

A Series of Early Lanthanide Chloranilate Frameworks with a Square Grid Topology
Carol Hua A B C , Hui Min Tay A , Qilin He B and T. David Harris B C
A B C , Hui Min Tay A , Qilin He B and T. David Harris B C
A School of Chemistry, The University of Melbourne, Parkville, Vic. 3010, Australia.
B Department of Chemistry, Northwestern University, Evanston, IL 60208, USA.
C Corresponding authors. Email: carol.hua@unimelb.edu.au; dharris@northwestern.edu
Australian Journal of Chemistry 72(10) 778-785 https://doi.org/10.1071/CH19193
Submitted: 30 April 2019 Accepted: 7 June 2019 Published: 4 July 2019
Abstract
A series of lanthanide chloranilate frameworks containing a (4,4)-net with LaIII, CeIII, NdIII, SmIII, and EuIII have been synthesised and structurally characterised. Two structure types of square grids were obtained for these frameworks. Type 1 consists of the formula (Et4N)[Ln(can)2(H2O)] (Ln = LaIII, CeIII, NdIII; H2can = chloranilic acid) and crystallised in the tetragonal space group I4/m, featuring a nine-coordinate lanthanide ion with a coordinated water molecule and four chloranilate ligands. Type 2, (Et4N)[Ln(can)2] (SmIII and EuIII) crystallised in the I4/mcm space group, and contains an eight-coordinate lanthanide ion without a coordinated water molecule. A single-crystal-to-single-crystal transformation was carried out for (Et4N)[Nd(can)2(H2O)] on removal of the coordinated aqua ligand.
Introduction
Coordination polymers (CPs) and metal–organic frameworks (MOFs) have attracted significant research attention in the past three decades, becoming one of the fastest-growing areas in materials science.[1] The possibility of constructing an extended crystalline framework via the self-assembly of metal nodes with organic rods was proposed in seminal papers by Robson et al., who reported the first deliberate synthesis of a CP containing tetrahedral CuII nodes linked by 4,4′,4″,4″′-tetracyanotetraphenylmethane units.[2,3] The use of this rigid organic linker afforded a framework with open channels through which solvent molecules could freely diffuse, and it was recognised to be potentially useful for separation and catalysis. This pioneering work laid the foundation for the crystal engineering of an extensive family of CPs constructed from numerous combinations of organic linkers and metal nodes.[4] The flexibility in the choice of both the organic and inorganic components enables the structure and properties of the frameworks to be readily modified with a reasonably high degree of control.[5] These combined advantages of synthetic programmability and high structural porosity make CPs ideal materials for a wide range of applications, including gas storage and separation,[6–8] chiral recognition,[9–11] drug delivery,[12–14] and chemical sensing.[15,16]
Benzoquinoid molecules obtained from the deprotonation of 2,5-dihydroxy-1,4-benzoquinone (dhbq) and its 3,6-disubstituted derivatives are bis(bidentate) ligands containing anionic chelating groups, which form strong and predictable bonds to metal centres, thereby promoting the formation of robust porous coordination frameworks.[17–20] A notable feature of this family of ligands is the readiness with which they undergo redox chemistry to access multiple oxidation states; the −2 quinoid can undergo a one-electron reduction to a −3 radical semiquinoid or a two-electron reduction to a −4 catecholate. This presents the possibility of using benzoquinoid ligands as building units for novel redox-active frameworks. In addition, the radical semiquinoid form of the ligand has been shown to effectively mediate magnetic and electronic coupling between metal centres when serving as a bridging ligand,[17,21–25] fuelling a growing interest in the use of benzoquinoid ligands to construct materials that exhibit long-range magnetic order or electrical conductivity.[26–29] The potential of this approach was illustrated in a porous anionic framework containing FeIII bridged by chloranilate ligands in a mixture of −2 and −3 oxidation states, which simultaneously exhibits magnetic ordering below 80 K and electrical conductivity of up to electrical conductivity (σ) = 1.4(7) × 10−2 S cm−1.[27] Post-synthetic reduction of the chloranilate ligands in the framework to the −3 radical form resulted in a single-crystal-to-single-crystal transformation to yield an increased magnetic ordering temperature of 105 K, one of the highest reported for a porous solid.[30]
The combination of benzoquinoid-type ligands with transition metal ions gives rise to coordination frameworks with a range of different topologies. Commonly observed structural types include 1D zig-zag and linear chains,[31–35] honeycomb 2D sheets with a (6,3) topology,[36–41] and a (4,3) double chain.[42] The dominant topology in benzoquinoid-containing 3D frameworks is a (10,3)-a net, which consists of two interpenetrating chiral frameworks of opposite handedness.[18,24,43] A smaller number of studies have been undertaken on the synthesis of lanthanide–benzoquinoid frameworks, which are dominated by 2D sheets with a (6,3)-topology in which each lanthanide ion is coordinated to three chelating benzoquinoid-type ligands and two or three solvent molecules.[44–50] Varying degrees of distortion are obtained under different synthetic conditions, and this distortion alters the size and shape of the cavities. Less common topologies, including a (4,3)-double chain and 3D diamond-like framework, have also been observed.[42,47,53]
Another unusual structure type accessible to lanthanide-benzoquinoid frameworks is an anionic 2D square grid framework.[49,51] Unlike in the previously reported (6,3) frameworks, each lanthanide ion connects four chelating and bridging benzoquinoid ligands to afford a (4,4)-net. A series of square grid frameworks with the general composition (Et4N)[MIII(C6O4X2)2] have been reported with a wide variety of lanthanides and pseudo-lanthanides.[51] An important structural feature of these frameworks is the position of the tetraethylammonium counterions, which not only provide charge balance but also play an integral structure-directing role as pillars between adjacent sheets. Hydrogen bonds between the methylene hydrogens of the cation and the coordinated oxygens of the benzoquinoid-type ligands serve to align the sheets to create large square channels. A lower-symmetry square grid framework compound of composition (H3O)[Dy(C6O4(CN)Cl)2(H2O)]·4H2O has also been reported, in which each DyIII ion has a nine-coordinate monocapped square antiprism geometry, with an H2O molecule occupying the capped position.[49]
Herein, we report the synthesis of square grid structures with a (4,4)-net for the early lanthanides LaIII to EuIII. Two different structure types are observed; the type 1 square grids with LaIII, CeIII, and NdIII, which feature a water molecule coordinated to the metal centre, and type 2 square grids for SmIII, and EuIII, which are analogous to the structures of composition (Et4N)[MIII(C6O4X2)2] previously reported.[51] Removal of the coordinated water molecule from the type 1 NdIII square grid is demonstrated in a single-crystal-to-single-crystal transformation to form a type 2 square grid. The contribution of these early lanthanide chloranilate square grids, based on LaIII to EuIII, nearly completes the series of LnIII chloranilate square grids with the tetraethylammonium counterion.
Results and Discussion
The early lanthanide-containing square grid compounds were synthesised by the reaction of chloranilic acid (H2can) with Ln(NO3)3·xH2O (Ln = LaIII, CeIII, NdIII, SmIII, and EuIII) and tetraethylammonium chloride (Et4NCl) in a mixture of DMF and water at 130°C. A purple–red suspension was initially obtained for all samples, which yielded thin purple square plates on heating at 130°C for 16 h. The synthesis of the early lanthanide chloranilate square grids differs from that previously reported for the square grids with GdIII to LuIII. The square grids with GdIII to LuIII were synthesised via an ambient-temperature diffusion of a solution of chloranilic acid with LiO2CCH3 in acetone layered onto a solution of the metal nitrate salt and tetraethylammonium chloride salt in water.[51]
The higher reaction temperature and change in solvent mixture appear to be necessary for the synthesis of the square grids, where the slow decomposition of the DMF to dimethylamine and formic acid, enhanced by the small amount of water added, likely contributes to the formation of the early lanthanide square grid frameworks. Although the synthesis of the square grids is possible using pure DMF as the solvent, the quality of the crystals obtained is lower, and in some cases, co-crystallisation of the lanthanide honeycomb framework is obtained.
Square grids of the formula (Et4N)[Ln(can)2(H2O)] (Ln = La (1), Ce (2), Nd (3)) crystallised in the tetragonal space group I4/m (Fig. 1 and Fig. S1, Supplementary Material). Square grids 1, 2, and 3 feature a nine-coordinate capped square antiprismatic lanthanide centre as determined using the SHAPE program,[52] with one coordinated water molecule and chloranilate ligands bound in a bidentate manner (Fig. 1a). Full details of the SHAPE analysis are given in Table S3 (Supplementary Material). The La, Ce, and Nd square grids all feature two different Ln–O bond lengths, with a significantly shorter Ln–O bond length to the coordinated water molecule than to the oxygens of the can2− ligand (e.g. La–O2H = 2.444(9) Å, La–O = 2.553(4), 2.572(4) Å). A decrease in both the Ln–O2H and Ln–O bond lengths is observed on moving from La to Nd, which corresponds to the contracting lanthanide ionic radius across the series (Table 1). The nine-coordinate lanthanide centre is in contrast to the previously reported lanthanide square grid structures, which feature an eight-coordinate lanthanide centre with four coordinated chloranilate ligands.[51] The inclusion of the water molecule likely arises due to the larger size of the early lanthanide ions La, Ce, and Nd, when compared with the later lanthanides, owing to the lanthanide contraction across the f-block metals. The inclusion of the water molecule does not markedly change the coordination environment about the lanthanide ion, with the four chloranilate ligands still within the same plane (Fig. 1b).
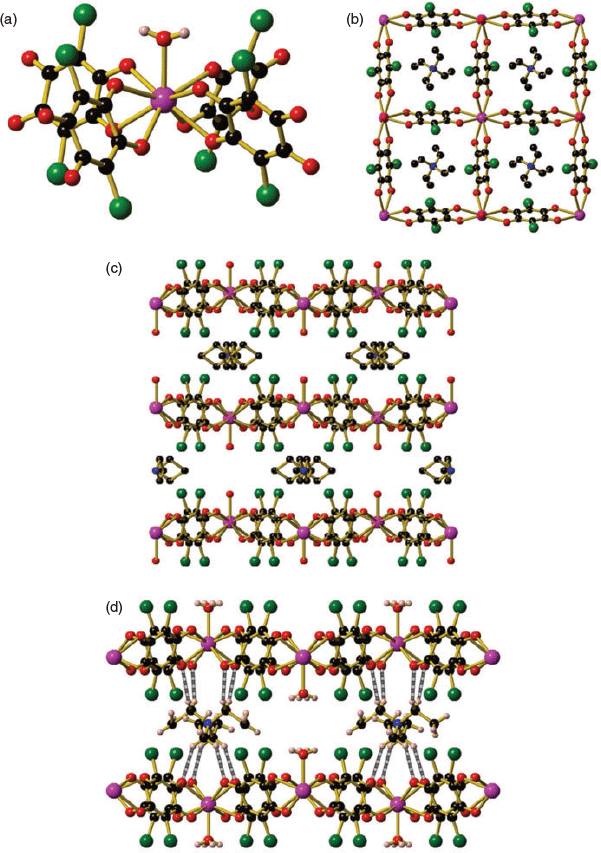
|

|
When the square grid structure is viewed from the side-on direction (along the a or b axes) for 1, 2, and 3, the space below the lanthanide centres is alternately occupied by a tetraethylammonium counterion and a coordinated water molecule (Fig. 1c). The second tetraethylammonium counterion required for charge balance is found within the square channels (Fig. 1b). The thermal gravimetric analyses (TGA) of compounds 1 and 2 are nearly identical, with a 16 % mass loss between 80 and 150°C, which can be attributed to 1.5 DMF molecules. A plateau in the TGA trace is observed between 150 and 325°C before decomposition occurs above 375°C (Fig. S3, Supplementary Material).
The alternation of the tetraethylammonium and coordinated water molecules results in two different Ln–Ln interlayer distances between adjacent 2D sheets (Table 1). A longer interlayer distance is found when the tetraethylammonium cation is present when compared with a coordinated water molecule (e.g. 10.834 versus 9.007 Å respectively for Et4N+ and H2O in 1), which results in a very slight zig-zag alternation of the 2D square grid when viewed along the a and b axes (Fig. 1c). The larger Ln–Ln interlayer distance in the presence of the larger tetraethylammonium cation is unsurprising when compared with the much smaller water molecule.
The tetraethylammonium counterions feature favourable intermolecular interactions including hydrogen bonding and halogen–hydrogen interactions between the hydrogens of the tetraethylammonium counterion and the chloranilate ligands. Inspection of the ‘cavity’ formed by four chlorines of the chloranilate ligand in adjacent layers of the 2D square grid structure shows the ideal fit of the tetraethylammonium counterion (Fig. 1d). The intermolecular interactions between the tetraethylammonium counterion and the framework structure are crucial in dictating the formation of the square grid, which was emphasised by a recent report with CeIII and ErIII chloranilate systems where the use of different alkyl ammonium counterions resulted in the formation of exclusively 2D honeycomb systems.[53]
The use of SmIII and EuIII resulted in the formation of square grids (Et4N)[Ln(can)2] (Ln = SmIII (4), EuIII (5)), which crystallised in the tetragonal space group I4/mcm (Fig. 2 and Fig. S2, Supplementary Material). The higher-symmetry space group of I4/mcm, compared with I4/m observed for 1, 2, and 3, is due to the absence of the coordinated water molecule, which imparts an additional two planes of symmetry. Compounds 4 and 5 are isostructural with the previously reported lanthanide chloranilate square grid structures with DyIII to LuIII.[51] The lanthanide centre in 4 and 5 is eight-coordinate in a square antiprism geometry (Table S4, Supplementary Material), with the four chloranilate ligands bound in a bidentate fashion. A decrease in the Ln–O bond distance occurs on moving from Nd to Eu (Nd–O = 2.450(5), Sm–O = 2.4165(13), Eu–O = 2.398(2) Å) corresponding with the decrease in ionic radius of the lanthanide ion (Table 1). The ligands are all in the same plane, forming a 2D square grid structure with a (4,4)-net (Fig. 2a). Channels through the structure can be seen when viewed along the c axis, where disordered solvent occupies the void space (Fig. 2b). The tetraethylammonium counterion is present both above and below the lanthanide centres (Fig. 2c) and features extensive intermolecular interactions, such as hydrogen bonding between the CH2 hydrogens of Et4N+ with the oxygens of the can2− ligand and halogen–hydrogen interactions between the CH3 hydrogens of Et4N+ with the chlorines on the can2− ligand (Fig. 2d). A gradual 11 % mass loss from 25 to 275°C in the TGA for compound 4 can be attributed to the liberation of one DMF molecule before decomposition is observed above 375°C (Fig. S4, Supplementary Material). A 16 % mass loss is observed between 25 and 275°C in the TGA for compound 5 that corresponds to the loss of one water molecule and 3.5 DMF molecules. In an analogous manner to compound 4, decomposition of compound 5 occurs above 475°C (Fig. S5, Supplementary Material).
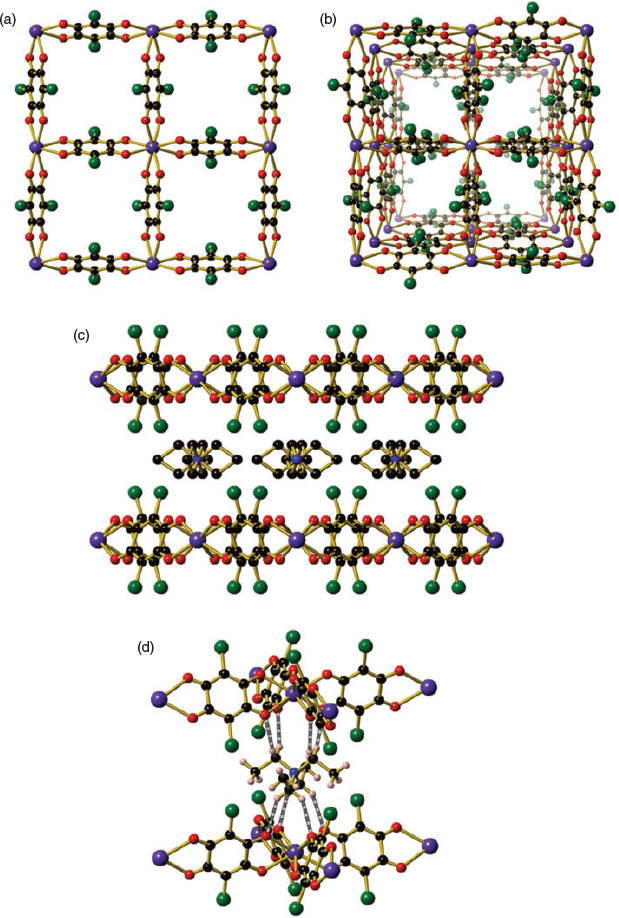
|
On moving across the f-block metals from La to Eu, several trends are observed for the unit cell parameters that are attributable to the contraction in the lanthanide ion. The Ln–O bond length decreases from 1 to 5, resulting in a decrease in the length of the a and b cell axes that directly corresponds to the size of the square cavity in the 2D sheet (Table 1). The interlayer distance between 2D sheets is correlated with the change in the c axis, with an increase from 19.8412(8) for 1 to 20.117(4) Å for 5. The increase in the interlayer distance reflects the contraction in the lanthanide ion size from LaIII to EuIII, where the interaction of the tetraethylammonium cation with the 2D sheets weakens slightly owing to the constriction of space. The small changes in the cell dimensions from 1 to 5 are reflected in the powder X-ray diffraction (XRD) patterns of the square grids (Fig. S6, Supplementary Material). Distinct shifts of the (0 0 2) peak at 8.395° for 1 to 8.834° 2θ for 5 correspond to the decreasing distance between the 2D sheets, whereas an increase for the (1 1 0) peak at 9.843° for 1 to 10.247° for 5 is due to the decrease in the size of the square cavities within the 2D sheet.
Void spaces are present in 1, 2, and 3 of 14.3 to 15.1 %, which are smaller than 4 and 5 with void spaces of 23.3 and 22.8 % respectively. The presence of the tetraethylammonium cation within the square channels contributes to the decreased void space of 1, 2, and 3. The void spaces obtained for 4 and 5 are comparable with those previously observed for chloranilate square grids with GdIII to LuIII of 24.7 to 26.9 %.[51] The slightly smaller void spaces for 4 and 5 (23.3 and 22.8 % respectively) when compared with the later lanthanides from GdIII to LuIII (24.7 and 26.9 %)[51] can be attributed to the larger lanthanide ion size.
The water molecule coordinated to the NdIII centre for (Et4N)[Nd(can)2(H2O)] (3) was able to be removed on gentle heating of the compound at 60°C under a pressure of 5000 Pa. A single-crystal-to-single-crystal transformation occurred where (Et4N)[Nd(can)2(H2O)] (3) was converted to (Et4N)[Nd(can)2] (3a) (Fig. 3). The tetraethylammonium counterion located within the square channels moves to a position between the 2D sheets. Compound 3a is isostructural with 4 and 5, where an increase in the symmetry of the space group from I4/m (3) to I4/mcm (3a) was observed. Attempts to remove the coordinated water molecule from 1 and 2 were unsuccessful even on heating at much higher temperatures under vacuum. The decreasing size of the lanthanide ion likely has a large influence on the successful removal of the coordinated water molecule. The ease of removal of the coordinated water molecule from 3 may result from the Nd analogue being at the ‘tipping point’ between the nine- and eight-coordinate structure types.
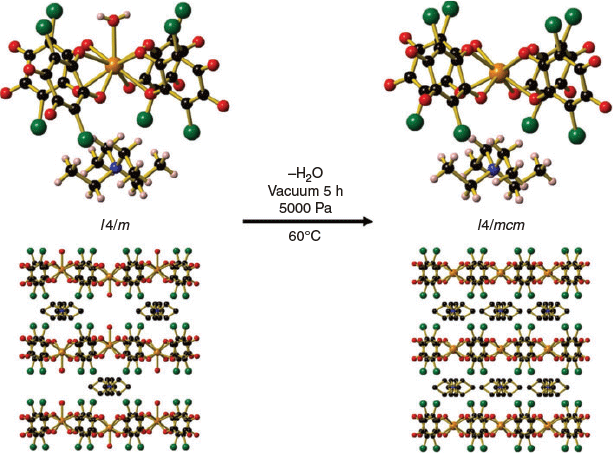
|
In contrast to the TGA of 1 and 2, which featured one sharp mass loss below 175°C, several stepwise mass losses were observed for 3 (Fig. S7, Supplementary Material). The first mass loss between 25 and 150°C is due to the liberation of one DMF molecule. The second mass loss between 150 and 195°C is due to the loss of one water molecule and reflects the successful single-crystal-to-single-crystal transformation observed for compound 3. The third mass loss between 195 to 265°C is due to the loss of a further DMF molecule.
A decrease in the interlayer distance from 10.925 to 10.037 Å (interlayer M M) and a decrease in the size of the square within each layer (intralayer M–M) from 8.974 to 10.8.804 Å is observed on the transformation of 3 to 3a. The change in the interlayer and intralayer M–M distances for the bulk sample of 3 and 3a was confirmed by the powder XRD pattern, with corresponding shifts in the (0 0 2) peak from 8.69 (3) to 8.74° (3a) for the interlayer M–M distance and 9.88 (3) to 10.04° (3a) in the (1 1 0) peak for the intralayer M–M distance (Fig. S8, Supplementary Material).
M) and a decrease in the size of the square within each layer (intralayer M–M) from 8.974 to 10.8.804 Å is observed on the transformation of 3 to 3a. The change in the interlayer and intralayer M–M distances for the bulk sample of 3 and 3a was confirmed by the powder XRD pattern, with corresponding shifts in the (0 0 2) peak from 8.69 (3) to 8.74° (3a) for the interlayer M–M distance and 9.88 (3) to 10.04° (3a) in the (1 1 0) peak for the intralayer M–M distance (Fig. S8, Supplementary Material).
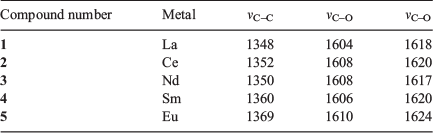
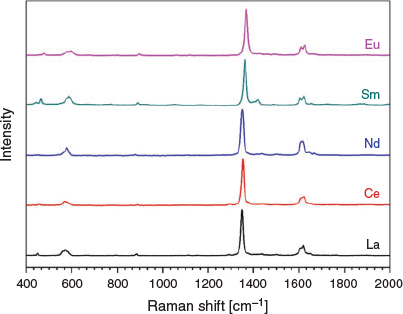
Raman spectroscopy was conducted on each of the square grid structures with a laser excitation of 433 nm. It was of interest to investigate the Raman spectra of compounds 1–5 as the energy of the C–C stretching band in can2− is highly diagnostic of the redox state of the ligand.[30] The C–C bond becomes stronger on reduction of the chloranilate ligand to its 3− redox state as it gains greater aromaticity, resulting in a Raman stretch at higher wavenumbers. The C–C stretch occurs between 1348 and 1369 cm−1 in compounds 1 to 5, which agrees with the C–C bond distances (1.530 to 1.547 Å) in the crystal structure, indicating that the chloranilate ligand is in its 2− state (Table 2 and Fig. 4). Two C–O stretches are observed, which correspond to the two different types of C–O bonds present. The C–O stretch with more single bond character is located at ~1608 cm−1 whereas the C–O stretch with greater double bond character is located at ~1620 cm−1, consistent with the 2− quinone redox state of the chloranilate ligand present in the square grids. No significant difference is present among compounds 1 to 5.
|
|
Conclusions
Six new lanthanide chloranilate square grids, spanning two structure types, were synthesised in this work. Type 1 contains a nine-coordinate lanthanide centre with a coordinated water molecule, (Et4N)[Ln(can)2(H2O)] (Ln = La, Ce, Nd), whereas type 2 contains an eight-coordinate lanthanide centre, (Et4N)[Ln(can)2] (Ln = Sm, Eu). A conversion of the Nd type 1 to a type 2 square grid was achieved in a single-crystal-to-single-crystal transformation, resulting in a contraction of the interlayer and intralayer M–M distances. Raman spectroscopy of the lanthanide square grids confirmed the presence of the chloranilate ligand in its 2− redox state.
Experimental
General Considerations
All chemicals and solvents were used as obtained and without further purification.
Synthesis of Frameworks
General Procedure
The lanthanide nitrate hexahydrate (0.114 mmol), Et4NCl (246 mg, 1.49 mmol), and chloranilic acid (47.5 mg, 0.227 mmol) were dissolved in a mixture of DMF (10.0 mL) and water (0.338 mL) in vials to yield a purple–red suspension. The vials were shaken until all the solid was suspended, then placed in an oven at 130°C for 16 h to yield purple square-shaped crystals. The crystals were washed with DMF (2 × 5 mL) before being allowed to dry in air.
(Et4N)[La(can)2(H2O)] (1). Yield 37.0 mg, 46 %. νmax (attenuated total reflectance (ATR))/cm−1 1680 (νC–O), 1662 (νC–O), 1373 (νC–C). Anal. Calc. for C20H21Cl4LaNO9·1.53C3H7NO: C 36.33, H 4.06, N 4.56; found: C 36.33, H 3.764, N 4.93 %.
(Et4N)[Ce(can)2(H2O)] (2). Yield 36.1 mg, 45 %. νmax (ATR)/cm−1 1683 (νC–O), 1664 (νC–O), 1373 (νC–C). Anal. Calc. for C20CeH21Cl4NO9·1.53C3H7NO: C 36.28, H 4.05, N 4.36; found C 36.28, H 3.707, N 4.78 %.
(Et4N)[Nd(can)2(H2O)] (3). Yield 33.2 mg, 41 %. νmax (ATR)/cm−1 1676 (νC–O), 1645 (νC–O), 1375 (νC–C). Anal. Calc. for C20H21Cl4NNdO9·2.03C3H7NO: C 36.66, H 4.27, N 4.97; found C 36.91, H 3.953, N 4.97 %.
(Et4N)[Sm(can)2] (4). Yield 29.8 mg, 38 %. νmax (ATR)/cm−1 1676 (νC–O), 1377 (νC–C). Anal. Calc. for C20H20Cl4NO8Sm·1.29C3H7NO·0.26H2O: C 36.13, H 3.75, N 4.04; found C 36.13, H 3.422, N 4.04 %.
(Et4N)[Eu(can)2] (5). Yield 66.1 mg, 83 %. νmax (ATR)/cm−1 1676 (νC–O), 1375 (νC–C). Anal. Calc. for C20EuH20Cl4NO8·1.1C3H7NO: C 36.04, H 3.60, N 3.79; found C 36.08, H 3.72, N 3.89 %.
Synthesis of (Et4N)[Nd(can)2] (3a). (Et4N)[Nd(can)2(H2O)] (3) (20 mg, 0.0173 mmol) was placed under vacuum (5000 Pa) and heated at 60°C for 5 h before being slowly cooled to room temperature to yield a light purple solid (18 mg, 95 %). Anal. Calc. for C20H20Cl4NNdO8: C 34.89, H 2.93, N 2.03; found C 34.53, H 3.04, N 2.24 %.
Physical Characterisation Techniques
Infrared Spectroscopy
Infrared spectra were collected on a Bruker Alpha ATR instrument. A small amount (~2 mg) was placed on the ATR crystal and the data collected using 16 scans with 4 cm−1 resolution over the range of 4000–400 cm−1. The infrared spectra of compounds 1–5 are shown in Fig. S9 (Supplementary Material).
Powder X-Ray Diffraction Analyses (PXRD)
A powder sample of each of the compounds was sandwiched between two pieces of Kapton tape in the 3-mm hole of a metallic mask. Powder X-ray diffraction data were collected at ambient temperature on a STOE-Stadi MP powder diffractometer equipped with an asymmetric curved germanium monochromator (CuKα1 radiation, λ 1.54056 Å) and one-dimensional silicon strip detector (MYTHEN2 1K from Dectris), and the sample was measured in transmission geometry in a rotating holder. The line-focussed Cu X-ray tube was operated at 40 kV and 40 mA. Intensity data from 5 to 50° 2θ were collected over a period of 5 min. The instrument was calibrated against a National Institute of Standards and Technology (NIST) silicon standard (640d). The bulk experimental PXRD patterns of compounds 1–5 against their calculated patterns are shown in Figs S10–S13 (Supplementary Material).
Raman Measurements
Crystals of each sample were deposited on a glass slide and Raman spectra collected using a Horiba LabRam HR Evolution confocal microscope. Individual crystals were excited with a 473 nm continuous-wave diode laser (Cobolt Blues) at 263-μW power equipped with a long working distance 50× microscope objective (NA = 0.50; Nikon) and 1800 grooves mm−1 grating. The crystals were irradiated with the laser for 1 s.
Thermal Gravimetric Analysis
TGA was measured on a Mettler TGA/SDTA851 instrument using 5–10 mg of sample from 25 to 400°C using an aluminium pan and heated at a rate of 5°C min−1 under a nitrogen atmosphere.
Crystallography
Single crystals of 1, 2, 3, 3a, 4, and 5 were coated with deoxygenated Paratone-N oil and mounted on a MicroMountsTM rod, before being frozen under a stream of cold N2 during the data collection. Data were collected either on a Bruker Kappa APEX-II CCD diffractometer employing a monochromated CuKα 1 μS microfocus source (compound 1), Bruker Kappa APEX-II CCD diffractometer employing monochromated MoKα 1 μS microfocus source (compounds 3, 3a, 4, 5) or XtaLAB Synergy, Dualflex, HyPix diffractometer employing mirror-monochromated CuKα radiation generated from a microfocus sealed X-ray tube (compound 2).
The data integration and reduction were undertaken with SAINT and XPREP[54] (compounds 1, 3, 3a, 4, 5) or CrysAlisPro (compound 2) and subsequent computations were carried out with the WinGX[54] and Olex2[55] graphical user interface. All structures were solved by direct methods with SHELXT[56] and extended and refined with SHELXL.[57] The non-hydrogen atoms in the asymmetric unit were modelled with anisotropic displacement parameters. A riding atom model with group displacement parameters was used for the hydrogen atoms. An empirical absorption correction determined with SADABS[58,59] (compounds 1, 3, 3a, 4, 5) or CrysAlisPro (compound 2) was applied to the data. Crystallographic data for these compounds at 100 K are given in Table S1 (Supplementary Material). At 100 K, the solvent molecules in compounds 1–5 were severely disordered and could not be modelled. These species were therefore treated as a diffuse contribution to the overall scattering without specific atom positions using the solvent-masking procedure implemented in OLEX2.[55] A twin component was present in compound 2, which was deconvoluted using the CrysAlisPro software and subsequently refined as a single twin component. A summary of the crystallographic parameters is shown in Tables S1 and S2 (Supplementary Material).
The Cambridge Crystallographic Data Centre (CCDC) numbers 1909480–1909485 contain the supplementary crystallographic data for this paper.
Supplementary Material
Crystallographic and structural details, SHAPE analysis, powder XRD, TGA, and ATR-FTIR spectra are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This work was partially supported by the National Science Foundation (grant no. DMR-1351959) and a McKenzie Postdoctoral Fellowship awarded by the University of Melbourne. We thank Associate Professor Brendan Abrahams for helpful discussions. C.H. gratefully acknowledges the American–Australian Association for a Dow Chemical Co. Fellowship and the Australian Government for an Endeavour Research Fellowship.
References
[1] H. Furukawa, K. E. Cordova, M. O’Keeffe, O. M. Yaghi, Science 2013, 341,| Crossref | GoogleScholarGoogle Scholar | 23990564PubMed |
[2] B. F. Hoskins, R. Robson, J. Am. Chem. Soc. 1990, 112, 1546.
| Crossref | GoogleScholarGoogle Scholar |
[3] R. Robson, B. F. Abrahams, S. R. Batten, R. W. Gable, B. F. Hoskins, J. Liu, in Supramolecular Architecture (Eds T. Bein) 1992, Vol. 499, pp. 256–273 (American Chemical Society: Washington, DC).
[4] S. R. Batten, N. R. Champness, Philos. Trans. Royal Soc. A 2017, 375,
| Crossref | GoogleScholarGoogle Scholar |
[5] H.-C. Zhou, J. R. Long, O. M. Yaghi, Chem. Rev. 2012, 112, 673.
| Crossref | GoogleScholarGoogle Scholar | 22280456PubMed |
[6] B. Li, B. Chen, in Lanthanide Metal–Organic Frameworks (Ed. P. Cheng) 2015, pp. 75–107 (Springer: Heidelberg).
[7] D. S. Ahmed, G. A. El-Hiti, E. Yousif, A. A. Ali, A. S. Hameed, J. Polym. Res. 2018, 25, 75.
| Crossref | GoogleScholarGoogle Scholar |
[8] J. Yu, L. H. Xie, J. R. Li, Y. Ma, J. M. Seminario, P. B. Balbuena, Chem. Rev. 2017, 117, 9674.
| Crossref | GoogleScholarGoogle Scholar | 28394578PubMed |
[9] K. K. Bisht, B. Parmar, Y. Rachuri, A. C. Kathalikattil, E. Suresh, CrystEngComm 2015, 17, 5341.
| Crossref | GoogleScholarGoogle Scholar |
[10] H. C. Hoffmann, S. Paasch, P. Müller, I. Senkovska, M. Padmanaban, F. Glorius, S. Kaskel, E. Brunner, Chem. Commun. 2012, 10484.
| Crossref | GoogleScholarGoogle Scholar |
[11] J. D. Martell, L. B. Porter-Zasada, A. C. Forse, R. L. Siegelman, M. I. Gonzalez, J. Oktawiec, T. Runčevski, J. Xu, M. Srebro-Hooper, P. J. Milner, K. A. Colwell, J. Autschbach, J. A. Reimer, J. R. Long, J. Am. Chem. Soc. 2017, 139, 16000.
| Crossref | GoogleScholarGoogle Scholar | 28991466PubMed |
[12] M. A. Chowdhury, J. Biomed. Mater. Res. A 2017, 105, 1184.
| Crossref | GoogleScholarGoogle Scholar | 28033653PubMed |
[13] W. Chen, C. Wu, Dalton Trans. 2018, 2114.
| Crossref | GoogleScholarGoogle Scholar | 29369314PubMed |
[14] T. Simon-Yarza, A. Mielcarek, P. Couvreur, C. Serre, Adv. Mater. 2018, 30,
| Crossref | GoogleScholarGoogle Scholar | 29876985PubMed |
[15] K. Lu, T. Aung, N. Guo, R. Weichselbaum, W. Lin, Adv. Mater. 2018, 30,
| Crossref | GoogleScholarGoogle Scholar | 29971835PubMed |
[16] Q. Zhang, C. F. Wang, Y. K. Lv, Analyst 2018, 143, 4221.
| Crossref | GoogleScholarGoogle Scholar | 30090910PubMed |
[17] M. L. Mercuri, F. Congiu, G. Concas, S. A. Sahadevan, Magnetochemistry 2017, 3, 17.
| Crossref | GoogleScholarGoogle Scholar |
[18] B. F. Abrahams, T. A. Hudson, L. J. McCormick, R. Robson, Cryst. Growth Des. 2011, 11, 2717.
| Crossref | GoogleScholarGoogle Scholar |
[19] B. F. Abrahams, A. M. Bond, T. H. Le, L. J. McCormick, A. Nafady, R. Robson, N. Vo, Chem. Commun. 2012, 11422.
| Crossref | GoogleScholarGoogle Scholar |
[20] M. E. Ziebel, L. E. Darago, J. R. Long, J. Am. Chem. Soc. 2018, 140, 3040.
| Crossref | GoogleScholarGoogle Scholar | 29400059PubMed |
[21] A. Dei, D. Gatteschi, L. Pardi, U. Russo, Inorg. Chem. 1991, 30, 2589.
| Crossref | GoogleScholarGoogle Scholar |
[22] K. Heinze, G. Huttner, L. Zsolnai, A. Jacobi, P. Schober, Chem. – Eur. J. 1997, 3, 732.
| Crossref | GoogleScholarGoogle Scholar |
[23] I.-R. Jeon, J. G. Park, D. J. Xiao, T. D. Harris, J. Am. Chem. Soc. 2013, 135, 16845.
| Crossref | GoogleScholarGoogle Scholar |
[24] R. Murase, B. F. Abrahams, D. M. D’Alessandro, C. G. Davies, T. A. Hudson, G. N. L. Jameson, B. Moubaraki, K. S. Murray, R. Robson, A. L. Sutton, Inorg. Chem. 2017, 56, 9025.
| Crossref | GoogleScholarGoogle Scholar | 28723082PubMed |
[25] M. Atzori, S. Benmansour, G. Mínguez Espallargas, M. Clemente-León, A. Abhervé, P. Gómez-Claramunt, E. Coronado, F. Artizzu, E. Sessini, P. Deplano, A. Serpe, M. L. Mercuri, C. J. Gómez García, Inorg. Chem. 2013, 52, 10031.
| Crossref | GoogleScholarGoogle Scholar | 23968133PubMed |
[26] S. Benmansour, A. Abhervé, P. Gómez-Claramunt, C. Vallés-García, C. J. Gómez-García, ACS Appl. Mater. Interfaces 2017, 9, 26210.
| Crossref | GoogleScholarGoogle Scholar | 28715894PubMed |
[27] I.-R. Jeon, B. Negru, R. P. Van Duyne, T. D. Harris, J. Am. Chem. Soc. 2015, 137, 15699.
| Crossref | GoogleScholarGoogle Scholar |
[28] L. E. Darago, M. L. Aubrey, C. J. Yu, M. I. Gonzalez, J. R. Long, J. Am. Chem. Soc. 2015, 137, 15703.
| Crossref | GoogleScholarGoogle Scholar | 26573183PubMed |
[29] S. Halis, A. K. Inge, N. Dehning, T. Weyrich, H. Reinsch, N. Stock, Inorg. Chem. 2016, 55, 7425.
| Crossref | GoogleScholarGoogle Scholar | 27427885PubMed |
[30] J. A. DeGayner, I.-R. Jeon, L. Sun, M. Dincă, T. D. Harris, J. Am. Chem. Soc. 2017, 139, 4175.
| Crossref | GoogleScholarGoogle Scholar | 28230984PubMed |
[31] B. F. Abrahams, A. D. Dharma, B. Dyett, T. A. Hudson, H. Maynard-Casely, C. J. Kingsbury, L. J. McCormick, R. Robson, A. L. Sutton, K. F. White, Dalton Trans. 2016, 1339.
| Crossref | GoogleScholarGoogle Scholar | 26733002PubMed |
[32] M. Jurić, K. Molčanov, D. Žilić, B. Kojić-Prodić, RSC Adv. 2016, 6, 62785.
| Crossref | GoogleScholarGoogle Scholar |
[33] L.-M. Zheng, H. W. Schmalle, R. Huber, S. Decurtins, Polyhedron 1996, 15, 4399.
| Crossref | GoogleScholarGoogle Scholar |
[34] J. A. DeGayner, K. Wang, T. D. Harris, J. Am. Chem. Soc. 2018, 140, 6550.
| Crossref | GoogleScholarGoogle Scholar | 29747503PubMed |
[35] M. K. Kabir, M. Kawahara, K. Adachi, S. Kawata, T. Ishii, S. Kiaagawa, Mol. Cryst. Liq. Cryst. 2002, 376, 65.
| Crossref | GoogleScholarGoogle Scholar |
[36] M. K. Kabir, M. Kawahara, H. Kumagai, K. Adachi, S. Kawata, T. Ishii, S. Kitagawa, Polyhedron 2001, 20, 1417.
| Crossref | GoogleScholarGoogle Scholar |
[37] T. T. Luo, Y. H. Liu, H. L. Tsai, C. C. Su, C. H. Ueng, K. L. Lu, Eur. J. Inorg. Chem. 2004, 4253.
| Crossref | GoogleScholarGoogle Scholar |
[38] L. Liu, L. Li, J. A. DeGayner, P. H. Winegar, Y. Fang, T. D. Harris, J. Am. Chem. Soc. 2018, 140, 11444.
| Crossref | GoogleScholarGoogle Scholar | 30063830PubMed |
[39] A. Abhervé, S. Mañas-Valero, M. Clemente-León, E. Coronado, Chem. Sci. 2015, 6, 4665.
| Crossref | GoogleScholarGoogle Scholar | 28717480PubMed |
[40] S. Benmansour, C. Gómez-García, Polymers 2016, 8, 89.
| Crossref | GoogleScholarGoogle Scholar |
[41] C. J. Kingsbury, B. F. Abrahams, D. M. D’Alessandro, T. A. Hudson, R. Murase, R. Robson, K. F. White, Cryst. Growth Des. 2017, 17, 1465.
| Crossref | GoogleScholarGoogle Scholar |
[42] B. F. Abrahams, J. Coleiro, K. Ha, B. F. Hoskins, S. D. Orchard, R. Robson, J. Chem. Soc., Dalton Trans. 2002, 1586.
| Crossref | GoogleScholarGoogle Scholar |
[43] S. Benmansour, C. Vallés-García, P. Gómez-Claramunt, G. Mínguez Espallargas, C. J. Gómez-García, Inorg. Chem. 2015, 54, 5410.
| Crossref | GoogleScholarGoogle Scholar | 25965415PubMed |
[44] S. Benmansour, I. Pérez-Herráez, G. López-Martínez, C. J. Gómez García, Polyhedron 2017, 135, 17.
| Crossref | GoogleScholarGoogle Scholar |
[45] K. Nakabayashi, S. I. Ohkoshi, Acta Crystallogr. Sect. E Struct. Rep. Online 2010, 66, m1300.
| Crossref | GoogleScholarGoogle Scholar | 21587439PubMed |
[46] C. Robl, Mater. Res. Bull. 1987, 22, 1483.
| Crossref | GoogleScholarGoogle Scholar |
[47] S. Benmansour, I. Pérez-Herráez, C. Cerezo-Navarrete, G. López-Martínez, C. Martínez Hernández, C. J. Gómez-García, Dalton Trans. 2018, 6729.
| Crossref | GoogleScholarGoogle Scholar | 29713717PubMed |
[48] S. Benmansour, A. Hernández-Paredes, C. J. Gómez-García, J. Coord. Chem. 2018, 71, 845.
| Crossref | GoogleScholarGoogle Scholar |
[49] P. Gómez-Claramunt, S. Benmansour, A. Hernández-Paredes, C. Cerezo-Navarrete, C. Rodríguez-Fernández, J. Canet-Ferrer, A. Cantarero, C. Gómez-García, Magnetochemistry 2018, 4, 6.
| Crossref | GoogleScholarGoogle Scholar |
[50] S. Benmansour, A. Hernández-Paredes, C. J. Gómez-García, Magnetochemistry 2018, 4, 58.
| Crossref | GoogleScholarGoogle Scholar |
[51] C. J. Kingsbury, B. F. Abrahams, J. E. Auckett, H. Chevreau, A. D. Dharma, S. Duyker, Q. He, C. Hua, T. A. Hudson, K. S. Murray, W. Phonsri, V. K. Peterson, R. Robson, K. F. White, Chem. – Eur. J. 2019, 25, 5222.
| Crossref | GoogleScholarGoogle Scholar | 30729591PubMed |
[52] M. Llunell, D. Casanova, J. Cicera, P. Alemany, S. Alvarez, SHAPE version 2.1 2013 (Universitat de Barcelona: Barcelona).
[53] K. Bondaruk, C. Hua, Cryst. Growth Des. 2019, 19, 3338.
[54] L. Farrugia, J. Appl. Cryst. 1999, 32, 837.
| Crossref | GoogleScholarGoogle Scholar |
[55] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Cryst. 2009, 42, 339.
| Crossref | GoogleScholarGoogle Scholar |
[56] G. Sheldrick, Acta Crystallogr. Sect. A 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[57] G. M. Sheldrick, Acta Crystallogr. Sect. C 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[58] R. H. Blessing, Acta Crystallogr. Sect. A 1995, A51, 33.
| Crossref | GoogleScholarGoogle Scholar |
[59] L. Krause, R. Herbst-Irmer, G. M. Sheldrick, D. Stalke, J. Appl. Cryst. 2015, 48, 3.
| Crossref | GoogleScholarGoogle Scholar |