpy)] and (c) [Ir(bt)2(Hdcbpy)]2 in a dichloromethane solution (25 μM, 350 nm excitation, dotted line), solid-state powder (380 nm excitation, dashed line) and synthesised calcium(II) MOF (380 nm excitation, solid line).
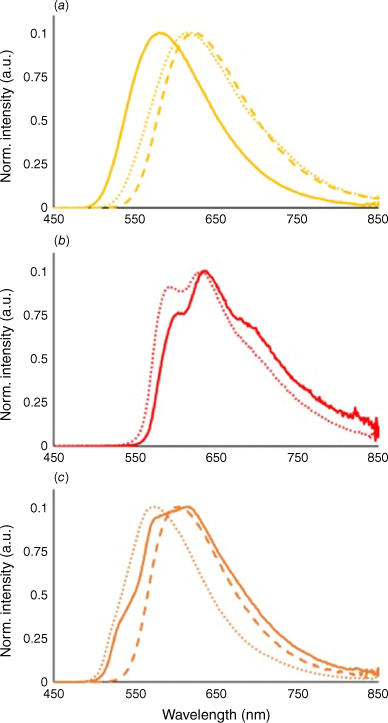
Incorporating [Ir(piq)2(Hdcbpy)] into the rigid framework of 2 counteracted the observed emission quenching of the complex in the solid state (Fig. 5b, solid line). Vibronic resolution was still observed, albeit with different ratios in peak intensity, and a slight bathochromic shift in emission relative to the complex in solution (λMOF 636 nm, ΔMOF 6 nm). A similar bathochromic shift (λMOF 615 nm, ΔMOF 9 nm) and greater vibronic structure were also observed in the emission spectra of 3 (Fig. 5c, solid line), suggesting an increased contribution from the 3LC excited state of [Ir(bt)2(Hdcbpy)]. Importantly, the additional dcbpy ligand in 3 does not affect emission from the iridium(III) luminophore. By contrast, framework 1 exhibited a pronounced hypsochromic shift in emission relative to the complex in either solution or the solid state (λMOF 580 nm, ΔMOF −42 nm, Fig. 5a, solid line). We assign this large change in emission energy to destabilisation of the LUMO caused by the electron rich carboxylates following deprotonation of the ancillary ligand. Similar hypsochromic emission shifts are well known in ruthenium(II) polypyridine complexes containing H2dcpby,32,33 and are also observed in MOFs combining iridium(III) metalloligands with magnesium(II) and cadmium(II).23,29 Higher energy emission may also occur due to rigidochromic effects in the highly ordered 1framework.34 The 3MLLCT excited state in [Ir(ppy)2(dcbpy)]− results in a LUMO that resides almost exclusively on the ancillary 2,2′-bipyridine ligand, making luminescent properties highly sensitive to changes in electron density at this position. The dominant 3LC excited states in both 2 and 3 are more localised across the cyclometalating ligands and therefore less affected by calcium(II) binding.
Conclusion
In summary, we have demonstrated the effect of altering the cyclometalating ligand in iridium(III) linkers for constructing calcium(II) metal organic frameworks. The three synthesised frameworks exhibit a similar structure apart from differences in the intrinsic void space, which is principally controlled by the relative steric bulk of the cyclometalating linkers. Framework 3, comprising the [Ir(bt)2(dcbpy)]− metalloligand, crystallised with an additional molecule of dcbpy in the structure, providing a possible additional binding site that might see future utility in sensing or catalysis. The metalloligand frontier molecular orbitals strongly dictate the emission properties of the resulting calcium(II) MOFs. Increasing the conjugation of the cyclometalating ligand in [Ir(piq)2(Hdcbpy)] and [Ir(bt)2(Hdcbpy)] bathochromically shifted MOF emission energy relative to the archetypal [Ir(ppy)2(Hdcbpy)] complex. The 3MLLCT excited state of [Ir(ppy)2(Hdcbpy)] was sensitive to metal binding, undergoing a large hypsochromic shift in emission (ΔMOF −42 nm) when incorporated into the calcium(II) MOF. This result suggests that the emission energy of iridium(III) metalloligands may be further tuned by using different metals during MOF synthesis. The results of this project help direct further understanding of the emission properties of highly ordered solid-state materials and their varied application.
Experimental
General considerations
All chemicals and solvents were used as obtained and without further purification. NMR spectra were acquired on a Bruker Avance 400 spectrometer; 1H NMR spectra were acquired at 400 MHz. All spectra were recorded at 298 K and chemical shifts were referenced to residual solvent peaks and are quoted in parts per million (ppm). Electrospray ionisation–mass spectrometry (ESI-MS) experiments were performed using a Shimadzu LCMS-9030 quadrupole time-of-flight mass spectrometer. Fourier Transform infrared (FTIR) spectra were measured on a Bruker Alpha spectrometer between 4000 and 400 cm−1 with 4 cm−1 resolution and 32 scans and normalised as absorbance spectra (Supplementary Fig. S13–S15). TGA was conducted on a PerkinElmer TGA instrument using ceramic crucibles as sample holders under high-purity nitrogen at a flow rate of 20°C min−1. The samples were heated up to 450°C with a temperature increment of 10°C min−1. Microanalysis was carried out at the Chemical Analysis Facility – Elemental Analysis Service in the Department of Chemistry and Biomolecular Science at Macquarie University, Australia.
Single-crystal X-ray diffraction data were collected on the MX2 beamline at the Australian Synchrotron.35,36 Single crystals were transferred directly from the mother liquor into immersion oil and placed under a stream of nitrogen at 100 K. Crystal structures were solved by direct methods using the program SHELXT37 and refined using a full matrix least-squares procedure based on F2 (SHEXL38), within the Olex239 GUI program.
Iridium(III) complex synthesis
A mixture of [Ir(ppy)2Cl]2 (202 mg, 188 μmol) and H2dcbpy (92 mg, 377 μmol) was suspended in methanol (20 mL) before excess sodium acetate (~0.5 g) was added. The suspension was heated at 80°C for 16 h, in the dark under an inert atmosphere (N2). The mixture was cooled to ambient temperature and concentrated under reduced pressure, then redissolved in minimum 3 : 1 water : ethanol (~200 mL) and filtered through Celite. Solution pH was adjusted to ~3 with 1 M HCl solution, and the solution was cooled to 4°C and stirred for 2 h before the solid was collected by filtration. The precipitate was washed with water, then diethyl ether and dried at the pump to yield the product as an orange powder (255 mg, 91%). 1H NMR (400 MHz, [D6]DMSO): δ 9.13 (1H, s), 8.26 (1H, d, J = 8.0 Hz), 7.93 (4H, m) 7.62 (1H, d, J = 4.5 Hz), 7.12 (1H, m), 7.01 (1H, t, J = 7.2 Hz), 6.91 (1H, t, J = 7.2 Hz), 6.17 (1H, d, J = 7.2 Hz). HRMS m/z 745.137 ([C34H24N4O4Ir]+ requires 745.142).
A mixture of [Ir(piq)2Cl]2 (250 mg, 196 μmol) and H2dcbpy (96 mg, 393 μmol) was suspended in methanol (30 mL) before excess sodium acetate (~0.5 g) was added. The suspension was heated at 80°C for 16 h, in the dark under an inert atmosphere (N2). The mixture was cooled to ambient temperature and concentrated under reduced pressure, then redissolved in minimum 9 : 1 water : ethanol (~200 mL) and filtered through Celite. Solution pH was adjusted to ~3 with 1 M HCl solution, and the solution cooled to 4°C and stirred for 30 min before the solid was collected by filtration. The precipitate was washed with water, diethyl ether and then recrystallised from hot ethanol to yield the product as a dark red crystalline powder (322 mg, 97%). 1H NMR (400 MHz, [D6]DMSO): δ 9.13 (1H, s), 9.00 (1H, m), 8.37 (1H, d, J = 7.9 Hz), 8.07 (1H, m), 7.90 (3H, m), 7.78 (1H, d, J = 5.4 Hz), 7.54 (2H, q, J = 7.8 Hz), 7.13 (1H, t, J = 7.5 Hz), 6.91 (1H, t, J = 7.4 Hz), 6.18 (1H, d, J = 7.7 Hz). HRMS m/z 845.167 ([C42H28N4O4Ir]+ requires 845.173)
A mixture of [Ir(bt)2Cl]2 (150 mg, 116 μmol) and H2dcbpy (57 mg, 233 μmol) was suspended in methanol (20 mL) before excess sodium acetate (~0.5 g) was added. The suspension was heated at 80°C for 3 h, in the dark under an inert atmosphere (N2). The mixture was cooled to ambient temperature and concentrated under reduced pressure, then redissolved in minimum 5 : 1 water : ethanol (~200 mL) and filtered through Celite. Solution pH was adjusted to ~3 with 1 M HCl solution, and the solution cooled to 4°C and stirred for 2 h before the solid was collected by filtration. The precipitate was washed with water, diethyl ether and then recrystallised from hot ethanol to yield the product as a bright orange crystalline powder (159 mg, 80%). 1H NMR (400 MHz, [D6]DMSO): δ 9.05 (1H, s), 8.18 (1H, d, J = 8.1 Hz), 8.07 (1H, m), 8.01 (2H, m), 7.32 (1H, t, J = 7.7 Hz) 7.13 (1H, t, J = 7.5 Hz), 7.02 (1H, t, J = 7.6 Hz), 6.93 (1H, t, J = 7.5Hz), 6.26 (1H, d, J = 7.7 Hz), 6.06 (1H, d, J = 8.4 Hz). HRMS m/z 857.080 ([C38H24N4O4S2Ir]+ requires 857.086).
Calcium(II) framework synthesis
[Ir(ppy)2(Hdcbpy)] (2.0 mg, 0.00269 mmol) and Ca(NO3)2.4H2O (1.3 mg, 0.0538 mmol) were dissolved in a mixture of DMF and H2O (0.75/0.25 mL) and heated to 100°C for 48 h to yield yellow block crystals (4.1 mg, 89%). The yellow crystals were filtered, washed with DMF and air dried. Elemental analysis (%): calculated for C74H62CaIr2N10O12: C 51.99, H 3.77, N 8.41; found: C 51.96, H 3.696, N 8.41.
[Ir(piq)2(Hdcbpy)] (2.3 mg, 0.00269 mmol) and Ca(NO3)2.4H2O (1.3 mg, 0.0538 mmol) were dissolved in a mixture of DMF and H2O (0.75/0.25 mL) and heated to 100°C for 48 h to yield orange needles (3.2 mg, 62%). The orange crystals were filtered, washed with DMF and air dried. Elemental analysis (%): calculated for C84H50CaIr2N8O8·7.65(DMF): C 56.70, H 5.041, N 9.60; found: C 56.67, H 4.87, N 9.60.
[Ir(bt)2(Hdcbpy)] (2.3 mg, 0.00269 mmol), Hdcbpy (0.66 mg, 0.00269 mmol) and Ca(NO3)2.4H2O (1.3 mg, 0.0538 mmol) were dissolved in a mixture of DMF and H2O (0.75/0.25 mL) and heated to 100°C for 48 h to yield orange block crystals (4.7 mg, 82%). The orange crystals were filtered, washed with DMF and air dried. Elemental analysis (%): calculated for C30H20CaN4O7S2·0.97(DMF): C 50.99, H 3.09, N 7.58; found: C 51.01, H 3.078, N 7.58.
Optical spectroscopy
Solution spectra were measured using dichloromethane solutions of iridium(III) metalloligands (25 μM); absorbance and emission spectra were collected using a Cary 300 Bio UV−visible spectrophotometer or Cary Eclipse fluorescence spectrometer respectively. Metalloligands and calcium(II) MOFs were mounted on quartz slides and solid-state emission spectra collected using a Cary Eclipse fluorescence spectrometer with the appropriate attachment. All emission spectra were corrected for the variation in instrument sensitivity over the wavelength range of the measurements, using a correction factor established using a quartz halogen tungsten lamp of standard spectral irradiance. Spectra are the average of three successive scans, with the photomultiplier tube voltage adjusted between 400 and 700 V to ensure sufficient signal.
Supplementary material
1H NMR spectra, mass spectra, powder XRD, crystallographic and structural details, thermogravimetric analysis, absorbance spectroscopy and ATR-FTIR spectra. The crystallographic information files can be obtained from the Cambridge Structural Database: CCDC 2270980–2270982. Supplementary material is available online.
Acknowledgements
This paper was as an invited contributribution for the 2023 Awards Special Issue. Dr Connell was awarded the 2021 Organometallic Award. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.
References
1 Allendorf MD, Bauer CA, Bhakta RK, Houk RJT. Luminescent metal–organic frameworks. Chem Soc Rev 2009; 38: 1330-1352.
| Crossref | Google Scholar | PubMed |
2 Shu Y, Ye Q, Dai T, Xu Q, Hu X. Encapsulation of luminescent guests to construct luminescent metal–organic frameworks for chemical sensing. ACS Sensors 2021; 6: 641-658.
| Crossref | Google Scholar | PubMed |
3 Liu Y, Xie X-Y, Cheng C, Shao Z-S, Wang H-S. Strategies to fabricate metal–organic framework (MOF)-based luminescent sensing platforms. J Mater Chem C 2019; 7(35): 10743-10763.
| Crossref | Google Scholar |
4 Cui Y, Yue Y, Qian G, Chen B. Luminescent functional metal–organic frameworks. Chem Rev 2012; 112: 1126-1162.
| Crossref | Google Scholar | PubMed |
6 You Y, Cho S, Nam W. Cyclometalated iridium(III) complexes for phosphorescence sensing of biological metal ions. Inorg Chem 2014; 53: 1804-1815.
| Crossref | Google Scholar | PubMed |
7 Mills IN, Porras JA, Bernhard S. Judicious design of cationic, cyclometalated Ir(III) complexes for photochemical energy conversion and optoelectronics. Acc Chem Res 2018; 51: 352-364.
| Crossref | Google Scholar | PubMed |
8 Pitre SP, Overman LE. Strategic use of visible-light photoredox catalysis in natural product synthesis. Chem Rev 2022; 122: 1717-1751.
| Crossref | Google Scholar | PubMed |
9 Galliher MS, Roldan BJ, Stephenson CRJ. Evolution towards green radical generation in total synthesis. Chem Soc Rev 2021; 50: 10044-10057.
| Crossref | Google Scholar | PubMed |
10 Connell TU, Donnelly PS. Labelling proteins and peptides with phosphorescent d6 transition metal complexes. Coord Chem Rev 2018; 375: 267-284.
| Crossref | Google Scholar |
11 Xu G-X, Mak EC-L, Lo KK-W. Photofunctional transition metal complexes as cellular probes, bioimaging reagents and phototherapeutics. Inorg Chem Front 2021; 8: 4553-4579.
| Crossref | Google Scholar |
12 Li J, Djurovich PI, Alleyne BD, Yousufuddin M, Ho NN, Thomas JC, Peters JC, Bau R, Thompson ME. Synthetic control of excited-state properties in cyclometalated Ir(III) complexes using ancillary ligands. Inorg Chem 2005; 44: 1713-1727.
| Crossref | Google Scholar | PubMed |
13 Lowry MS, Bernhard S. Synthetically tailored excited states: phosphorescent, cyclometalated iridium(III) complexes and their applications. Chem Eur J 2006; 12: 7970-7977.
| Crossref | Google Scholar | PubMed |
16 Kim J-H, Kim S-Y, Cho Y-J, Son H-J, Cho DW, Kang SO. A detailed evaluation for the non-radiative processes in highly phosphorescent iridium(III) complexes. J Phys Chem C 2018; 122: 4029-4036.
| Crossref | Google Scholar |
17 DiLuzio S, Mdluli V, Connell TU, Lewis J, VanBenschoten V, Bernhard S. High-throughput screening and automated data-driven analysis of the triplet photophysical properties of structurally diverse, heteroleptic iridium(III) complexes. J Am Chem Soc 2021; 143: 1179-1194.
| Crossref | Google Scholar | PubMed |
18 You Y, Park SY. Phosphorescent iridium(III) complexes: toward high phosphorescence quantum efficiency through ligand control. Dalton Trans 2009; 1267-1282.
| Crossref | Google Scholar | PubMed |
19 Rota Martir D, Zysman-Colman E. Supramolecular iridium(III) assemblies. Coord Chem Rev 2018; 364: 86-117.
| Crossref | Google Scholar |
20 Ho ML, Chen YA, Chen TC, Chang PJ, Yu YP, Cheng KY, Shih CH, Lee GH, Sheu HS. Synthesis, structure and oxygen-sensing properties of iridium(III)-containing coordination polymers with different cations. Dalton Trans 2012; 41: 2592-2600.
| Crossref | Google Scholar | PubMed |
21 Huang S-L, Liu N, Ling Y, Luo H-K. IrIII-based octahedral metalloligands derived primitive cubic frameworks for enhanced CO2/N2 separation. Chem Asian J 2017; 12: 3110-3113.
| Crossref | Google Scholar | PubMed |
22 Li L, Zhang S, Xu L, Wang J, Shi L-X, Chen Z-N, Hong M, Luo J. Effective visible-light driven CO2 photoreduction via a promising bifunctional iridium coordination polymer. Chem Sci 2014; 5: 3808-3813.
| Crossref | Google Scholar |
23 Xu Y, Li L, Zhang S, Zhao S, Luo J. Three highly fluorescent iridium(III) unit based coordination polymers: coordinated solvent-dependent photoluminescence. Cryst Growth Des 2015; 16: 406-411.
| Crossref | Google Scholar |
24 Li L, Zhang S, Xu L, Han L, Chen ZN, Luo J. An intensely luminescent metal–organic framework based on a highly light-harvesting dyclo-metalated iridium(III) unit showing effective detection of explosives. Inorg Chem 2013; 52: 12323-12325.
| Crossref | Google Scholar | PubMed |
25 Li L, Zhang S, Xu L, Chen Z-N, Luo J. Highly sensitized near-infrared luminescence in Ir–Ln heteronuclear coordination polymers via light-harvesting antenna of Ir(III) unit. J Mater Chem C 2014; 2: 1698-1703.
| Crossref | Google Scholar |
26 Wang C, deKrafft KE, Lin W. Pt Nanoparticles@photoactive metal–organic frameworks: efficient hydrogen evolution via synergistic photoexcitation and electron injection. J Am Chem Soc 2012; 134: 7211-7214.
| Crossref | Google Scholar | PubMed |
27 Qiu L, Dong A, Zhang S, Wang S, Chang Z, Lu Y, Sui Z, Feng L, Chen Q. Fluorinated phenylpyridine iridium(III) complex based on metal–organic framework as highly efficient heterogeneous photocatalysts for cross-dehydrogenative coupling reactions. J Mater Sci 2020; 55: 9364-9373.
| Crossref | Google Scholar |
28 Zhu Y-Y, Lan G, Fan Y, Veroneau SS, Song Y, Micheroni D, Lin W. Merging photoredox and organometallic catalysts in a metal–organic framework significantly boosts photocatalytic activities. Angew Chem Int Ed 2018; 57: 14090-14094.
| Crossref | Google Scholar | PubMed |
29 Mo J-T, Wang Z, Zhu C-Y, Zhang Y, Pan M. Switching from oxygen quenching resistance to linear response by smart luminescent iridium(III)-based metal–organic frameworks. ACS Appl Mater Interfaces 2022; 14: 41208-41214.
| Crossref | Google Scholar | PubMed |
30 Lan G, Fan Y, Shi W, You E, Veroneau SS, Lin W. Biomimetic active sites on monolayered metal–organic frameworks for artificial photosynthesis. Nat Catal 2022; 5: 1006-1018.
| Crossref | Google Scholar |
31 Prajapati MJ, Yadav RAK, Swayamprabha SS, Dubey DK, Solanki JD, Jou J-H, Surati KR. Highly efficient solution-processed deep-red organic light-emitting diodes based on heteroleptic Ir(III) complexes with effective heterocyclic Schiff base as ancillary ligand. Org Electron 2020; 86: 105885.
| Crossref | Google Scholar |
32 Giordano PJ, Bock CR, Wrighton MS, Interrante LV, Williams RFX. Excited state proton transfer of a metal complex: determination of the acid dissociation constant for a metal-to-ligand charge transfer state of a ruthenium(II) complex. J Am Chem Soc 1977; 99(9): 3187-3189.
| Crossref | Google Scholar |
33 Qu P, Meyer GJ. Proton-controlled electron injection from molecular excited states to the empty states in nanocrystalline TiO2. Langmuir 2001; 17(21): 6720-6728.
| Crossref | Google Scholar |
34 Colombo MG, Hauser A, Guedel HU. Evidence for strong mixing between the LC and MLCT excited states in bis(2-phenylpyridinato-C2,N′)(2,2′-bipyridine)iridium(III). Inorg Chem 1993; 32(14): 3088-3092.
| Crossref | Google Scholar |
35 McPhillips TM, McPhillips SE, Chiu H-J, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-ice and the distributed control system: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 2002; 9: 401-406.
| Crossref | Google Scholar | PubMed |
36 Aragão D, Aishima J, Cherukuvada H, Clarken R, Clift M, Cowieson NP, Ericsson DJ, Gee CL, Macedo S, Mudie N, Panjikar S, Price JR, Riboldi-Tunnicliffe A, Rostan R, Williamson R, Caradoc-Davies TT. MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J Synchrotron Radiat 2018; 25: 885-891.
| Crossref | Google Scholar | PubMed |
37 Sheldrick GM. SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallogr A 2015; 71: 3-8.
| Crossref | Google Scholar | PubMed |
38 Sheldrick GM. Crystal structure refinement with SHELXL. Acta Crystallogr C 2015; 71: 3-8.
| Crossref | Google Scholar | PubMed |
39 Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr 2009; 42(2): 339-341.
| Crossref | Google Scholar |