

Assembly, Structure, and Properties of Six Coordination Polymers Based on 1,3,5-Tri-4-pyridyl-1,2-ethenylbenzene
Chen Cao A B , Tian-Yi Gu A , Jian-Guo Zhang A , Ming Dai C , Chun-Yan Ni A E , Zhi-Gang Yao D E and Jian-Ping Lang A B EA College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, China.
B State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China.
C Suzhou Clean Environment Institute, Jiangsu Sujing Group Co., Ltd, Suzhou 215122, China.
D Analysis and Testing Center, Soochow University, Suzhou 215123, China.
E Corresponding authors. Email: chunyan89.ok@163.com; zhgyao@suda.edu.cn; jplang@suda.edu.cn
Australian Journal of Chemistry 72(10) 751-761 https://doi.org/10.1071/CH19178
Submitted: 20 April 2019 Accepted: 31 May 2019 Published: 21 June 2019
Abstract
Six coordination polymers including [Cd(tpeb)X2]n (1: X = Br, 2: X = I), [Cu(tpeb)I]n (3), [Cd2I2(tpeb)2(1,4-bdc)]n (4), {[Co(tpeb)(dpa)]·MeCN}n (5), and {[Ni2(tpeb)3(oba)2]·solvent}n (6) were prepared from solvothermal reactions of 1,3,5-tri-4-pyridyl-1,2-ethenylbenzene (tpeb) with CdII, CuI, CoII, or NiII salts in the absence or presence of 1,4-benzenedicarboxylic acid (1,4-H2bdc), 2,2′-biphenyldicarboxylic acid (H2dpa), and 4,4′-oxybisbenzoic acid (H2oba). Compounds 1–3 have 1D chain structures while 4 holds a 2D wave-like network. Compound 5 adopts a 3D (3,5)-connected net with a Schläfli symbol of 33.48.513.63.7 and 6 possesses a 3D 5-connected net with a Schläfli symbol of 42.65.7.82. Compound 2 as a representative photocatalyst shows efficient degradation of rhodamine B in water while 5 displays highly selective sensing of p-nitrophenol in water through fluorescence quenching.
Introduction
In 1989, Richard Robson clearly defined the concept of coordination polymers (CPs) in which these compounds exhibit network structures of infinite periodicity, created by coordination bonds between organic ligands and metal ions.[1] Since then, research in this respect has gradually become one of the research hotspots in modern coordination chemistry and material chemistry.[2–7] At present, as a unique class of inorganic–organic hybrid material constructed from metal ions/clusters and organic bridging ligands, CPs have witnessed a rapid development in various scientific fields due to not only structural diversity and fascinating topologies but also potential applications in gas storage and separation, catalysis, luminescence, chemical sensing, and so on.[8–13] In recent years, various CPs have been employed to detect and degrade organic pollutants and hazardous substances in aqueous solutions.[14–17] As a new class of photocatalysts, CPs have been widely developed and utilised for the degradation of organic dyes under light irradiation due to their tunable metal centres, organic linkers, and their structures.[18–25] On the other hand, the popular detection of harmful materials basically relies on complicated instrumental methods such as ion mobility spectroscopy, X-ray dispersion, Raman spectroscopy, etc.[26–28] However, all these techniques are tedious, costly, and have limited portability and thus cheap and ready-to-use methods for the detection of dangerous compounds are greatly desired. Promising CP-based materials can supply a suitable route to tackle the above problems and have been extensively used for sensing organic pollutants with high sensitivity and selectivity.[29,30] To our knowledge, in spite of great efforts on the synthesis of CPs with good performances as photocatalysis or for chemical sensing, there is still a long way to go for these materials with the above functions being used in industry and even modern life.[31–33]
In the past decade, we have been interested in the rational assembly of functional CPs from multipyridyl ligands and their potential applications.[34,35] For example, a Y-shaped trivinyl-pyridyl ligand, 1,3,5-tri-4-pyridyl-1,2-ethenylbenzene (tpeb)[36,37] (Chart 1), was used to construct multidimensional CPs with various structures and properties.[38,39] Some M/tpeb/dicarboxylate CPs were prepared to selectively detect CrIII/CrVI species in water with very low detection limits[38] while others could be used to efficiently degrade congo red in water.[39] As an extension of the above project and our efforts on exploring more applications of metal complexes of the tpeb ligand, we performed its reactions with CdII (or CuI, CoII, NiII) salts in the absence or presence of 1,4-benzenedicarboxylic acid (1,4-H2bdc), 2,2′-biphenyldicarboxylic acid (H2dpa), or 4,4′-oxybisbenzoic acid (H2oba) under solvothermal conditions. Six coordination polymers, namely, [Cd(tpeb)X2]n (1: X = Br, 2: X = I), [Cu(tpeb)I]n (3), [Cd2I2(tpeb)2(1,4-bdc)]n (4), {[Co(tpeb)(dpa)]·MeCN}n (5), and {[Ni2(tpeb)3(oba)2]·solvent}n (6) were isolated and structurally characterised by elemental analysis, infrared spectra, thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD), and single-crystal X-ray diffraction. Their structures vary from 1D (1–3) to 2D (4) to 3D (5–6). Compounds 1–4 were employed as photocatalysts for the photodegradation of rhodamine B (RhB) in water, while 5 as a good luminescent probe was also investigated for sensing p-nitrophenol in water. Described below are their synthesis, structural characterisation, and properties.
|
Results and Discussion
Structure Description
[Cd(tpeb)X2]n (1: X = Br; 2: X = I) and [Cu(tpeb)I]n (3)
Complexes 1 and 2 are isostructural, and thus only the structure of 1 is described in detail. Complex 1 belongs to the triclinic Pī space group and its asymmetric unit consists of one [Cd(tpeb)Br2] molecule. Cd1 is four-coordinated by two N atoms (N2A and N1) from two different tpeb ligands and two distinct Br atoms (Cd1–? = 2.248(3)–2.264(3) Å and Cd1–Br = 2.538(6)–2.539(7) Å), which generates an irregular [CdN2Br2] tetrahedral geometry (Fig. 1a). The CdII ions are linked by tpeb ligands to give a 1D linear chain (Fig. 1a). The adjacent 1D chains are further interlinked by π–π interactions (4.238–4.258 Å) of pyridyl and phenyl rings to form a 3D supramolecular structure (Fig. S1, Supplementary Material).

|
Complex 3 also crystallises in the triclinic Pī space group and its asymmetric unit has two crystallographically independent [Cu(tpeb)I] molecules. Each CuI centre is three-coordinated by two N atoms (N3A and N1) from two different tpeb ligands and one I atom, (Cu1–? = 1.974(3)–1.997(3) Å, and Cu1–I = 2.4927(6) Å) (Fig. 1b). CuI units in 3 are interconnected to its equivalent ones by tpeb ligands to form a 1D chain approximately extending along the c axis. The two independent chains interdigitate each other through I⋯H–C interactions (3.143 Å) to a double H-bound chain (Fig. 1b). The neighbouring 1D chains are further interlinked by π–π interactions (3.499–3.716 Å) between pyridyl and phenyl rings to form a 3D supramolecular structure (Fig. 1b).
In all three cases, each tpeb has its third pyridyl group uncoordinated. The C=C bonds in adjacent tpeb ligands in 1 and 2 are found in both parallel alignment and anti-parallel alignment (Fig. S2, Supplementary Material). The distances between the parallel C=C bonds are 4.059 (1) and 4.138 (2) Å, while those between the anti-parallel C=C bonds are 3.542 (1) and 3.586 (2) Å. However, the C=C bonds in adjacent tpeb ligands in 3 are found to be all in an anti-parallel alignment with a distance of 3.752 Å. Unfortunately, many attempts to irradiate their single crystals using different light sources to provoke an expected [2 + 2] cycloaddition failed.
[Cd2I2(tpeb)2(1,4-bdc)]n (4)
Complex 4 crystallises in the triclinic Pī space group and its asymmetric unit contains half a [Cd2I2(tpeb)2(1,4-bdc)] unit. Cd1 is octahedrally coordinated by three atoms (N3B, N2A, and N1) from three different tpeb ligands, two O atoms (O1 and O2) from one 1,4-bdc2− ligand, and one I atom (Cd1–? = 2.362(3)–2.417(3) Å; Cd1–O = 2.278(3)–2.513(3) Å) (Fig. 2a). Two [CdI] units are bridged by a pair of tpeb ligands to form a rhombic [Cd2I2(tpeb)2] fragment. Each fragment is held together by edge-sharing to form a 1D cationic chain [Cd2I2(tpeb)2]n2n+ (Fig. 2b). Such a chain is interconnected to its equivalent ones by 1,4-bdc2− ligands to afford a 2D wave-like network (Fig. 2c). In this case, each tpeb ligand works as a 3-conneting bridge. The C=C bonds in adjacent tpeb ligands in 4 are found to be in a parallel arrangement and the distances between the parallel C=C bonds are 4.623, 5.368, and 5.738 Å, which exclude the possibility of photochemical [2 + 2] cycloaddition in 4.[40]

|
{[Co(tpeb)(dpa)]·MeCN}n (5)
Complex 5 crystallises in the monolinic P21/c space group and its asymmetric unit consists of one [Co(tpeb)(dpa)] molecule and one lattice MeCN molecule. Co1 is octahedrally coordinated by three carboxylate O atoms (O1, O2, and O4C) from two different bpa2− ligands and three N atoms (N1, N2B, and N3A) from three distinct tpeb ligands (Co1–? = 2.139(3)–2.173(3) Å and Co1–O = 2.033(3)–2.316(3) Å) (Fig. 3a). The CoII ions are linked by bpa2− ligands to generate the spiral chains extending approximately along the b axis (Fig. 3b). These spiral chains are further interconnected by tpeb ligands to afford a 3D framework (Fig. 3c). If each Co centre and each tpeb ligand are treated as a five-connected node and a three-connected node, respectively, the topology of 5 can be considered as a 3D (3,5)-connected net with a Schläfli symbol of 33.48.513.63.7 (Fig. 3d). The accessible volume of 5, calculated by PLATON,[41] is 451.4 Å3 (12.6 %) per unit cell volume when the MeCN solvent molecules are removed. The C=C bonds in adjacent tpeb ligands in 5 are in a parallel alignment. The distances between the parallel C=C bonds are 3.879 and 3.951 Å, which are within the requirements of a photochemical cycloaddition. However, 5 remained intact when it was exposed to UV light for several days.

|
{[Ni2(tpeb)3(oba)2]·solvent}n (6)
Complex 6 crystallises in the trigonal P3221 space group and its asymmetric unit contains half a [Ni2(tpeb)3(oba)2] molecule. Ni1 is also octahedrally coordinated by three carboxylate O atoms (O1A, O2, and O5B) from two different oba2− ligands and three N atoms (N1, N2A, and N4) from three distinct tpeb ligands (Ni1–? = 2.067(8)–2.114(7) Å and Ni1–O = 2.022(6)–2.152(6) Å) (Fig. 4a). The NiII ions are bridged by oba2− ligands to yield a spiral [Ni(oba)]n chain extending approximately along the b axis (Fig. 4b). Such spiral chains are linked by the tpeb ligands to generate a 3D framework (Fig. 4c). When each Ni centre is considered as a five-connecting node, the topology of 6 can be considered as a 3D 5-connected net with a Schläfli symbol of (42.65.7.82) (Fig. 4d). The accessible volume for 6, calculated by PLATON, is 769.0 Å3 (10.9 %) per unit cell volume when the solvent molecules are removed. The C=C bonds of adjacent tpeb ligands in 6 are in an anti-parallel alignment. The distances between the parallel C=C bonds are 3.717 and 3.951 Å, which did not provoke a photodimerisation between tpeb ligands upon UV light irradiation.

|
PXRD and Thermal Properties
In order to confirm the phase purities of complexes 1–6, PXRD was carried out on their polycrystalline power samples. The measured PXRD patterns were consistent with the simulated ones obtained from their corresponding crystal data, implying high phase purity of the bulk samples of 1–6 (Fig. S3, Supplementary Material). We also soaked each of the solid samples of 1–6 in water for 3 days and then centrifuged the remaining solid, which was washed with Et2O and dried under vacuum. The PXRD patterns of each remaining solid sample were consistent with those simulated from its single crystal data (Fig. S3, Supplementary Material). In addition, 1H NMR spectroscopy did not show any signals from the ligand (tpeb) and the related carboxylic acid. All these results revealed that CPs 1–6 have good stability in water and could be used for the applications described later in this article. To investigate the thermal stability of 1–6, thermogravimetric analysis (TGA) was performed under nitrogen atmosphere from 20 to 800°C. The results revealed that the complexes remained stable up to 388 (1), 393 (2), 395 (3), 248 (4), 310 (5), and 352°C (6) respectively (Figs S4 and S5, Supplementary Material).
Optical Properties
In order to investigate the degradation of organic dyes using complexes 1–4 as photocatalysts, we investigated their fluorescence properties in the solid state at room temperature (Fig. S6, Supplementary Material). Upon excitation at 380 nm, tpeb displayed an emission maximum at 416 nm, which may be attributed to a π*→π transition.[42] The photoluminescence emission bands of 1, 2, and 4 are 505, 475, and 495 nm (λex = 405 (1), 380 (2), and 395 nm (4)), respectively. However, complex 3 exhibited very weak fluorescence and could hardly be detected. In contrast to the tpeb ligand, the luminescent emission of 1, 2, and 4 presented a blue shift with a decreasing emission intensity, which may be ascribed to the heavy-atom effect from halogen atoms.[43] Complex 5 and H2bda in the solid state gave an emission band with maxima at 475 and 440 nm upon excitation at 320 nm, respectively (Fig. S7, Supplementary Material). Compared with that of the free tpeb, the emission band of 5 was remarkably red-shifted, which was probably due to the intraligand charge transition of tpeb.[44]
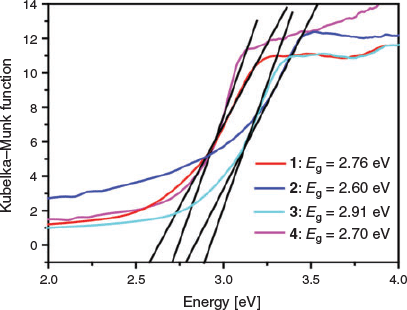
To evaluate the bandgaps, the optical diffuse reflection spectra of 1–4 were carried out at room temperature. The absorption data (α/S) were estimated from the reflectance based on the Kubelka–Munk equation (Eqn 1).[45]

In Eqn 1, α and S represent the absorption coefficient and scattering coefficient, respectively, and R equals the reflectivity at a given energy. The bandgap energy (Eg) values of 1–4 are 2.76 (1), 2.60 (2), 2.91 (3), and 2.70 eV (4), respectively, indicating that these complexes are potential photocatalysts for the decomposition of organic pollutants (Fig. 5).
|
Photocatalytic Activities
As we know, organic dyes such as RhB employed widely in the textile industry have been found to be harmful, stable, and hard to biodegrade.[14,15] Thus, it is imperative to breakdown these dye molecules into small non-toxic molecules or ions to minimise their environment pollution. In this work, the photocatalytic efficiencies of 1–4 were examined by decomposing a model organic dye, RhB, under UV-vis light irradiation at ambient temperature. The degradation process was characterised by the intensity of the characteristic ultraviolet absorption peak of RhB (553 nm). In addition, the simple photolysis experiment was also performed under the same conditions without any catalyst. The results revealed that only 30 % of RhB was degraded after 4 h when no catalyst existed in the system (Fig. S8, Supplementary Material). For the photodegradation of RhB in the presence of catalyst, the complete degradation times were 150 min for 1 (Fig. 6a), 105 min for 2 (Fig. 6b), and 210 min for 4 (Fig. 6d), respectively. However, the RhB was only degraded 15 % for complex 3 after 7 h (Fig. 6c), implying that 3 had no catalytic activity towards the decomposition of RhB. Thus compound 2 showed the best performance among these compounds.

|
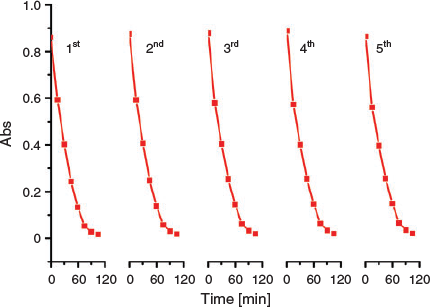
Compound 2 was chosen as a representative catalyst to check its recyclability by successive runs in the photocatalytic decomposition of RhB. It was isolated from the RhB degradation reaction system by centrifugation, washed and dried in air, and re-used in the next degradation run. After five runs for the RhB degradation, the photocatalytic degradation performance of 2 remained almost unchanged (Fig. 7). PXRD patterns of 2 obtained after five runs were consistent with those derived from its crystal data (Fig. S9, Supplementary Material), suggesting that its skeleton structure remained intact in the catalytic process and it could be used for repeated RhB degradation. The colour of the RhB solution faded after the degradation reaction. The colourless solution was extracted by ether and analysed by gas chromatography (GC). Notably, there were almost no organic species in the clarified solution. Therefore, it is assumed that RhB may be converted into CO2 and H2O after photocatalytic degradation.[46]
|
The mechanism for dye degradation using CP-based catalysts has been reported.[47,48] Generally, upon UV light irradiation, the electrons from the valence band of CPs are excited to transport to the conduction band, yielding a large number of holes (h+) to form corresponding electron (e−)–hole (h+) pairs. Meanwhile, O2 binds with e− to form O2−, which further binds with water to afford highly active •OH radicals. In addition, the interaction between the rest of H+ and OH− also yields radical •OH after excitation. During the degradation of a dye, these •OH radicals can act as active species to promote the reaction. In our cases, the photocatalytic efficiency of 1–4 may be related to the energy difference between the energy values of the irradiating light (λ = 365 nm, hv = 3.40 eV) and the bandgap values (2.76 (1), 2.60 (2), 2.91 (3), and 2.70 eV (4)). In contrast to the structures of 1 and 2, both of their skeleton structures are very similar, except for halogen atoms. Therefore, the photocatalytic activities of 1 and 2 may be related to their halogen atoms. Compared with 2–4, the central metal of the complexes may affect their photocatalytic activities.
Luminescent Sensing of p-Nitrophenol
In recent years, the selective detection of explosives, especially nitroaromatics, has attracted much attention due to homeland security, forensic science, and environment safety.[49] p-Nitrophenol (PNP) is one of the representative nitroaromatic explosives and it is of great importance that sensing PNP can be performed in a sensitive and rapid way.[50] In this work, we chose 5 as a representative complex to check its capability for luminescent sensing of PNP in water. The luminescence intensity of 5 can be gradually quenched with the addition of PNP aqueous solution to its suspension. To further explore the sensitivity of its sensing for PNP, the emissive responses were recorded by steadily increasing PNP contents in the emulsion of 5 dispersed in water. When the addition of PNP was increased to 40 μL, the emission intensities of the suspension were almost fully quenched (≥ 80 %) (Fig. 8a). Such a quenching manner was also observed even with the addition of 1 μL of PNP. The quenching efficiency can be quantitatively analysed using the Stern–Volmer (SV) equation, (I0/I) = KSV[M] + 1, where I0 and I are the fluorescence intensities before and after the addition of the analyte, respectively, KSV is the quenching constant, and [M] is the concentration of the analyte.[51] As shown in Fig. 8b, the relative luminescence intensity (I0/I) exhibited a good linear correlation with PNP content in the range of 0–70 μL, with R2 = 0.9904. The Stern–Volmer constants (KSV) were calculated to be 0.2537 ppm−1. In addition, the detection limit calculated by the formula 3σ/k (k: slope, σ: standard error) is ~0.0821 ppm, revealing the higher sensing sensitivity of 5 towards PNP relative to those observed in the literature.[52]

|
The possible sensing mechanism of 5 towards PNP was also examined. The UV-vis spectrum of PNP exhibited absorption bands in the range of 300 to 375 nm, which suggested that the competition between the absorption of PNP and the excitation of 5 (λex 320 nm) may be responsible for the selective quenching behaviour. The nitro group of PNP is a typical electron withdrawing substituent. Thus, when the sample of 5 dispersed in the PNP solution was irradiated by light with certain energy, the excited electron of 5 might be transported to PNP, thereby resulting in the fluorescence quenching phenomenon. According to the aforementioned analysis, the selective fluorescence sensing of 5 might be ascribed to both competition absorption and an electron transfer (ET) mechanism.[52]
Conclusions
In summary, six new transition metal coordination polymers 1–6 based on the tpeb ligand were prepared under solvothermal conditions. They showed interesting structural diversity from a 1D chain (1–3) to a 2D wave-like layer (4), to a 3D (3,5)-connected net (5) or 3D 5-connected net (6). Compounds 1, 2, and 4 showed good photocatalytic performances in the degradation of RhB in water. The representative photocatalyst 2 maintained a good photodegradation activity even after five cycles. Compound 5 was demonstrated to be a good luminescent probe for sensing PNP in water through fluorescence quenching. The results provide an interesting insight into the assembly of functional CPs and their potential applications in solving environment pollution.
Experimental
Materials and General Methods
Chemical reagents were obtained commercially and used as received without further purification. The tpeb ligand was prepared according to the literature method.[53] Elemental analyses (C, H, and N) were performed using a Carlo-Erba CHNO-S microanalyzer. PXRD measurements were recorded on a PANalytical X’PertPRO MPD system (PW3040/60). The simulated PXRD spectra were from the single crystal data and diffraction-crystal module of the Mercury (Hg) program available free of charge. IR spectra were obtained over 4000–400 cm−1 on a Nicolet IS-10 spectrometer with KBr pellets. TGA was carried out on a TA SDT-600 analyzer at a heating rate of 5°C min−1. UV-Vis absorption spectra were measured on a Varian Cary-50 spectrophotometer. The photoluminescence spectra and quantum yields were collected on a HORIBA PTI QuantaMaster 40 Spectrofluorometer.
Preparation of [Cd(tpeb)Br2]n (1)
To a thick Pyrex tube (10 mL) was added a mixture of tpeb (19.4 mg, 0.05 mmol) and CdBr2·4H2O (34.4 mg, 0.1 mmol) in 3 mL of H2O and MeCN (v/v, 4 : 1). The tube was then sealed and heated at 150°C for 24 h. After cooling to room temperature (RT) at a rate of 7°C h−1, pale yellow plates were obtained in 59 % yield (based on Cd). Anal. Calc. for C27H21N3Br2Cd: C 49.16, H 3.21, N 6.37. Found: C 50.12, H 3.61, N 5.98 %. νmax (KBr)/cm−1 3045 (m), 1775 (m), 1688 (s), 1618 (m), 1576 (m), 1441 (s), 1325 (m), 1189 (m), 1009 (s), 837 (m), 756 (s), 671 (m), 530 (m).
Preparation of [Cd(tpeb)I2]n (2)
Complex 2 was synthesised by a similar procedure to that used for the preparation of 1, except that CdBr2·4H2O (34.4 mg, 0.1 mmol) was replaced by CdI2 (36.6 mg, 0.1 mmol). Deep yellow plates were obtained in 46 % yield (based on Cd). Anal. Calc. for C27H21N3I2Cd: C 43.03, H 2.81, N 5.58. Found: C 43.72, H 3.09, N 5.91 %. νmax (KBr)/cm−1 3010 (m), 1748 (m), 1609 (s), 1512 (s), 1411 (m), 1401 (s), 1290 (m), 1175 (m), 1053 (s), 992 (m), 857 (m), 826 (s), 538 (s).
Preparation of [Cu(tpeb)I]n (3)
Under N2 atmosphere, a thick Pyrex tube (10 mL) containing a mixture of tpeb (19.4 mg, 0.05 mmol), CuI (19.0 mg, 0.1 mmol), and iodobenzene (0.5 mL) in 3 mL of H2O and MeCN (v/v, 4 : 1) was sealed and heated at 90°C for 72 h. After cooling to RT at a rate of 7°C h−1, orange plates were obtained in 67 % yield (based on Cu). Anal. Calc. for C27H21N3ICu: C 56.11, H 3.66, N 7.27. Found: C 55.60, H 4.13, N 7.32 %. νmax (KBr)/cm−1 3036 (m), 1741 (m), 1601 (s), 1509 (s), 1461 (m), 1366 (m), 1221 (s), 1149 (s), 1065 (s), 1033 (m), 953 (s), 907 (s), 724 (s), 679 (m), 509 (s).
Preparation of [Cd2I2(tpeb)(1,4-bdc)]n (4)
Complex 4 was synthesised by a similar procedure to that used for the preparation of 1, except that CdBr2·4H2O (34.4 mg, 0.1 mmol) was replaced by 1,4-H2bdc (16.6 mg, 0.1 mmol) and CdI2 (36.6 mg, 0.1 mmol). Yellow columnar crystals were obtained in 61 % yield (based on Cd). Anal. Calc. for C31H23N3O2ICd: C 52.53, H 3.27, N 5.93. Found: C 52.53, H 3.27, N 5.93 %. νmax (KBr)/cm−1 3040 (m), 1746 (m), 1603 (s), 1505 (s), 1456 (m), 1371(s), 1218 (s), 1141 (s), 1062 (s), 1029 (s), 959 (m), 901 (s), 853 (s), 737 (s), 657 (m), 524 (s).
Preparation of {[Co(tpeb)(dpa)]·MeCN}n (5)
A thick Pyrex tube (14 mL) containing a mixture of tpeb (38.8 mg, 0.1 mmol), H2dpa (24.2 mg, 0.1 mmol), and Co(NO3)2·6H2O (29.1 mg, 0.1 mmol) in 6 mL of H2O and MeCN (v/v, 2 : 1) was sealed and heated at 150°C for 2 h. After cooling to RT at a rate of 6°C h−1, red block crystals were obtained in 46 % yield (based on Co). Anal. Calc. for C43H32N4O4Co: C 70.97, H 4.43, N 7.70. Found: C 71.05, H 4.50, N 7.75 %. νmax (KBr)/cm−1 3423 (m), 3034 (w), 2240 (w), 1606 (s), 1554 (m), 1500 (m), 1399 (m), 1360 (s), 1203 (m), 1150 (w), 1100 (w), 1066 (w), 1014 (m), 1004 (m), 959 (m), 951 (m), 862 (m), 844 (m), 812 (w), 751 (m), 723 (m), 677 (m), 586 (w), 542 (m).
Preparation of {[Ni2(tpeb)3(oba)2]·solvent}n (6)
Complex 6 was synthesised by a similar procedure to that used for the preparation of 5, except that tpeb (38.8 mg, 0.1 mmol), H2dpa (24.2 mg, 0.1 mmol), and Co(NO3)2·6H2O (29.1 mg, 0.1 mmol) was replaced by tpeb (58.4 mg, 0.15 mmol), H2oba (25.8 mg, 0.1 mmol), and Ni(NO3)2·6H2O (29.1 mg,0.1 mmol). Light blue block crystals were obtained in 34 % yield (based on Ni). Anal. Calc. for C109H79N9O10Ni2: C 73.05, H 4.44, N 7.03. Found: C 73.33, H 4.56, N 7.18 %. νmax (KBr)/cm−1 3442 (m), 3031 (w), 1620 (s), 1602 (m), 1571 (w), 1550 (w), 1527 (w), 1494 (w), 1415 (m), 1370 (s), 1222 (s), 1160 (m), 1091 (w), 1064 (w), 1014 (w), 991 (m), 872 (w), 844 (w), 799 (w), 778 (w), 764 (w), 710 (w), 676 (w), 642 (w), 534 (w), 523 (w).
X-Ray Crystallographic Study
Single crystals of 1–6 suitable for X-ray analysis were obtained directly from the above procedures. All crystal data were collected on an Agilent Technologies Gemini A Ultra CCD and Rigaku Saturn 724 + CCD X-ray diffractometers with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) by ω scan mode. All structures were solved by direct methods and refined by full-matrix least-squares methods through the SHELXTL program.[54] The non-hydrogen atoms were located in successive difference Fourier syntheses and refined with anisotropic thermal parameters on F2. All hydrogen atoms of ligands were generated theoretically onto the specific atoms and refined isotropically with fixed thermal factors. For 6, one 4-pyridyl-1,2-ethenyl group (C39–C40–C41–C42–C43–C44–C45–N5) of tpeb was disordered over two positions by rotating about the centre of C36–C38 with an equal occupancy factor. The lattice solvent molecules in the channel of 6 were highly disordered and were removed by the SQUEEZE program in PLATON.[41] A summary of the pertinent crystal data and structural refinements for 1–6 is provided in Table 1. The selected bond lengths and angles for 1–6 are given in Table S1 (Supplementary Material).

|
Photocatalytic Activity Measurements
The photocatalytic activities of as-synthesised samples of complexes 1–4 were evaluated by the degradation of a model organic dye RhB under irradiation of a 400 W Hg lamp (365 nm). Each as-synthesised crystalline sample was obtained from the above preparation, which was firmly ground into a powder. In a typical process, 20 mg of a powder sample as a photocatalyst was added into 20 mL of RhB aqueous solution (0.14 g L−1). The RhB aqueous solution was stirred for 0.5 h in the dark before irradiation to reach adsorption equilibrium between the catalyst and solution. The mixture was then exposed to a 400 W Hg lamp (365 nm). The lamp and each sample were put in a water bath while the temperature was kept lower than 35°C to avoid any temperature effect. At regular time intervals, 1 mL of suspension was continually taken out of the reaction cell and filtered. The filtrate was diluted to 10 mL by deionised water. The resulting solution was analysed on a Varian Cary-50 UV-vis spectrophotometer.
Photoluminescence Sensing of PNP
A uniform and stable suspension of the model complex 5 was prepared by grinding its crystalline solid (2 mg) and suspending the powder in deionised water (2 mL) by ultrasound mixing for 1 h. An aqueous solution (1 g L−1) of PNP was injected into the suspension by a microsyringe and evenly mixed at room temperature. The luminescence intensities of the suspension were then measured on a spectrofluorometer at various time intervals.
Crystallographic Data
CCDC 1900658–1900661, 1902625, and 1902628 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Supplementary Material
The selected bond lengths and angles, TGA curves and as-synthesised and simulated PXRD patterns are available on the Journal’s website.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (Grant Nos. 21531006, and 21773163), the State Key Laboratory of Organometallic Chemistry of Shanghai Institute of Organic Chemistry (Grant No. 2018kf-05), the ‘Priority Academic Program Development’ of Jiangsu Higher Education Institutions, and the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201708). They are grateful for the useful comments and suggestions of the editor and the reviewers.
References
[1] B. F. Hoskins, R. Robson, J. Am. Chem. Soc. 1989, 111, 5962.| Crossref | GoogleScholarGoogle Scholar |
[2] S. R. Batten, R. Robson, Angew. Chem. Int. Ed. 1998, 37, 1460.
| Crossref | GoogleScholarGoogle Scholar |
[3] H. L. Li, M. Eddaoudi, M. O’Keeffe, O. M. Yaghi, Nature 1999, 402, 276.
| Crossref | GoogleScholarGoogle Scholar |
[4] B. F. Abrahams, S. R. Batten, M. J. Grannas, H. Hamit, B. F. Hoskins, R. Robson, Angew. Chem. Int. Ed. 1999, 38, 1475.
| Crossref | GoogleScholarGoogle Scholar |
[5] D. Liu, Z. G. Ren, H. X. Li, J. P. Lang, N. Y. Li, B. F. Abrahams, Angew. Chem. Int. Ed. 2010, 49, 4767.
| Crossref | GoogleScholarGoogle Scholar |
[6] B. Li, H. M. Wen, Y. Cui, W. Zhou, G. Qian, B. Chen, Adv. Mater. 2016, 28, 8819.
| Crossref | GoogleScholarGoogle Scholar | 27454668PubMed |
[7] X. H. Bu, J. L. Zuo, Sci. China Chem. 2016, 59, 927.
| Crossref | GoogleScholarGoogle Scholar |
[8] F. L. Li, Q. Shao, X. Huang, J. P. Lang, Angew. Chem. Int. Ed. 2018, 57, 1888.
| Crossref | GoogleScholarGoogle Scholar |
[9] T. Zhang, W. Lin, Chem. Soc. Rev. 2014, 43, 5982.
| Crossref | GoogleScholarGoogle Scholar | 24769551PubMed |
[10] F. L. Hu, Y. Mi, C. Zhu, B. F. Abrahams, P. Braunstein, J. P. Lang, Angew. Chem. Int. Ed. 2018, 57, 12696.
| Crossref | GoogleScholarGoogle Scholar |
[11] C. Cao, S. J. Liu, S. L. Yao, T. F. Zheng, Y. Q. Chen, J. L. Chen, H. R. Wen, Cryst. Growth Des. 2017, 17, 4757.
| Crossref | GoogleScholarGoogle Scholar |
[12] L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. V. Van Duyne, J. T. Hupp, Chem. Rev. 2012, 112, 1105.
| Crossref | GoogleScholarGoogle Scholar | 22070233PubMed |
[13] Z. C. Shao, M. J. Liu, J. Dang, C. Huang, W. J. Xu, J. Wu, H. W. Hou, Inorg. Chem. 2018, 57, 10224.
| Crossref | GoogleScholarGoogle Scholar |
[14] Q. Gao, J. Xu, X. H. Bu, Coord. Chem. Rev. 2019, 378, 17.
| Crossref | GoogleScholarGoogle Scholar |
[15] C. C. Wang, J. R. Li, X. L. Lv, Y. Q. Zhang, G. S. Guo, Energy Environ. Sci. 2014, 7, 2831.
| Crossref | GoogleScholarGoogle Scholar |
[16] F. Wang, F. L. Li, M. M. Xu, H. Yu, J. G. Zhang, H. T. Xia, J. P. Lang, J. Mater. Chem. A 2015, 3, 5908.
| Crossref | GoogleScholarGoogle Scholar |
[17] W. P. Lustig, S. Mukherjee, Z. N. Rudd, A. V. Desai, J. Li, S. K. Ghosh, Chem. Soc. Rev. 2017, 46, 3242.
| Crossref | GoogleScholarGoogle Scholar | 28462954PubMed |
[18] S. Li, Q. Xu, Energy Environ. Sci. 2013, 6, 1656.
| Crossref | GoogleScholarGoogle Scholar |
[19] S. Q. Li, Y. F. Chen, X. K. Pei, S. H. Zhang, X. Feng, J. W. Zhou, B. Wang, Chin. J. Chem. 2016, 34, 175.
| Crossref | GoogleScholarGoogle Scholar |
[20] M. M. Chen, H. X. Li, J. P. Lang, Eur. J. Inorg. Chem. 2016, 2508.
| Crossref | GoogleScholarGoogle Scholar |
[21] Y. Q. Chen, S. J. Liu, Y. W. Li, G. R. Li, K. H. He, Y. K. Qu, T. L. Hu, X. H. Bu, Cryst. Growth Des. 2012, 12, 5426.
| Crossref | GoogleScholarGoogle Scholar |
[22] X. W. Lei, C. Y. Yue, J. Q. Zhao, Y. F. Han, J. T. Yang, R. R. Meng, C. S. Gao, H. Ding, C. Y. Wang, W. D. Chen, M. C. Hong, Inorg. Chem. 2015, 54, 10593.
| Crossref | GoogleScholarGoogle Scholar | 26505902PubMed |
[23] D. X. Li, C. Y. Ni, M. M. Chen, M. Dai, W. H. Zhang, W. Y. Yan, H. X. Qi, Z. G. Ren, J. P. Lang, CrystEngComm 2014, 16, 2158.
| Crossref | GoogleScholarGoogle Scholar |
[24] Y. P. Wu, X. Q. Wu, J. F. Wang, J. Zhao, W. W. Dong, D. S. Li, Q. C. Zhang, Cryst. Growth Des. 2016, 16, 2309.
| Crossref | GoogleScholarGoogle Scholar |
[25] W. Meng, Z. Q. Xu, J. Ding, D. Q. Wu, X. Han, H. W. Hou, Y. T. Fan, Cryst. Growth Des. 2014, 14, 730.
| Crossref | GoogleScholarGoogle Scholar |
[26] M. M. Liu, G. Li, Z. H. Cheng, New J. Chem. 2015, 39, 8484.
| Crossref | GoogleScholarGoogle Scholar |
[27] Y. F. Zhang, X. J. Bo, A. Nsabimana, C. Han, M. Li, L. P. Guo, J. Mater. Chem. A 2015, 3, 732.
| Crossref | GoogleScholarGoogle Scholar |
[28] L. E. Kreno, N. G. Greenelth, O. K. Farha, J. T. Hupp, R. P. V. Duyne, Analyst 2014, 139, 4073.
| Crossref | GoogleScholarGoogle Scholar | 24949495PubMed |
[29] Y. Li, S. S. Zhang, D. T. Song, Angew. Chem. 2013, 125, 738.
| Crossref | GoogleScholarGoogle Scholar |
[30] S. L. Yao, S. J. Liu, X. M. Tian, T. F. Zheng, C. Cao, C. Y. Niu, Y. Q. Chen, J. L. Chen, H. P. Huang, H. R. Wen, Inorg. Chem. 2019, 58, 3578.
| Crossref | GoogleScholarGoogle Scholar | 30821447PubMed |
[31] T. Zhang, W. B. Li, Chem. Soc. Rev. 2014, 43, 5982.
| Crossref | GoogleScholarGoogle Scholar | 24769551PubMed |
[32] Y. B. Huang, J. Liang, X. S. Wang, R. Cao, Chem. Soc. Rev. 2017, 46, 126.
| Crossref | GoogleScholarGoogle Scholar | 27841411PubMed |
[33] B. L. Chen, S. C. Xiang, G. D. Qian, Acc. Chem. Res. 2010, 43, 1115.
| Crossref | GoogleScholarGoogle Scholar |
[34] D. Liu, J. P. Lang, B. F. Abrahams, J. Am. Chem. Soc. 2011, 133, 11042.
| Crossref | GoogleScholarGoogle Scholar | 21718000PubMed |
[35] F. L. Hu, Y. Mi, C. Zhu, B. F. Abrahams, P. Braunstein, J. P. Lang, Angew. Chem. Int. Ed. 2018, 57, 12696.
| Crossref | GoogleScholarGoogle Scholar |
[36] M. H. Mir, J. X. Ong, G. K. Kole, G. K. Tan, M. J. McGlinchey, Y. Wu, J. J. Vittal, Chem. Commun. 2011, 47, 11633.
| Crossref | GoogleScholarGoogle Scholar |
[37] Y. Liu, K. Q. Ye, Y. Wang, Q. C. Zhang, X. H. Bu, P. Y. Feng, Dalton Trans. 2017, 46, 1481.
| Crossref | GoogleScholarGoogle Scholar | 28074203PubMed |
[38] T. Y. Gu, M. Dai, D. J. Young, Z. G. Ren, J. P. Lang, Inorg. Chem. 2017, 56, 4668.
| Crossref | GoogleScholarGoogle Scholar |
[39] J. G. Zhang, W. J. Gong, Y. S. Guan, H. X. Li, D. J. Young, J. P. Lang, Cryst. Growth Des. 2018, 18, 6172.
| Crossref | GoogleScholarGoogle Scholar |
[40] G. M. J. Schmidt, J. Pure Appl. Chem. 1971, 27, 647.
| Crossref | GoogleScholarGoogle Scholar |
[41] A. L. Spek, J. Appl. Cryst. 2003, 36, 7.
| Crossref | GoogleScholarGoogle Scholar |
[42] S. L. Wang, F. L. Hu, J. Y. Zhou, Y. Zhou, Q. Huang, J. P. Lang, Cryst. Growth Des. 2015, 15, 4087.
| Crossref | GoogleScholarGoogle Scholar |
[43] Y. P. Xiang, Y. B. Zhao, N. Xu, S. L. Gong, F. Ni, K. L. Wu, J. J. Luo, G. H. Xie, Z. H. Lu, C. L. Yang, J. Mater. Chem. C 2017, 5, 12204.
| Crossref | GoogleScholarGoogle Scholar |
[44] M. D. Allendorf, C. A. Bauer, R. K. Bhakta, R. J. T. Houk, Chem. Soc. Rev. 2009, 38, 1330.
| Crossref | GoogleScholarGoogle Scholar | 19384441PubMed |
[45] C. N. Lv, M. M. Chen, W. H. Zhang, D. X. Li, M. Dai, J. P. Lang, CrystEngComm 2015, 17, 1935.
| Crossref | GoogleScholarGoogle Scholar |
[46] T. T. Liu, T. T. Hu, C. J. Hu, J. P. Lang, Inorg. Chem. Commun. 2018, 90, 26.
| Crossref | GoogleScholarGoogle Scholar |
[47] C. C. Chen, W. H. Ma, J. C. Zhao, Chem. Soc. Rev. 2010, 39, 4206.
| Crossref | GoogleScholarGoogle Scholar |
[48] M. Li, L. Liu, L. Zhang, X. F. Lv, J. H. Ding, W. Hou, Y. T. Fan, CrystEngComm 2014, 16, 6408.
| Crossref | GoogleScholarGoogle Scholar |
[49] Z. C. Hu, B. J. Deibert, J. Li, Chem. Soc. Rev. 2014, 43, 5815.
| Crossref | GoogleScholarGoogle Scholar |
[50] G. Y. Wang, C. Song, D. M. Kong, W. J. Ruan, Z. Chang, Y. Li, J. Mater. Chem. A 2014, 2, 2213.
| Crossref | GoogleScholarGoogle Scholar |
[51] Y. T. Yan, W. Y. Zhang, F. Zhang, F. Cao, R. F. Yang, Y. Y. Wang, L. Hou, Dalton Trans. 2018, 47, 1682.
| Crossref | GoogleScholarGoogle Scholar | 29327748PubMed |
[52] X. J. Liu, X. Wang, J. L. Xu, D. Tian, R. Y. Chen, J. Xu, X. H. Bu, Dalton Trans. 2017, 46, 4893.
| Crossref | GoogleScholarGoogle Scholar | 28304037PubMed |
[53] A. J. Amoroso, A. M. W. Thompson, C. J. P. Maher, J. A. McCleverty, M. D. Ward, Inorg. Chem. 1995, 34, 4828.
| Crossref | GoogleScholarGoogle Scholar |
[54] C. B. Hubschle, G. M. Sheldrick, B. Dittrich, J. Appl. Cryst. 2011, 44, 1281.
| Crossref | GoogleScholarGoogle Scholar |