

A Copper Complex of a Thiosemicarbazone-Pyridylhydrazone Ligand Containing a Vinylpyridine Functional Group as a Potential Imaging Agent for Amyloid-β Plaques*
Lachlan E. McInnes A , Asif Noor A , Peter D. Roselt B , Catriona A. McLean C , Jonathan M. White A and Paul S. Donnelly A DA School of Chemistry and Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Melbourne, Vic. 3010, Australia.
B Cancer Imaging, Peter MacCallum Cancer Centre, Melbourne, Vic. 3000, Australia.
C Department of Anatomical Pathology, The Alfred Hospital, Vic. 3181, Australia, and The Florey Institute of Neuroscience and Mental Health, University of Melbourne, Melbourne, Vic. 3010, Australia.
D Corresponding author. Email: pauld@unimelb.edu.au
Australian Journal of Chemistry 72(10) 827-834 https://doi.org/10.1071/CH19311
Submitted: 8 July 2019 Accepted: 9 August 2019 Published: 25 September 2019
Abstract
Complexes containing positron-emitting radionuclides of copper have the potential to be of use for diagnostic imaging with positron emission tomography. Alzheimer’s disease is characterised by the presence of amyloid-β plaques in the brain. A new thiosemicarbazone-pyridyl hydrazone tetradentate ligand with a pyridyl-4-vinylpyridine functional group was prepared with the aim of making a copper complex that binds to amyloid-β plaques to assist in the diagnosis of Alzheimer’s disease. The ligand forms a charge neutral complex with copper(ii) that was characterised by X-ray crystallography and the electrochemical behaviour of the complex was investigated by cyclic voltammetry. The new ligand can be radiolabelled with positron-emitting copper-64 at room temperature in excellent radiochemical yields. The new complex interacts with synthetic amyloid-β fibrils and binds amyloid-β plaques present in post-mortem Alzheimer’s disease brain tissue.
Introduction
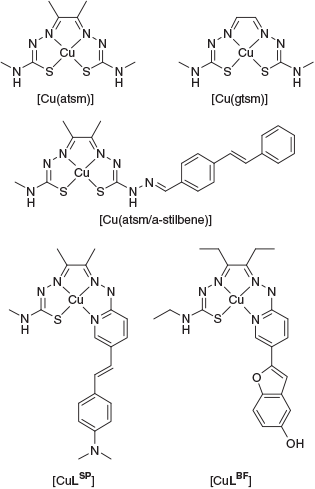
Ligands containing thiosemicarbazone functional groups can coordinate metal ions as anionic, resonance stabilised N,S donors where the sulfur can act as a non-reducing thiolate-like donor. Early work on bis(thiosemicarbazone) ligands included examples by Robson, McFadyen, and Schaap where they isolated a range of binuclear copper(ii) and nickel(ii) complexes derived from a binucleating ligand, 2-hydroxy-5-methylisopthalaldehyde bis(thiosemicarbazone).[1,2] Continued interest in bis(thiosemicarbazone) ligands has focussed on their potential to coordinate positron-emitting isotopes of copper in the development of tracers for positron emission tomography (PET). PET relies on the administration of a tracer that has been radiolabelled with a positron-emitting isotope and there are positron-emitting isotopes of copper that are available with a diverse range of half-lives. Two of these isotopes, copper-60 (t1/2 = 20 min) and copper-62 (t1/2 = 9.7 min) have relatively short radioactive half-lives. There are also two positron-emitting isotopes with longer half-lives, copper-61 (t1/2 = 3.4 h) and copper-64 (t1/2 = 12.7 h). The charge neutral, lipophilic complex [Cu(atsm)] (atsm = diacetyl-bis-(4-methyl-3-thiosemicarbazone)) (Fig. 1) has been investigated as a hypoxia imaging agent as well as for probing oxidative stress in amyotrophic lateral sclerosis.[3–9] In addition, the structurally related compound [Cu(gtsm)] (gtsm = glyoxal-bis(4-methyl-3-thiosemicarbazone)) has been investigated as a probe for measuring altered copper trafficking in the brain during Alzheimer’s disease.[10]
|
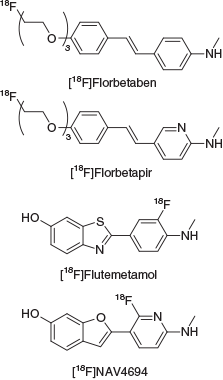
Neurodegeneration associated with the onset of Alzheimer’s disease (AD) leads to a decline in cognition, memory, attention, and motor function. A key pathological hallmark of the disease is the presence of extracellular protein deposits called amyloid plaques. These plaques are comprised mostly of aggregated amyloid-β peptide (Aβ), which contains between 39 and 43 amino acids and is derived from the amyloid precursor protein.[11–15] Visualising these deposits in the brain using PET imaging can provide an estimation of the Aβ burden in patients, which can assist in differential diagnosis of AD from other neurodegenerative diseases with overlapping symptoms. Imaging of Aβ plaque burden could also serve as a tool to help monitor the effectiveness of emerging therapeutic agents.[16,17] Recent advances have led to three Aβ plaque PET imaging agents radiolabelled with fluorine-18 being approved for clinical use; [18F]florbetapir, [18F]flutemetamol, and [18F]florbetaben and a fourth agent, [18F]NAV4694, is in Phase 3 clinical trials for AD imaging (Fig. 2).[18–22] These conjugated, aromatic, hydrophobic molecules bind to Aβ fibrils and plaques through a combination of non-covalent, hydrophobic, and π–π interactions with the aromatic residues located on the surface of aggregated Aβ fibrils and plaques.[23–25]
|
The various positron-emitting isotopes of copper offer alternatives to fluorine-18 labelled tracers and a copper-based radiopharmaceutical to image Aβ plaques is an attractive prospect. A successful candidate radiopharmaceutical must cross the blood–brain barrier (BBB), bind strongly to Aβ plaques, remain sufficiently stable to accumulate a reasonable signal at the target, and any radioactive metabolites must not be BBB permeable. Previously, we have used the bis(thiosemicarbazone) framework to append a targeting group for Aβ plaques. The compound, [Cu(atsm/a-stilbene)] (Fig. 1), features a stilbene functional group appended to the framework through a hydrazone linkage and showed good BBB penetration and selective uptake in an animal model of AD (APP/PS1 transgenic mice) compared with a wild-type control.[26] An alternative approach is to incorporate the targeting group within the chelating domain, thereby negating the need for a linker and reducing the molecular weight of the tracer. To that end we have investigated hybrid thiosemicarbazone-pyridylhydrazone ligands. This class of ligand can be radiolabelled with copper-64 under mild conditions to give a single radioactive species. Incorporation of functional groups such as styryl-pyridine, [CuLSP] (Fig. 1) or pyridyl-benzofuran, [CuLBF] (Fig. 1) into the ligand framework can give complexes that bind to Aβ plaques.[27–29] In this work, we incorporated a 4-vinylpyridine functional group to investigate whether the complex binds to Aβ plaques with an additional pyridyl hydrogen bond acceptor at the expense of the electron donating dimethlylamino and hydroxy groups present in [CuLSP] and [CuLBF].
Results and Discussion
2-Bromo-5-(2-(pyridin-4-yl)vinyl)pyridine (5) was synthesised using a Wadsworth–Emmons–Horner reaction between 2-bromopyridinyl-4-methylenediethylphosphonate, which was generated in situ from 2-bromo-5-(chloromethyl)pyridine (3) and 4-pyridinecarboxaldehyde (4). The resulting 2-bromo-5-(2-(pyridin-4-yl)vinyl)pyridine (5) was then converted into the substituted hydrazine 6 by treatment with hydrazine hydrate.[30] The ligand H2L1 was afforded by the condensation of the hydrazine with the asymmetric diacetyl-mono-4-methyl-3-thiosemicarbazone (7) (Scheme 1). The ligand was characterised by NMR spectroscopy, electrospray ionisation-orbital time of flight mass spectrometry (ESI/O-TOF MS), and elemental analysis.

|
The ligand [H2L1] reacts with Cu(OAc)2·H2O rapidly in methanol to yield a dark purple complex (Scheme 1). The resulting [CuL1] gave a single peak when analysed by reversed phase HPLC (RP-HPLC) and analysis by ESI-MS gave a signal with the expected copper isotope pattern at a m/z corresponding to [CuIIL1 + H]+. The conditional stability constant of the complex at pH 7.4 was determined to be (6.69 ± 3) × 10−18 M which is the same order of magnitude as the previously reported values for this class of ligand.[28,29] Analysis of the UV-visible spectroscopy of [CuL1] revealed an absorption at λmax 350 nm and λ 600 nm. The ligand H2L1 showed an emission maxima at λem 471 nm (λex 382 nm), but this emission was significantly quenched by the presence of copper(ii) in [CuL1] (λem 411 nm, λex 331 nm) (Fig. 3).

|
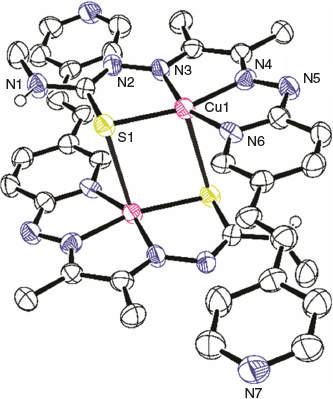
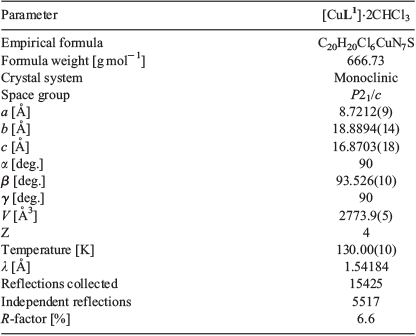
Crystals of sufficient quality for X-ray analysis were grown by diffusion vapours between a solution of [CuL1] in chloroform and pentane (Fig. 4 and Table 1). The complex crystallises with the CuII in a distorted square planar N3S environment tending towards five-coordinate square pyramidal geometry due to a weak axial interaction between the CuII and a sulfur of an adjacent ligand (Cu–S′ 2.932 Å), and the CuII being marginally out of the N3S plane (~0.126 Å). Of the N3S plane, the Cu–S bond is the longest (2.2633(16) Å), which is similar to the Cu–S bond found in Cu(atsm) and previously reported thiosemicarbazone-hydrazinopyridyl copper complexes.[27–29,31,32] Of the nitrogen donors, Cu–Nthiosemicarbazanato (Cu–N3, 1.950(5) Å) and Cu–Nhydrazone (Cu–N4, 1.951(5) Å) were found to be shorter than Cu–Npyridyl (Cu–N6, 1.958(5) Å). The C–S bond distance of the thiosemicarbazanato-arm (S–C1, 1.753(6) Å) suggests a more thiolate-like rather than thione-like character.
|
|
The biological activity and stability of copper bis(thiosemicarbazone) complexes can, in part, be related to the CuII/I reduction potential.[33–35] The reducing environment inside cells can lead to reduction of the copper(ii) to copper(i) leading to transfer of the copper to high affinity copper binding proteins.[35] This instability can lead to dissociation of radioactive copper in vivo, resulting in poorer quality images due to increased background signal. The electrochemical properties of [CuL1] were investigated by cyclic voltammetry in anhydrous N,N-dimethylformamide (1 mM analyte) with tetrabutylammonium hexafluorophosphate (0.1 M) as a supporting electrolyte. Under these conditions, quasi-reversible reduction of [CuL1] was found at E0′ = –1.18 V (versus Fc+/Fc) which corresponds to the CuII/I redox process (Fig. 5a), which is similar to previously reported values for [Cu(atsm)] (E0′ = –1.17 V versus Fc+/Fc) under similar conditions.[36] An irreversible oxidation is seen at E0′ = 0.01 V (versus Fc+/Fc) (Fig. 5b) that could be due to either a CuIII/II process or a ligand-based process.
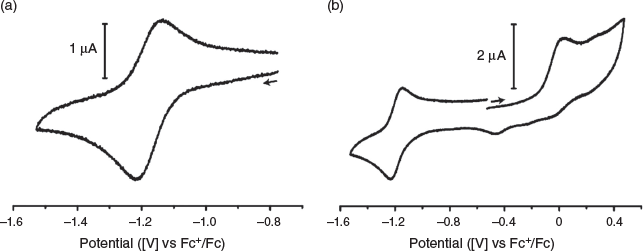
|
The copper-64 complex, [64Cu][CuL1], could be prepared at room temperature (sodium acetate 0.1 M, pH 5, 30 min). The radiolabelled complex was characterised by RP-HPLC comparing the UV-trace of [CuL1] and the scintillation detection of [64Cu][CuL1] (Fig. 6). The straight-forward radiolabelling with copper-64 suggests that this ligand could be easily adapted into a kit-based formulation that has been so central to the success of 99mTc-labelled radiopharmaceuticals.
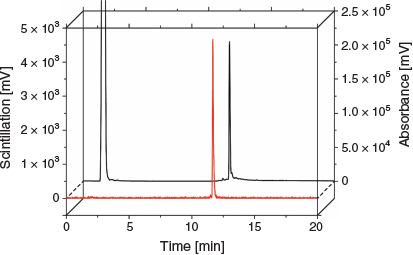
|
The affinity of [CuL1] for synthetic Aβ1–40 was investigated through fluorescence assays using the dye thioflavin T (ThT).[37] ThT binds to amyloid fibrils resulting in a characteristic increase in fluorescence at λ 480 nm.[38] This assay is useful for screening compounds but does not reliably report on molecules that bind to different sites on Aβ fibrils as does ThT.[23] Furthermore, fibril morphology is variable and dependent on the method used for the formation of fibrils.[39,40] Using this assay, the Ki of [CuL1] was found to be 4.3 ± 1.6 μM (Fig. 7), which suggests only a relatively moderate affinity of the complex towards Aβ1–40.[41,42]
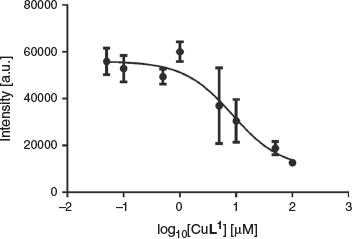
|
The fluorescence of H2L1 was used to investigate its potential to bind to Aβ plaques present in post-mortem brain tissue from clinically diagnosed AD subjects. Brain tissue (7 μM thick) from the frontal cortex of the AD subject was blocked for non-specific binding with bovine serum albumin and then treated with H2L1. The tissue was washed and then analysed by epi-fluorescent microscopy. Aβ plaques are typically 60 μm in diameter, so 7 μm serial sections often contain components of the same plaque. Comparison of the fluorescence from the tissue that had been treated with H2L1 with the adjacent tissue section that had been stained immunohistochemically with an Aβ specific antibody (1E8) revealed good co-localisation suggesting that the ligand has some selectivity for Aβ (Fig. 8).

|
Conclusion
A new thiosemicarbazone-pyridyl hydrazone tetradentate ligand with a pyridyl-4-vinylpyridine functional group was prepared. The ligand forms a charge neutral complex with copper(ii), [CuL1], that was characterised by X-ray crystallography. The copper(ii) complex is stable with a conditional stability constant of (6.69 ± 3) × 10−18 M and an investigation of the electrochemical behaviour of the complex by cyclic voltammetry revealed a quasi-reversible CuII/I couple at E0′ = −1.18 V (versus Fc+/Fc). The new ligand can be radiolabelled with positron-emitting copper-64 at room temperature to give [64Cu][CuL1] with high radiochemical purity. The [CuL1] complex interacts with synthetic Aβ fibrils and binds to amyloid-β plaques present in post-mortem Alzheimer’s disease brain tissue.
Experimental
General Procedures
All reagents and solvents were acquired from commercial sources and used without further purification. Elemental analysis for C, H, and N was carried out by The Campbell Microanalytical Laboratory, The University of Otago, Dunedin, New Zealand. NMR spectra were recorded on a Varian FT-500 NMR (Varian, California, USA) (1H at 500 MHz and 13C{1H} at 125.7 MHz) at 297 K and referenced to internal solvent residue. High resolution mass spectra were recorded on a Thermo Scientific Exactive Plus OrbiTrap LC/MS (Thermo Fisher Scientific, Massachusetts, USA) and calibrated to internal references.
Analytical RP-HPLC traces were acquired using an Agilent 1200 HPLC system equipped with an Alltech Hypersil BDS C18 analytical HPLC column (4.6 × 150 mm, 5 μm) with a flow rate of 1 mL min−1 and UV absorbance detection at 280 and 400 nm. Retention times (Rt, min) were recorded using a gradient elution of 5–100 % B in A (A = 0.1 % trifluoroacetic acid (TFA), B = acetonitrile with 0.1 % TFA).
UV-Vis spectra were recorded on a Shimadzu UV1650-PC spectrometer (Shimadzu, Kyoto, Japan) from 800 to 250 nm. Emission and excitation spectra were recorded on a Varian CARY eclipse fluorescence spectrophotometer in quartz cuvettes.
Cyclic voltammograms were recorded using a Metrohm (Switzerland) AUTOLAB PGSTAT100 and analysed using Autolab GPES V4.9 software. Measurements were carried out using 1 mM of analyte in anhydrous DMF with tetrabutylammonium hexafluorophosphate (0.1 M) as supporting electrolyte. A glassy carbon working electrode was used along with a Pt wire counter electrode and a pseudo leakless Ag/Ag+ working electrode (EDAQ, New South Wales, Australia). Measurements were referenced versus ferrocene (Fc+/Fc, E0′ = 0 V), where E0′ refers to the midpoint of the reduction (Epc) and oxidation (Epa) peaks of a quasi- or fully reversible process where E0′ = (Epc + Epa)/2.
All X-ray crystal structure data was collected on an Oxford Diffraction SuperNova CCD diffractometer using Cu-Kα radiation, and the temperature during collection was maintained at 130.00(10) K using an Oxford Cryosystems cooling device. The structure was solved by direct methods using SHELXT and refined using least-squares methods using SHELXL.[43,44] Thermal ellipsoid plots were generated using ORTEP-3 integrated within the WINGX suite of programs.[45] [CuL1]·2CHCl3 CCDC 1936998.
[64Cu]CuCl2 was obtained from either the Sir Charles Gardiner Hospital Radiopharmaceutical Production and Development Centre (Perth, WA) or from Austin Health Department of Molecular Imaging and Therapy (Heidelburg, Victoria) as a formulation in 0.1 M HCl.
Radiolabelling of [64Cu][CuL1] was achieved by the addition of a DMSO stock solution of H2L1 (2 μL, 1 mg mL−1) to a solution of [64Cu]CuCl2 (10 MBq, 0.1 M HCl) that had been buffered with sodium acetate (0.1 M, pH 5). The reaction was incubated at room temperature for 30 min and an aliquot removed for characterisation by radio-RP-HPLC. Radio-RP-HPLC was performed using a Shimadzu SPD-10ATvP HPLC system equipped with a Phenomenex Luna C18 100 Å column (4.6 × 150 mm, 5 μm) with a 1 mL min−1 flow rate. Chromatograms were acquired with a UV-vis detector (254 and 350 nm) and scintillation detector in series. Retention times are recorded in minutes using a gradient elution method of 5–100 % B in A over 15 min followed by holding at 100 % B for a further 3 min. Buffer A was 0.05 % TFA, buffer B was 0.05 % TFA in acetonitrile.
Synthetic Aβ1–40 was prepared using a Liberty microwave peptide synthesiser from CEM Corporation (USA). For coupling, 5 equivalents of Fmoc amino acid and O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU), plus 10 equivalents of N,N-diisopropylethylamine (DIEA) were used. Residues 28–42 were coupled once at 75°C for 5 min and residues 1–27 were double coupled under the same conditions except for Fmoc-His(Trt)-OH which was double coupled at 50°C. Fmoc cleavage was performed using a solution of 0.1 M oxyma pure in 20 % piperidine/DMF at 90°C for 1 min, except for residues 23–27 which were deprotected for 10 min. Cleavage of the Mmt protecting group at Lys-28 was affected via four multiple treatments with 1 % TFA in DCM. After synthesis, the resin-bound peptide was deprotected and cleaved with a cocktail of 2 % water/2 % thioanisole/1 % triisopropylsilane in TFA for 3 h. The resin was filtered and the volume of TFA reduced to ~2 mL via nitrogen aspiration, followed by a precipitation step and two further washes with diethyl ether. Crude material was purified using RP-HPLC on an Agilent 1100 instrument, which was equipped with a degasser, auto-sampler, fraction collector, and multichannel UV-vis detector. Mobile phases were A = 10 mM NH4OAc in water, pH 9.2 and B = 10 mM NH4OAc in 80 % acetonitrile/20 % water, pH 9.2. A Phenomonex Jupiter 10μ C4, 300 Å, 4.6 × 150 mm column was used for purification using a gradient elution of 20–60 % buffer B in A over 40 min with a 5 mL min−1 flow rate with detection at λ 230 nm. Isolated fractions were lyophilised on a Christ freeze dryer and the identity of the peptide confirmed by mass spectrometry. The purity was found to be > 95 % by HPLC.
The ThT binding assay was performed as follows. Lyophilised Aβ1–40 was dissolved in 1,1,1,3,3,3-hexafluoropropan-2-ol to a theoretical concentration of 1 mg mL−1 and sonicated in an ice bath for 1 h. The mixture was then allowed to incubate at room temperature for 1 h. The solvent was then allowed to evaporate overnight. Drying was completed by centrifugal vacuum for 10 min to obtain a clear peptide film which was either used fresh or stored at −80°C for future experiments. Synthetic amyloid fibrils were obtained by first dissolving the obtained peptide film (100 µg) in NaOH (60 mM, 20 µL), and then vortexing the solution before sonicating in an ice bath for 10 min. The mixture was then diluted with MilliQ water (70 µL) followed by phosphate buffered saline (PBS) (× 10, 10 µL). The mixture was then centrifuged (14000 g) for 5 min at 4°C and the supernatant transferred to a fresh centrifuge tube. The concentration of Aβ1–40 was determined by making a 1 : 50 dilution and measuring the absorbance at 214 nm by UV-vis spectroscopy and determined using ϵ214 = 92 = 1462 M−1 cm−1. The solution was then allowed to incubate for 3 days at 37°C with constant agitation to induce fibril formation. The assay was performed by incubating Aβ1–40 (1 µM) with a constant concentration of ThT (2 µM) in a medium of 20 % DMSO in PBS. Increasing concentrations of [CuL1] in a DMSO stock solution were added to give increasing final concentrations of the compound in the assay (50 nM – 100 µM). Mixtures were thoroughly mixed before being added to a 96 well plate and analysed using a FLUOstar Omega (BMG Labtech) using a excitation filter of 440 nm and an emission filter of 480 nm and performed in triplicate and blanked to a well containing the assay components excluding Aβ1–40. The results were analysed using GraphPad Prism 6 (Graph Pad) using a one site–fit Ki model using a Kd value of 6.18 µM for ThT that had been determined under the same assay conditions (20 % DMSO in PBS).
Paraffin preserved brain tissue blocks were provided by the Victorian Brain Bank Network.
Brain tissue sections (8 μm) were deparaffined (xylene, 3 × 3 min) followed by rehydration (2 min soakings in 100, 90, 70, and 0 % v/v ethanol/water). The hydrated sections were then washed in PBS (5 min). Autofluoresence of the tissue was quenched by treatment with potassium permanganate (0.25 % w/v in PBS, 20 min) followed by washing with PBS (2 × 2 min). The brown coloured tissue was then washed with potassium metabisulfite and oxalic acid (both 1 % w/v in PBS, 6 min). The tissue sections were then washed with PBS (3 × 2 min) followed by blocking with bovine serum albumin (BSA, 2 % w/v in PBS, 10 min), rinsed briefly with PBS and then covered with [CuL1] (10 mM in 30 % DMSO in PBS, 10 min). The sections were then washed with BSA solution again to remove non-specifically bound complex. The sections were then washed with PBS (3 × 2 min), dipped in distilled water, and mounted with non-fluorescent mounting medium (DAKO).
(6-Bromopyridin-3-yl)methanol (2)
This compound was synthesised as previously reported and gave spectra that corresponded well with literature values.[30]
A solution of 6-bromonicotinaldehyde (1) (5.10 g, 27.4 mmol) was dissolved in anhydrous ethanol (150 mL) under an atmosphere of nitrogen gas. To this mixture was added sodium borohydride (1.25 g, 33.0 mmol). The reaction mixture was stirred at room temperature for 24 h. The pH of the reaction mixture was then adjusted to pH 2 by the addition of HCl (1 M) and then to pH 10 by the addition of a saturated solution of NaHCO3. The solvent was removed under reduced pressure and the residue extracted with ethyl acetate (100 mL + 50 mL). The combined organic extracts were washed with brine (2 × 75 mL), dried over MgSO4, and evaporated to dryness under reduced pressure. The resultant solid was purified by column chromatography (SiO2, 30 % ethyl acetate/petroleum spirits) to yield an off-white solid (3.11 g, 16.5 mmol, 60 % yield). m/z (ESI/O-TOF) [C6H6BrNO+H]+ 187.97093, calcd 18797055. δH (400 MHz, CDCl3) 8.32 (d, 4JHH 2.3, 1H, ArH), 7.59 (dd, 3JHH 8.2, 4JHH 2.4, 1H, ArH), 7.47 (d, 3JHH 8.2, 1H, ArH), 4.70 (s, 2H, CH2), 2.37 (s, 1H, OH).
2-Bromo-5-(chloromethyl)pyridine (3)
This compound was synthesised as previously reported and analysis matched with literature values.[30]
(6-Bromopyridin-3-yl)methanol (2) (2.93 g, 11.3 mmol) was dissolved in dichloromethane (40 mL) and treated with an excess of thionyl chloride (7 mL). The reaction was monitored by TLC (33 % ethyl acetate in petroleum spirits; Rf 0.81) and once complete, volatiles were removed by evaporation. The residue was then suspended in saturated NaHCO3 (50 mL) and extracted with ethyl acetate (2 × 50 mL). The organic extracts were combined, dried over MgSO4, and evaporated to dryness to yield a brown oil, which yielded a crystalline white solid upon standing. The solid was suspended in pentane and isolated by filtration, washed with pentane, and air-dried to give a crystalline colourless solid (2.13 g, 10.3 mmol, 91 % yield). δH (400 MHz, CDCl3) 8.37 (d, 4JHH 1.9, 1H, ArH), 7.60 (dd, 3JHH 8.2, 4JHH 2.4, 1H, ArH), 7.50 (d, 3JHH 8.2, 1H, ArH), 4.54 (s, 2H, CH2).
(E)-2-Bromo-5-(2-(pyridin-4-yl)vinyl)pyridine (5)
A solution of 2-bromo-5-(chloromethyl)pyridine (3) (1.05 g, 5.09 mmol) in triethylphosphite (9 mL) was heated to reflux for 4 h. After that time the solvent was removed by evaporation and the resultant residue dissolved in anhydrous tetrahydrofuran (30 mL). To this, 4-nicotinaldehyde (4) (521 mg, 4.86 mmol) was then added, followed by sodium hydride (60 % w/w in mineral oil, 617 mg, 15.4 mmol). The reaction was then stirred for 16 h at room temperature under an atmosphere of nitrogen gas. The reaction was then quenched by the addition of saturated NaHCO3 (25 mL) and then extracted with ethyl acetate (2 × 25 mL). The combined organic extracts were dried over MgSO4, filtered, and the filtrate evaporated to dryness under reduced pressure to yield a yellow solid that was suspended in pentane, isolated by filtration, and air-dried to yield a yellow powder (436 mg, 1.67 mmol, 34 % yield). m/z (ESI/O-TOF) [C12H9BrN2+H]+ 261.00249; calcd 261.00219. δH (500 MHz, CDCl3) 8.61 (m, 2H, PyrH), 8.49 (d, 4JHH 2.5, 1H, PyrH), 7.72 (dd, 3JHH 8.3, 4JHH 2.6, 1H, PyrH), 7.51 (d, 3JHH 8.3, 1H, PyrH), 7.36 (m, 2H, PyrH), 7.21 (d, 3JHH 16.4, 1H, CH=CH), 7.07 (d, 3JHH 16.4, 1H, CH=CH). δC (126 MHz, CDCl3) 150.6 (PyrC), 149.1 (PyrC), 143.6 (PyrC), 141.9 (PyrC), 135.5 (PyrC), 131.4 (PyrC), 129.1 (PyrC), 128.4 (PyrC), 128.2 (CH=CH), 121.1 (CH=CH).
(E)-2-Hydrazino-5-(2-(pyridin-4-yl)vinyl)pyridine (6)
A suspension of (E)-2-bromo-5-(2-(pyridine-4-yl)vinyl)pyridine (5) (372 mg, 1.42 mmol) in hydrazine monohydrate (4 mL) and ethanol (8 mL) was heated at reflux for 16 h. The reaction mixture was then allowed to cool to room temperature and the solvent volume was reduced by approximately half by evaporation, resulting in the precipitation of a yellow solid that was isolated by filtration, washed with water, and air-dried to yield a yellow powder (55 mg, 0.26 mmol, 18 % yield). m/z (ESI/O-TOF) [C12H12N4 + H]+ 213.11373; calcd 213.11347. δH (500 MHz, DMSO-d6) 8.48 (m, 2H, PyrH), 8.18 (d, 4JHH 2.1, 1H, PyrH), 7.84 (m, 2H, NH + PyrH), 7.46 (m, 2H, PyrH), 7.41 (d, 3JHH 16.4, 1H, CH=CH), 6.97 (d, 3JHH 16.4, 1H, CH=CH), 6.75 (d, 3JHH 8.8, 1H, PyrH), 4.24 (s, 2H, NH2). δC (126 MHz, DMSO-d6) 161.7 (PyrC), 149.9 (PyrC), 148.3 (PyrC), 144.9 (PyrC), 133.9 (PyrC), 130.6 (CH=CH), 121.2 (CH=CH), 120.9 (PyrC), 120.3 (PyrC), 106.4 (PyrC).
Diacetyl-mono-4-methyl-3-thiosemicarbazone (7)
This compound was synthesised as previously reported.[46]
A solution of 2,3-butanedione (2.40 mL, 27.6 mmol) in water (50 mL) was acidified with HCl (conc., 5 drops) and cooled in an ice bath. To this, 4-methyl-3-thiosemicarbazide (2.65 g, 25.2 mmol) was added portion-wise over the course of 10 min, resulting in the precipitation of a white solid. The reaction mixture was allowed to warm to room temperature and the mixture was extracted with chloroform (2 × 40 mL) and the combined organic extracts dried over MgSO4, filtered, and concentrated. The solution was then diluted with pentane, precipitating a colourless solid that was collected by filtration, washed with pentane, and air-dried to yield a white solid (3.68 g, 21.2 mmol, 84 % yield). m/z (ESI/O-TOF) [C6H11N3OS+H]+ 174.06963; calcd 174.06956. δH (400 MHz, DMSO-d6) 10.63 (s, 1H, NNH(C=S)N), 8.62 (d, 3JHH 3.9, 1H, CH3NH), 3.05 (d, 3JHH 4.6, 3H, CH3NH), 2.42 (s, 3H, CH3), 1.96 (s, 3H, CH3).
Diacetyl-2-((E)-5-(2-(6-hydrazinopyridin-3-yl)vinyl)pyridine)-(4-ethyl-3-thiosemicarbazone) (H2L1)
A suspension of diacetyl-mono-4-methyl-3-thiosemicarbazone (7) (48 mg, 0.28 mmol) and (E)-2-hydrazino-5-(2-(pyridine-4-yl)vinyl)pyridine (6) (46 mg, 0.22 mmol) in ethanol (4 mL) was acidified with acetic acid (4 drops) and heated at reflux for 2 h, resulting in the precipitation of an orange solid. The reaction was allowed to cool to room temperature and the solid isolated by filtration, which was then washed with diethyl ether and air-dried to yield an orange powder (52 mg, 0.14 mmol, 64 % yield). Anal. Calc. for C18H21N7S: C 58.83, H 5.76, N 26.68 %. Found: C 57.23, H 5.76, N 26.01. m/z (ESI/O-TOF) [C18H21N7S+H]+ 368.18530; calcd 368.16519. Rt (RP-HPLC) 8.79 min. δH (500 MHz, DMSO-d6) 10.20 (s, 1H, PyrNH), 10.16 (s, 1H, NNH(C=S)N), 8.53 (dd, 3JHH 4.6, 4JHH 1.5, 2H, PyrH), 8.42 (d, 4JHH 2.0, 1H, PyrH), 8.32 (q, 3JHH 4.3, 1H, CH3NH), 8.04 (dd, 3JHH 8.8, 4JHH 2.2, 1H, PyrH), 7.53–7.49 (m, 3H, PyrH + CH=CH), 7.31 (d, 3JHH 8.8, 1H, PyrH), 7.16 (d, 3JHH 16.5, 1H, CH=CH), 3.04 (d, 3JHH 4.6, 3H, CH3NH), 2.24 (s, 6H, CH3C=N). δC (126 MHz, DMSO-d6) 178.4 (C=S), 156.9 (PyrC), 149.9 (PyrC), 148.5 (C=N), 147.8 (PyrC), 145.3 (C=N), 144.7 (PyrC), 135.4 (PyrC), 130.1 (CH=CH), 124.4 (PyrC), 123.5 (CH=CH), 120.5 (PyrC), 107.2 (PyrC), 31.2 (CH3NH), 11.5 (CH3C=N), 11.0 (CH3C=N).
[Cu(diacetyl-2-((E)-5-(2-(6-hydrazinopyridin-3-yl)vinyl)pyridine)-(4-ethyl-3-thiosemicarbazone))] ([CuL1])
A suspension of diacetyl-2-((E)-5-(2-(6-hydrazinopyridin-3-yl)vinyl)pyridine)-(4-ethyl-3-thiosemicarbazone) (H2L1) (23 mg, 0.06 mmol) in methanol (4 mL) was alkalised with triethylamine (4 drops). To this, copper acetate monohydrate (19 mg, 0.10 mmol) was added and the reaction heated to reflux for 2 h. The reaction was then allowed to cool to room temperature and diluted with water. The mixture was filtered, washed with water, and air-dried to yield a dark coloured solid (24 mg, 0.06 mmol, quant. yield). Anal. Calc. for C18H19CuN7S: C 47.41, H 4.86, N 21.50 %. Found: C 47.19, H 4.36, N 20.91. m/z (ESI/O-TOF) [C18H19CuN7S+2H]2+ 215.04301, [C18H19CuN7S+H]+ 429.07867; calcd 215.04321, 429.07914. Rt (RP-HPLC) 9.28 min. Crystals of sufficient quality for X-ray analysis were grown from exchange of vapours of pentane and a solution of the complex dissolved in chloroform.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors recognise financial support from the Australian Research Council. The authors thank the Victorian Brain Bank Network for the provision of human brain tissue and Prof. Rodney J. Hicks (Peter MacCallum Cancer Centre) for radiochemistry laboratory facilities.
References
[1] W. D. McFadyen, R. Robson, H. Schaap, Inorg. Chem. 1972, 11, 1777.| Crossref | GoogleScholarGoogle Scholar |
[2] W. D. McFadyen, R. Robson, H. A. Schaap, J. Coord. Chem. 1978, 8, 59.
| Crossref | GoogleScholarGoogle Scholar |
[3] M. A. Green, D. L. Klippenstein, J. R. Tennison, J. Nucl. Med. 1988, 29, 1549.
| 3261785PubMed |
[4] M. A. Green, Int. J. Rad. Appl. Instrum. B 1987, 14, 59.
| Crossref | GoogleScholarGoogle Scholar | 3583756PubMed |
[5] M. A. Green, C. J. Mathias, M. J. Welch, A. H. McGuire, D. Perry, F. Fernandez-Rubio, J. S. Perlmutter, M. E. Raichle, S. R. Bergmann, J. Nucl. Med. 1990, 31, 1989.
| 2266398PubMed |
[6] Y. Fujibayashi, H. Taniuchi, Y. Yonekura, H. Ohtani, J. Konishi, A. Yokoyama, J. Nucl. Med. 1997, 38, 1155.
| 9225812PubMed |
[7] A. L. Vāvere, J. S. Lewis, Dalton Trans. 2007, 4893.
| Crossref | GoogleScholarGoogle Scholar | 17992274PubMed |
[8] B. M. Paterson, P. S. Donnelly, Chem. Soc. Rev. 2011, 40, 3005.
| Crossref | GoogleScholarGoogle Scholar | 21409228PubMed |
[9] M. Ikawa, H. Okazawa, T. Tsujikawa, A. Matsunaga, O. Yamamura, T. Mori, T. Hamano, Y. Kiyono, Y. Nakamoto, M. Yoneda, Neurology 2015, 84, 2033.
| Crossref | GoogleScholarGoogle Scholar | 25904686PubMed |
[10] M. T. Fodero-Tavoletti, V. L. Villemagne, B. M. Paterson, A. R. White, Q. X. Li, J. Camakaris, G. O’Keefe, R. Cappai, K. J. Barnham, P. S. Donnelly, J. Alzheimers Dis. 2010, 20, 49.
| Crossref | GoogleScholarGoogle Scholar | 20164590PubMed |
[11] D. J. Selkoe, Neuron 1991, 6, 487.
| Crossref | GoogleScholarGoogle Scholar | 1673054PubMed |
[12] D. J. Selkoe, Cell 2013, 154, 468.
| Crossref | GoogleScholarGoogle Scholar | 23870132PubMed |
[13] G. G. Glenner, C. W. Wong, in Amyloidosis (Eds J. Marrink, M. H. Van Rijswijk) 1986, pp. 227–242 (Springer Netherlands: Dordrecht).
[14] J. A. Hardy, G. A. Higgins, Science 1992, 256, 184.
| Crossref | GoogleScholarGoogle Scholar | 1566067PubMed |
[15] C. L. Masters, G. Simms, N. A. Weinman, G. Multhaup, B. L. McDonald, K. Beyreuther, Proc. Natl. Acad. Sci. USA 1985, 82, 4245.
| Crossref | GoogleScholarGoogle Scholar | 3159021PubMed |
[16] A. Nordberg, Lancet Neurol. 2004, 3, 519.
| Crossref | GoogleScholarGoogle Scholar | 15324720PubMed |
[17] B. Dubois, H. H. Feldman, C. Jacova, H. Hampel, J. L. Molinuevo, K. Blennow, S. T. DeKosky, S. Gauthier, D. Selkoe, R. Bateman, S. Cappa, S. Crutch, S. Engelborghs, G. B. Frisoni, N. C. Fox, D. Galasko, M.-O. O. Habert, G. A. Jicha, A. Nordberg, F. Pasquier, G. Rabinovici, P. Robert, C. Rowe, S. Salloway, M. Sarazin, S. Epelbaum, L. C. de Souza, B. Vellas, P. J. Visser, L. Schneider, Y. Stern, P. Scheltens, J. L. Cummings, Lancet Neurol. 2014, 13, 614.
| Crossref | GoogleScholarGoogle Scholar | 24849862PubMed |
[18] C. A. Mathis, B. J. Bacskai, S. T. Kajdasz, M. E. McLellan, M. P. Frosch, B. T. Hyman, D. P. Holt, Y. Wang, G.-F. Huang, M. L. Debnath, W. E. Klunk, Bioorg. Med. Chem. Lett. 2002, 12, 295.
| Crossref | GoogleScholarGoogle Scholar | 11814781PubMed |
[19] W. E. Klunk, H. Engler, A. Nordberg, Y. Wang, G. Blomqvist, D. P. Holt, M. Bergström, I. Savitcheva, G. F. Huang, S. Estrada, B. Ausén, M. L. Debnath, J. Barletta, J. C. Price, J. Sandell, B. J. Lopresti, A. Wall, P. Koivisto, G. Antoni, C. A. Mathis, B. Långström, Ann. Neurol. 2004, 55, 306.
| Crossref | GoogleScholarGoogle Scholar | 14991808PubMed |
[20] H. F. Kung, S. R. Choi, W. Qu, W. Zhang, D. Skovronsky, J. Med. Chem. 2010, 53, 933.
| Crossref | GoogleScholarGoogle Scholar | 19845387PubMed |
[21] L. Zhu, K. Ploessl, H. F. Kung, Chem. Soc. Rev. 2014, 43, 6683.
| Crossref | GoogleScholarGoogle Scholar | 24676152PubMed |
[22] A. Jureus, B. M. Swahn, J. Sandell, F. Jeppsson, A. E. Johnson, P. Johnstrom, J. A. Neelissen, D. Sunnemark, L. Farde, S. P. Svensson, J. Neurochem. 2010, 114, 784.
| Crossref | GoogleScholarGoogle Scholar | 20477945PubMed |
[23] K. K. Skeby, J. Sørensen, B. Schiøtt, J. Am. Chem. Soc. 2013, 135, 15114.
| Crossref | GoogleScholarGoogle Scholar | 23859103PubMed |
[24] G. Kuang, N. A. Murugan, Y. Tu, A. Nordberg, H. Ågren, J. Phys. Chem. B 2015, 119, 11560.
| Crossref | GoogleScholarGoogle Scholar | 26266837PubMed |
[25] N. A. Murugan, C. Halldin, A. Nordberg, B. Långström, H. Ågren, J. Phys. Chem. Lett. 2016, 7, 3313.
| Crossref | GoogleScholarGoogle Scholar | 27498616PubMed |
[26] S. Lim, B. M. Paterson, M. T. Fodero-Tavoletti, G. J. O’Keefe, R. Cappai, K. J. Barnham, V. L. Villemagne, P. S. Donnelly, Chem. Commun. 2010, 46, 5437.
| Crossref | GoogleScholarGoogle Scholar |
[27] A. R. Cowley, J. R. Dilworth, P. S. Donnelly, J. M. White, Inorg. Chem. 2006, 45, 496.
| Crossref | GoogleScholarGoogle Scholar | 16411680PubMed |
[28] J. L. Hickey, S. Lim, D. J. Hayne, B. M. Paterson, J. M. White, V. L. Villemagne, P. Roselt, D. Binns, C. Cullinane, C. M. Jeffery, R. I. Price, K. J. Barnham, P. S. Donnelly, J. Am. Chem. Soc. 2013, 135, 16120.
| Crossref | GoogleScholarGoogle Scholar | 24070589PubMed |
[29] L. E. McInnes, A. Noor, K. Kysenius, C. Cullinane, P. Roselt, C. A. McLean, F. C. K. Chiu, A. K. Powell, P. J. Crouch, J. M. White, P. S. Donnelly, Inorg. Chem. 2019, 58, 3382.
| Crossref | GoogleScholarGoogle Scholar | 30785268PubMed |
[30] D. J. Hayne, J. M. White, C. A. McLean, V. L. Villemagne, K. J. Barnham, P. S. Donnelly, Inorg. Chem. 2016, 55, 7944.
| Crossref | GoogleScholarGoogle Scholar | 27459001PubMed |
[31] P. J. Blower, T. C. Castle, A. R. Cowley, J. R. Dilworth, P. S. Donnelly, E. Labisbal, F. E. Sowrey, S. J. Teat, M. J. Went, Dalton Trans. 2003, 4416.
| Crossref | GoogleScholarGoogle Scholar |
[32] A. R. Cowley, J. R. Dilworth, P. S. Donnelly, E. Labisbal, A. Sousa, J. Am. Chem. Soc. 2002, 124, 5270.
| Crossref | GoogleScholarGoogle Scholar | 11996559PubMed |
[33] J. L. Dearling, J. S. Lewis, G. E. Mullen, M. J. Welch, P. J. Blower, J. Biol. Inorg. Chem. 2002, 7, 249.
| Crossref | GoogleScholarGoogle Scholar | 11935349PubMed |
[34] P. S. Donnelly, A. Caragounis, T. Du, K. M. Laughton, I. Volitakis, R. A. Cherny, R. A. Sharples, A. F. Hill, Q.-X. Li, C. L. Masters, K. J. Barnham, A. R. White, J. Biol. Chem. 2008, 283, 4568.
| Crossref | GoogleScholarGoogle Scholar | 18086681PubMed |
[35] Z. Xiao, P. S. Donnelly, M. Zimmermann, A. G. Wedd, Inorg. Chem. 2008, 47, 4338.
| Crossref | GoogleScholarGoogle Scholar | 18412332PubMed |
[36] B. M. Paterson, C. Cullinane, P. J. Crouch, A. R. White, K. J. Barnham, P. D. Roselt, W. Noonan, D. Binns, R. J. Hicks, P. S. Donnelly, Inorg. Chem. 2019, 58, 4540.
| Crossref | GoogleScholarGoogle Scholar | 30869878PubMed |
[37] H. Levine, Protein Sci. 1993, 2, 404.
| Crossref | GoogleScholarGoogle Scholar | 8453378PubMed |
[38] K. Gade Malmos, L. M. Blancas-Mejia, B. Weber, J. Buchner, M. Ramirez-Alvarado, H. Naiki, D. Otzen, Amyloid 2017, 24, 1.
| Crossref | GoogleScholarGoogle Scholar | 28393556PubMed |
[39] S. A. Hudson, H. Ecroyd, T. W. Kee, J. A. Carver, FEBS J. 2009, 276, 5960.
| Crossref | GoogleScholarGoogle Scholar | 19754881PubMed |
[40] L. P. Jameson, N. W. Smith, S. V. Dzyuba, ACS Chem. Neurosci. 2012, 3, 807.
| Crossref | GoogleScholarGoogle Scholar | 23173064PubMed |
[41] H. Watanabe, A. Kawasaki, K. Sano, M. Ono, H. Saji, Bioorg. Med. Chem. 2016, 24, 3618.
| Crossref | GoogleScholarGoogle Scholar | 27301677PubMed |
[42] N. Bandara, A. K. Sharma, S. Krieger, J. W. Schultz, B. H. Han, B. E. Rogers, L. M. Mirica, J. Am. Chem. Soc. 2017, 139, 12550.
| Crossref | GoogleScholarGoogle Scholar | 28823165PubMed |
[43] G. Sheldrick, Acta Crystallogr. A 2008, 64, 112.
| Crossref | GoogleScholarGoogle Scholar | 18156677PubMed |
[44] G. Sheldrick, Acta Crystallogr. C 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[45] L. Farrugia, J. Appl. Cryst. 2012, 45, 849.
| Crossref | GoogleScholarGoogle Scholar |
[46] B. M. Paterson, J. A. Karas, D. B. Scanlon, J. M. White, P. S. Donnelly, Inorg. Chem. 2010, 49, 1884.
| Crossref | GoogleScholarGoogle Scholar | 20055473PubMed |
* This manuscript is dedicated to Professor Richard Robson in recognition of his enormous contributions to inorganic chemistry.