
Navigating phylogenetic conflict and evolutionary inference in plants with target-capture data
E. M. Joyce A * , A. N. Schmidt-Lebuhn B , H. K. Orel C , F. J. Nge D , B. M. Anderson E , T. A. Hammer F G and T. G. B. McLay C H I *
A * , A. N. Schmidt-Lebuhn B , H. K. Orel C , F. J. Nge D , B. M. Anderson E , T. A. Hammer F G and T. G. B. McLay C H I *
A
B
C
D
E
F
G
H
I
Abstract
Target capture has rapidly become a preferred approach for plant systematic and evolutionary research, marking a step change in the generation of data for phylogenetic inference. Although this advancement has facilitated the resolution of many relationships, phylogenetic conflict continues to be reported and is often attributed to genome duplication, reticulation, incomplete lineage sorting or rapid speciation – common processes in plant evolution. The proliferation of methods for analysing target-capture data in the presence of these processes can be overwhelming for many researchers, especially students. In this review, we break down the causes of conflict and guide researchers through a target-capture bioinformatic workflow, with a particular focus on robust phylogenetic inference in the presence of conflict. Through the workflow, we highlight key considerations for reducing artefactual conflict, managing paralogs and assessing conflict, and discuss current methods for investigating causes of conflict. Although we draw from examples in the Australian flora, this review is broadly relevant for any researcher working with target-capture data. We conclude that conflict is often inherent in plant phylogenomic datasets, and, although further methodological development is needed, when conflict is carefully investigated, target-capture data can provide unprecedented insight into the extraordinary evolutionary histories of plants.
Keywords: Angiosperms353, deep coalescence, discordance, GAP, Genomics for Australian Plants, hybridisation, HybSeq, incomplete lineage sorting, incongruence, PAFTOL, paralogy, polyploidy, polytomy, target enrichment.
Introduction
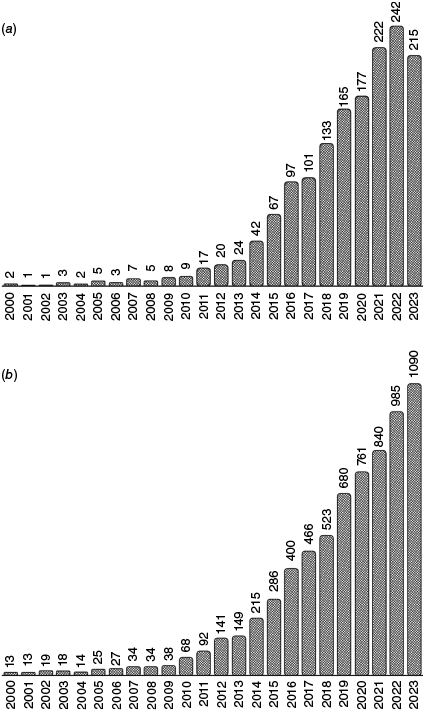
Target-capture sequencing (also referred to as target enrichment and HybSeq) has rapidly become a preferred approach for phylogenetic inquiry. In Australian plant systematics, a multitude of data types is used (Nauheimer et al. 2019; Fowler et al. 2020; Gunn et al. 2020, 2024; Orel et al. 2023, 2024) but the ongoing trend points to a greater adoption of target-capture sequencing (Fig. 1). Briefly, target-capture sequencing works by breaking genomic DNA into short fragments, capturing fragments that belong to loci of interest (‘target loci’) by binding (‘hybridising’) these to pre-designed baits (also referred to as ‘probes’) and subsequently amplifying those fragments while washing away superfluous fragments. The remaining DNA fragments of targeted loci (often called ‘enriched’ or ‘hybridised’ libraries) are subsequently sequenced and these short sequences (‘reads’) are reassembled through a bioinformatic pipeline to reconstruct the sequences of the targeted loci (for more detail see e.g. Andermann et al. 2020). The rapid uptake of this technique in phylogenetic research is due to the many advantages that target-capture data offer, including the ability to sequence a high number of loci with large amounts of phylogenetic information, compatibility across datasets using the same baits and the ability to obtain targeted loci from degraded material such as herbarium specimens (Hart et al. 2016; Shee et al. 2020). This has been further expedited through the establishment of initiatives such as the Plant and Fungal Trees of Life (PAFTOL) project in 2016 (Baker et al. 2021; https://www.kew.org/science/our-science/projects/plant-and-fungal-trees-of-life), and Genomics for Australian Plants (GAP; https://www.genomicsforaustralianplants.com) in 2017. These ventures coordinated efforts of researchers and institutions to use the Angiosperms353 bait kit (Johnson et al. 2019) to sequence 353 single- or low-copy nuclear loci conserved in angiosperms, facilitating the generation of the most densely sampled and data-rich nuclear phylogeny of angiosperms to date (Zuntini et al. 2024). Other universal bait kits have also been developed during the past 5 years, such as the GoFlag bait kit for flagellate plants (Breinholt et al. 2021) and the OzBaits kit for Australian plants (Waycott et al. 2021). Custom bait kits for particular groups are commonly used for finer-scale phylogenetic investigation (e.g. Compositae1061, Vatanparast et al. 2018; Siniscalchi et al. 2021) or for groups with low recovery of Angiosperms353 loci. This has culminated in the production of an unprecedented volume of data for plant phylogenomic research across taxonomic levels within the Australian flora, as detailed in Table 1.
Number of academic papers in Google Scholar published each year from 2000 to 2023 matching the search terms: ‘target capture’ OR ‘target enrichment’ OR ‘Hyb-Seq’ AND ‘DNA’ AND ‘plant’. (a) Matches also including the search term ‘Australia’. (b) Matches not including the search term ‘Australia’. Obtained using the Python script of Strobel (2018).
| Plant group | Baits kit | Assembly method | Tree inference method (concatenated or coalescent) | Authors | DOI | |
|---|---|---|---|---|---|---|
| Caladenia and Diurideae (Orchidaceae) | Custom baits (up to 1000+ loci) | Custom pipeline, HybPiper | Both | Peakall et al. (2021) | 10.1111/1755-0998.13327 | |
| Eucalypts (Myrtaceae) | Custom baits (101 low-copy nuc exons) | Custom pipeline | Both | Crisp et al. (2024) | 10.1111/jse.13047 | |
| Eucalyptus (Myrtaceae) | Custom baits (568 nuc genes, including Angiosperms353 and OzBaits) | HybPiper-nf, HybPhaser | Both | McLay et al. (2023) | 10.1016/j.ympev.2023.107869 | |
| Calandrinia (Montiaceae) | Custom baits for Caryophyllales | Custom pipeline | Both | Hancock et al. (2018) | 10.1002/ajb2.1110 | |
| Cryptandra (Rhamnaceae) | OzBaits | Custom pipeline | Concatenated | Nge et al. (2024) | 10.1093/botlinnean/boad051 | |
| Pomaderris (Rhamnaceae) | OzBaits | Custom pipeline | Concatenated | Nge et al. (2021b) | 10.1016/j.ympev.2021.107085 | |
| Calytrix (Myrtaceae) | OzBaits | Custom pipeline | Both | Nge et al. (2022) | 10.1002/ajb2.1790 | |
| Adenanthos (Proteaceae) | OzBaits | Custom pipeline | Both | Nge et al. (2021a) | 10.3389/fevo.2020.616741 | |
| Crinum (Amaryllidaceae) | OzBaits | Custom pipeline | Concatenated | Simpson et al. (2022) | 10.1071/SB21038 | |
| Halophila (Hydrocharitaceae) | OzBaits | Custom pipeline | Concatenated | Van Dijk et al. (2023) | 10.3390/d15010111 | |
| Pogonolepis (Asteraceae) | Angiosperms353 | HybPiper | Concatenated | Schmidt-Lebuhn (2022) | 10.1071/SB22010 | |
| Anthemideae tribe (Asteraceae) | Angiosperms353 | HybPiper-nf | Concatenated | Schmidt-Lebuhn and Grealy (2024) | 10.1071/SB23012 | |
| Gnaphalieae tribe (Asteraceae) | Custom baits (Compositae 1061) | HybPiper | Both | Schmidt-Lebuhn and Bovill (2021) | 10.1002/tax.12510 | |
| Hakea (Proteaceae) | Custom bait kit (450 nuc loci) | Custom pipeline | Both | Cardillo et al. (2017) | 10.1111/evo.13276 | |
| Thelypteridaceae | GoFlag (451 nuc loci) | HybPiper, HybPhaser | Both | Bloesch et al. (2022) | 10.1016/j.ympev.2022.107526 | |
| Cunoniaceae | Angiosperms353 | HybPiper | Coalescent | Pillon et al. (2021) | 10.1002/ajb2.1688 | |
| Zanthoxyloideae subfamily (Rutaceae; Sapindales) | Angiosperms353 | HybPiper, HybPhaser | Both | Joyce et al. (2023) | 10.3389/fpls.2023.1063174 | |
| Eriostemon group (Rutaceae) | Angiosperms353 | HybPiper-nf, HybPhaser | Both | Orel et al. (2025) | 10.1002/tax.13308 | |
| Aglaia (Meliaceae) | Angiosperms353 | HybPiper, HybPhaser | Concatenated | Cooper et al. (2023) | 10.54102/ajt.p8to6 | |
| Celmisiinae tribe | Angiosperms353 | HybPiper-nf | Both | Nicol et al. (2024) | 10.1016/j.ympev.2024.108064 | |
| Hibbertia (Dilleniaceae) | Angiosperms353, OzBaits (nuc), OzBaits (cp) | CAPTUS | Both | Hammer et al. (2025) | 10.1071/SB24009 | |
| Drosera (Droseraceae) | Angiosperms353, OzBaits (nuc), OzBaits (cp) | CAPTUS | Both | Williamson et al. (2025) | 10.1071/SB24016 | |
| Chamelaucieae tribe (Myrtaceae) | Angiosperms353 | HybPiper | Both | Nge et al. (2025) | 10.1071/SB24014 | |
| Minuria (Asteraceae) | Angiosperms353 | HybPiper-nf | Both | Schmidt-Lebuhn et al. (2024b) | 10.1071/SB23028 |
Ongoing work on several other Australian plant groups as part of GAP Stage 2 using Angiosperms353 baits can be found here: https://www.genomicsforaustralianplants.com/phylogenomics/. ‘Nuclear’ is abbreviated as ‘nuc’; ‘chloroplast’ is abbreviated as ‘cp’.
Although target capture has aided the resolution of many previously elusive plant relationships (e.g. Larridon et al. 2021; Pillon et al. 2021; Schmidt-Lebuhn and Grealy 2024), it has proven to not be the ‘silver bullet’ for resolving the evolutionary history of many plant groups as well-supported bifurcating trees, with many studies reporting the presence of phylogenetic ‘conflict’. Conflict, also often referred to as ‘discordance’ or ‘incongruence’, refers to when phylogenies of individual loci do not share the same topology with either the species tree or each other. Such conflict can be the result of contamination during laboratory work, artefacts introduced by researchers during data analysis, or inherent biases in target-capture data (Steenwyk et al. 2023; Frost et al. 2024). Alternatively, conflict can be the product of real biological processes that cause evolutionary histories of genes and lineages to deviate from each other or a bifurcating tree. Such processes, such as whole-genome duplication (WGD) events, reticulation and incomplete lineage sorting (ILS) have long been known to be common and important events in the evolution of plants but were difficult to detect in phylogenetic studies prior to the broad adoption of high-throughput sequencing techniques. Currently, the findings of a growing number of target-capture datasets demonstrate that these processes are pervasive in plants, manifesting as conflict. For example, in the Australian flora, conflict in target-capture datasets has been attributed to WGD events in Adenanthos (Nge et al. 2021a), Pomaderris (Nge et al. 2021b), Calytrix (Nge et al. 2022), Cryptandra (Nge et al. 2024), Senecio (Schmidt-Lebuhn et al. 2024a), Celmisiinae (Nicol et al. 2024) and many lineages in Sapindales (Joyce et al. 2023). Conflict due to reticulation has been detected in Adansonia (Karimi et al. 2020) and Thelypteridaceae (Bloesch et al. 2022), and reticulation in concert with deep coalescence in Adenanthos (Nge et al. 2021a) and Eucalyptus (McLay et al. 2023).
As illustrated by these examples, conflict in target-capture data, if handled carefully, can yield insight into key biological processes in the evolutionary history of plants. However, the recent, rapid proliferation of software and pipelines for target-capture analysis can be confusing for those new to the field, especially students. Faced with this abundance of methods, it can difficult to determine how to best design an analytical pipeline that minimises artefactual conflict and enables the investigation of any biological processes underlying conflict. In this review, we explain what causes phylogenetic conflict, and describe the key steps in a target-capture bioinformatic pipeline that should be considered when phylogenetic conflict is present. Although this is not an exhaustive review of all software available, we highlight practical considerations for reconstructing and interpreting a phylogeny from the point of having raw reads to: (1) locus extraction, (2) paralogy reconciliation, (3) phylogenomic reconstruction of gene trees and species trees, (4) conflict assessment, and (5) investigating patterns and underlying causes of conflict. Through the steps of the pipeline we draw attention to key points where conflict can be introduced, make recommendations on how to minimise artefactual conflict, and summarise current approaches for testing for the biological processes of WGD, reticulation, ILS and simultaneous or rapid speciation that may underlie any remaining phylogenetic conflict. We subsequently highlight remaining issues with current methods and areas in need of further research.
What is phylogenetic conflict and what are the causes?
Phylogenetic conflict, also often referred to as ‘discordance’ or ‘incongruence’, refers to situations in which subsets of phylogenetic data (such as gene trees, quartets or sites) do not share the same topology with either the species tree or each other (Lanfear and Hahn 2024). This often leads to species trees being ‘unresolved’ with poorly supported topologies, and is commonly viewed as a hindrance to conclusive evolutionary inference (see ‘4. Conflict assessment’ for more detail). However, providing that data are correctly handled, conflict can present an opportunity to gain new insight into the evolutionary history of a lineage. Assuming that data are free from contamination, conflict in a phylogenetic dataset can be attributed to two main sources: biological processes that cause individual loci to have topologies that are different from each other and the species tree or that cause the lineage’s evolutionary history to deviate from a bifurcating pattern (‘Biological sources of conflict’) and errors introduced into the dataset by the researcher through the analytical pipeline (‘Artefactual conflict’).
Biological sources of conflict
There are four main issues that can cause conflict in a phylogenetic dataset, that are the result of real, biological processes: (1) paralogy, (2) reticulation, (3) deep coalescence and ILS, and (4) simultaneous speciation or rapid radiation (Fig. 2). The processes that can lead to each issue are summarised below. Most are common in evolution, particularly in plants, and therefore care should be taken to design an analytical pipeline that can detect these processes. Detecting biological conflict can provide valuable insight into the evolutionary history of a lineage and can enrich the inferences made during scientific enquiry.
Possible scenarios for gene evolution during species diversification. (a) Congruence between the species tree (pink bars) and gene tree (narrow lines). (b) Paralogy with one gene duplication and no gene losses. Red and blue indicate two ortholog groups. (c) Paralogy with one gene duplication followed by gene losses (or failure to capture or assemble gene copies) that left no evidence of paralogy. (d) Introgression (reticulation). (e) Allopolyploid hybridogenic speciation (reticulation). (f) Deep coalescence. The dotted line marked ‘ILS’ indicates transient incomplete lineage sorting in an ancestral lineage, i.e. the two alleles present in the middle lineage (large population) are not monophyletic (one is more closely related to an allele in the sister lineage). (g) True multifurcation due to simultaneous speciation.
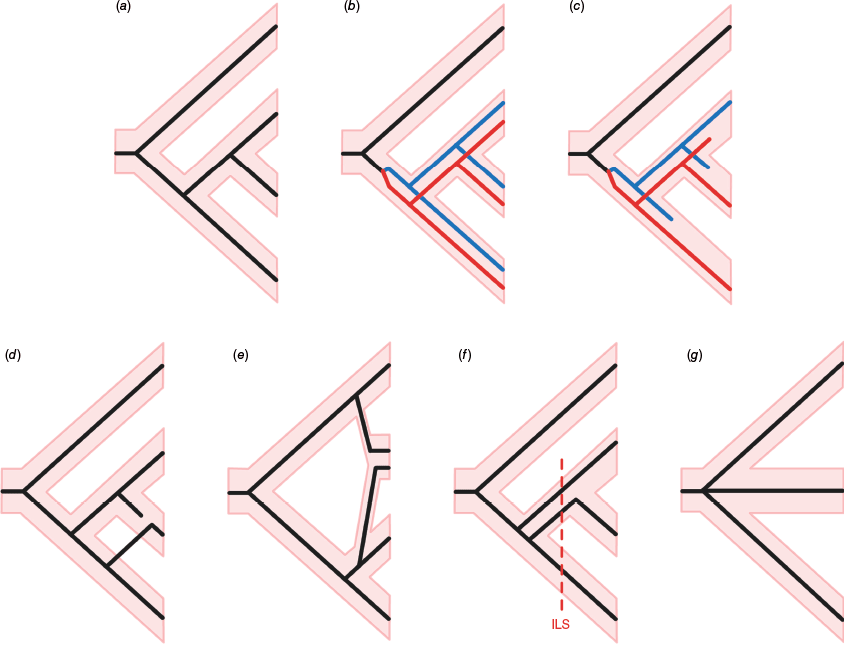
Generally in target-capture sequencing, single-copy loci are targeted to ensure sequences that share a common ancestor (‘orthologs’) are analysed. However, even when attempting to target single-copy loci, paralogy is commonly encountered, whereby multiple copies of the same locus are present in a phylogenetic dataset. Paralogy is caused either by gene duplication or WGD followed by lineage diversification. WGD events involve the doubling of an organism’s entire genetic material within the same species (autopolyploidy) or during interspecies hybridisation (allopolyploidy, also see below) (del Pozo and Ramirez-Parra 2015). These are known to be common and important sources of diversity in the evolution of land plants but present challenges for phylogenomic analysis (Clark and Donoghue 2018; Morales-Briones et al. 2021).
Divergent evolution of the resulting gene copies will lead to differences that are inherited by descendent species and can cause retained gene copies in the same individual to be placed in separate clades in phylogenetic analyses. Copies from the same clade (or ortholog group) of the resulting gene family phylogeny represent orthologs (descendants of the same copy) but copies in separate clades are paralogs (descendants of different copies). Treating paralogs as orthologs can mislead phylogenetic analysis (Struck 2013), and therefore avoiding the analysis of paralogs (see section ‘Step 2. Paralog reconciliation’) is usually important. In an ideal case, paralogy would be easily recognised by observing sister clades in a gene tree that both contain the same complement of samples (Fig. 2b) and for WGD, this pattern would be replicated across all genes. In these cases, WGD is easily detected in a phylogenetic dataset if paralogs are appropriately handled.
Although duplicated gene copies can be retained (and often specialise in function – a major source of evolutionary novelty; Flagel and Wendel 2009), more often the locus will rediploidise over time (lose one or more of the copies), leading to a more ambiguous pattern of gene duplications and losses (Fig. 2c) (Bomblies 2020; Mason and Wendel 2020). This means that some loci (although the exact proportion in plants is unknown, in yeasts this is estimated to be ~10% of loci; Scannell et al. 2006) will be present as single-copy loci but are acutally ‘hidden paralogs’ (also referred to as pseudo-orthologs) rather than orthologs. Therefore, even with careful handling of paralogs in a target-capture dataset, hidden paralogy may be an unavoidable and undetectable source of conflict in a dataset, especially in smaller datasets (Xiong et al. 2022). In contrast, some theoretical models suggest that hidden paralogs have negligible effect on species tree inference in specific biological scenarios (Smith and Hahn 2022). More studies are needed to understand the impact of hidden paralogs in phylogenetic inference in plants.
Reticulation is caused by a variety of processes such as introgression and allopolyploid speciation that are often colloquially lumped together as ‘hybridisation’. Introgression at the same ploidy level occurs when partially fertile hybrids between two species back-cross with parental species, leading to the movement of genetic material between the parents. It is increasingly recognised as a major driver of plant evolution, serving as a source of additional genetic variation in species and potentially even facilitating adaptation to novel stresses or habitats (Suarez-Gonzalez et al. 2018; Edelman and Mallet 2021). Conversely, introgression of maladaptive alleles during incipient speciation can lead to strong selective pressure towards reproductive isolation (Ostevik et al. 2016). In a phylogenetic context, introgression leads to incongruence between gene tree and species tree because the transfer of alleles between species follows a reticulate rather than bifurcating pattern (Fig. 2d).
A special case of introgression is organelle capture, with chloroplast capture particularly relevant to plant phylogenetics because of the field’s traditional reliance on chloroplast loci. Evidence across many taxa indicates that organelles are more easily transferred across lineages than nuclear genes (Stegemann et al. 2012), resulting in cases where plastid phylogenies are incongruent with morphological, nuclear ribosomal and low copy nuclear data (e.g. Schmidt-Lebuhn and Bovill 2021). Perhaps the best-known Australian example where phylogenetic inference has been confounded by frequent chloroplast capture is in eucalypts (McKinnon et al. 1999; Nevill et al. 2014). Organelle capture can sometimes be inferred from conflict in the topology of species trees generated with plastid, morphological and nuclear data, in combination with tests for introgression in the nuclear dataset (e.g. Nge et al. 2021a; McLay et al. 2023).
Allopolyploid speciation occurs when a hybrid between two species that is normally sterile due to an inability to produce functional gametes undergoes WGD, often through the production of unreduced gametes (Fig. 2e). This allows meiosis to be successful in the offspring, as the chromosomes can subsequently pair with the duplicates. Allopolyploid speciation is a major factor in plant evolution (Soltis et al. 2009; Aïnouche and Wendel 2014; Alix et al. 2017; Clark and Donoghue 2018). Allopolyploid speciation produces phylogenetic conflict in a dataset in two ways: it results in a non-bifurcating pattern of gene inheritance due to the crossing of two species, and it doubles gene copies. As in WGD or gene duplication without hybridisation, allopolyploidy results in paralogs in a phylogenomic dataset and the gene copies can specialise or rediploidise through gene losses (Bomblies 2020).
Under coalescent theory, incongruence between gene trees and between an individual gene tree and the species tree is expected even in the absence of paralogy or reticulation. The underlying process is referred to as either ‘deep coalescence’ or ‘ILS’, and has been understood for decades (Pamilo and Nei 1988; Maddison 1997). Ancestral species with large effective population sizes are able to maintain a high diversity of alleles. At a lineage split in an ancestral species, both daughter species inherit a random sample of this diversity. If a gene lineage splits simultaneously with the species split, over time genetic drift will lead to the extinction of relictual ancestral alleles and the remaining alleles in each species will be monophyletic (i.e. completely sorted), and the gene tree will be concordant with the species tree. However, if effective population sizes remain large and species lineage splits follow quickly upon each other, ancestral alleles that diverged prior to the species splits will not yet have been lost (i.e. they are incompletely sorted) and may be inherited. In this case, the gene tree will not reflect the species phylogeny (Fig. 2f). This pattern is referred to as deep coalescence when looking deeper in the tree (Rannala et al. 2020), because the gene lineages coalesce more deeply in the phylogeny than the species lineages in which these are evolving. The same pattern is called ILS when looking towards the tips of the tree because persistent ancestral alleles are stochastically inherited (i.e. incompletely sorted into species; Rannala et al. 2020). This is a common cause of phylogenetic conflict, though the detection is dependent on the lineages sampled.
Simultaneous speciation, whereby multiple species evolve at the same time rather than in a bifurcating manner, can also be a source of conflict in a phylogenetic tree. Simultaneous speciation is considered to occur rarely but most commonly through allopatric, non-adaptive speciation, whereby a population is separated into more than two isolated geographic areas (e.g. through vicariance, mountain building or glaciation) and the individuals in each area evolve into separate lineages (Matsubayashi and Yamaguchi 2022; e.g. Dillenberger and Kadereit 2017). However, for multiple species to evolve simultaneously through sympatric adaptive radiation events (Bolnick 2006) and combinatorial mechanisms (Marques et al. 2019) is also theoretically possible. In phylogenetic terms, the simultaneous evolution of multiple lineages is a ‘hard polytomy’, with multifurcating branches, rather than bifurcating branches (Maddison 1989; Hoelzer and Meinick 1994). When forced to be represented as bifurcations, each gene tree may have random, conflicting topologies between lineages originating by simultaneous speciation simply because the pattern of mutation does not comply with a bifurcating pattern. However, the challenge with inferring simultaneous speciation is differentiating it from cases of rapid radiations that do follow a bifurcating pattern of evolution. In rapid radiations, little time and few mutations may separate divergent lineages, and the lack of information makes these relationships particularly difficult to reconstruct. These often manifest as ‘soft polytomies’ (Maddison 1989), in which there is uncertainty as to whether conflict is a result of a lack of information to resolve the true, bifurcating relationships of a rapid radiation or a genuine case of simultaneous speciation (DeSalle et al. 1994; Whitfield and Lockhart 2007; Orel et al. 2023; Zhang Q et al. 2023).
Artefactual conflict is discordance between subsets of phylogenetic data that arises from inappropriate bioinformatic choices, leading to errors, anomalies, biases, or noise in phylogenetic results (e.g. Frost et al. 2024). Artefactual conflict can be introduced at any step of the bioinformatic pipeline but is especially common during locus extraction, paralog reconciliation and phylogenetic tree inference. For example, if loci are not assembled from the raw reads with sufficiently stringent parameters, incorrect sequences can be assembled for some loci, leading to the inference of an incorrect evolutionary history for that loci that could be in conflict with other loci. Such conflict is artificial, and may lead to inaccurate results and noise that reduces phylogenetic resolution and prevents the detection of real biological conflict. As such, carefully considering the parameters and assumptions underlying each step in the analytical pipeline, and checking and cleaning the output (especially alignments, homolog trees and gene trees) are important (see also the quality control steps marked with an asterisk in Fig. 3). Below, we set out the major steps in a phylogenomic analytical workflow: (1) locus extraction, (2) paralogy reconciliation, (3) phylogenomic reconstruction of gene trees and species trees, (4) conflict assessment and (5) investigating patterns and underlying causes of conflict (Fig. 3), and highlight key considerations at each step for reducing artefactual conflict.
Overview of the five major steps for a phylogenomic workflow with target-capture data outlined in this review, from raw reads to (1) locus extraction, (2) paralogy reconciliation, (3) species tree inference, (4) conflict assessment and (5) conflict investigation. Thick grey arrows indicate the general direction of the workflow. Within each step of the workflow, the main approaches are summarised and coloured arrows indicate which approaches are compatible from each step. Circles with asterisks (*) indicate particularly important points where quality control should be conducted on the output of the previous step to avoid the introduction of artefactual conflict (e.g. by checking and cleaning alignments and gene tree topologies). For more details on each step, see the relevant section of this review.
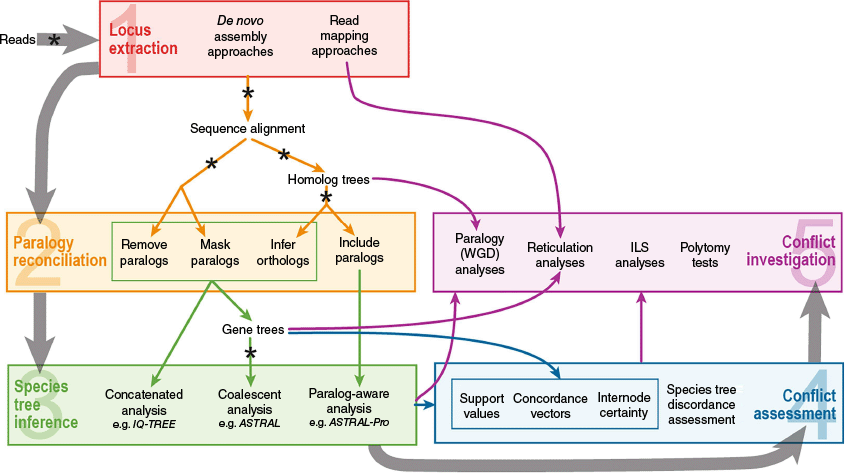
Key steps for evolutionary inference in the presence of phylogenetic conflict
Step 1. Locus extraction
Following the sequencing of enriched libraries and quality control, the targeted loci must be assembled and extracted from the reads (Fig. 3). Many methods are now available for this purpose. One of the oldest and most commonly used locus extraction pipelines in plant phylogenomics is HybPiper (Johnson et al. 2016) that was developed specifically for the retrieval and assembly of target-capture data using Angiosperms353 (see Table 2). HybPiper uses a read-mapping approach to align raw sequencing reads to reference gene sequences and subsequently assembles these reads into contigs for both exons and the flanking intron regions (Johnson et al. 2016). HybPiper was greatly improved through the course of the GAP project and versions since version 2.0 are much easier to use as either a Python package or container, with improvements in read-mapping, locus assembly and recovery reporting (Jackson et al. 2023). Another program that implements a read-mapping approach is HybPhyloMaker (Fér and Schmickl 2018) that was also written for target-capture recovery in plant phylogenomics but in addition implements phylogenetic reconstruction. An alternative set of methods begins by de novo assembly of all sequencing reads and subsequent retrieval of the target loci using reference gene sequences instead. Such software includes SECAPR (Andermann et al. 2020), PHYLUCE (that is more frequently used in animal phylogenomics with ultra-conserved elements; Faircloth 2016) and the recently developed CAPTUS (Ortiz et al. 2023). Assembly first methods have the advantage of being able to cluster and extract many off-target loci without a reference target file, facilitating the extraction of additional nuclear and plastid loci for phylogenetic analysis (e.g. Ortiz et al. 2023). Both read-mapping and assembly first assembly methods require a well-designed reference gene sequence file that includes sufficient coverage of the target genes across the phylogenetic scale of interest. For Angiosperms353, recovery can be improved by expanding the default target file to encompass more phylogenetic breadth (McLay et al. 2021). Although comparisons of some locus extraction pipelines have been published (e.g. Zhang Z et al. 2022; Raza et al. 2023), a comprehensive comparison of the performance of these methods across lineages and data qualities has not been conducted; as such, multiple methods could be tested on datasets to determine the most optimal and practical pipeline.
| Tool | Use | Output | Citation and URL | |
|---|---|---|---|---|
| (1) Locus extraction | ||||
| HybPiper | Locus assembly and extraction | Sequence files for each locus, assembly reporting, paralogy reporting, exons and intron sequences | Johnson et al. (2016), Jackson et al. (2023); https://github.com/mossmatters/HybPiper; https://github.com/chrisjackson-pellicle/hybpiper-nf | |
| HybPhyloMaker | Locus assembly and extraction, plus alignment, trees | Sequence files for each locus, assembly reporting, paralogy reporting, exons and intron sequences, alignments, gene trees, species trees | Fér and Schmickl (2018); https://github.com/tomas-fer/HybPhyloMaker | |
| SECAPR | Locus assembly and extraction | Sequence files for each locus, assembly reporting, paralogy reporting, exons and intron sequences, phased loci | Andermann et al. (2018); https://github.com/AntonelliLab/seqcap_processor | |
| PHYLUCE | Locus assembly and extraction, typically UCEs | Sequence files for each locus, assembly reporting, alignments | Faircloth (2016); https://github.com/faircloth-lab/phyluce | |
| CAPTUS | Locus assembly and extraction | Sequence files for each locus, assembly reporting, paralogy reporting, exons and intron sequences, plus organellar sequences, alignments | Ortiz et al. (2023); https://github.com/edgardomortiz/Captus | |
| NewTargets | Expanding target file phylogenetic breadth using available genomic resources | An expanded target file, curated to end-user needs for improved recovery | McLay et al. (2021); https://github.com/chrisjackson-pellicle/NewTargets | |
| (1.1) Locus extraction: Post-assembly assessment and alignment filtering | ||||
| AMAS | Alignment assessment and manipulation (e.g. sample removal) | Various alignment formats, individual locus or concatenated alignments plus partition files | Borowiec (2016); https://github.com/marekborowiec/AMAS/ | |
| trimAl | Alignment trimming based on several parameters (e.g. gappiness, informativeness, overlap) | Trimmed alignments in user specified format | Capella-Gutiérrez et al. (2009); https://vicfero.github.io/trimal/ | |
| ClipKit | Alignment trimming based on several parameters (e.g. gappiness, informativeness, codon position) | Trimmed alignments in user specified format | Steenwyk et al. (2020); https://github.com/JLSteenwyk/ClipKIT | |
| CIAlign | Alignment trimming based on several parameters (e.g. gappiness, informativeness, length, alignment quality) | Trimmed alignments in user specified format | Tumescheit et al. (2022); https://github.com/KatyBrown/CIAlign | |
| opTrimal | Assessment of optimal alignment trimming thresholds | Untrimmed alignments in user specified format | Shee et al. (2020); https://github.com/keblat/bioinfo-utils/blob/master/docs/advice/scripts/optrimAl.txt | |
| TAPER | Identification of alignment errors | Trimmed or untrimmed alignments in user specified format | Zhang C et al. (2021) | |
| (2) Paralogy reconciliation | ||||
| PPD | Uses sequence identity and heterozygous sites to identify and remove paralogs | Alignments with detected paralogs removed | Zhou W et al. (2022); https://github.com/Bean061/putative_paralog | |
| ParalogWizard | Refined reassembly of loci to identify divergent sequences (i.e. paralogs or alleles) and perform orthology inference | Alignments sorted into orthologous groups | Ufimov et al. (2022); https://github.com/rufimov/ParalogWizard | |
| CAPTUS | Identification of paralogous sequences during pipeline by comparing to a reference sequence | Paralogous sequences can be sorted into different alignments with user-defined parameters, including ‘best’ and ‘similarity’, or all copies can be kept or removed | Ortiz et al. (2023); https://github.com/edgardomortiz/Captus | |
| HybPhaser | Infer parental lineages of putatively hybridogenic lineages | Phased alignments with paralogs removed | Nauheimer et al. (2021); https://github.com/LarsNauheimer/HybPhaser | |
| ParaGone | Implements Yang and Smith’s (2014) collection of methods for resolving paralogy using gene tree topologies | Paralogy resolved alignments and gene trees from each Y&S algorithm | Jackson et al. (2023); https://github.com/chrisjackson-pellicle/paragone-nf | |
| (3) Species tree inference: Paralog-aware phylogenetic tree reconstruction | ||||
| ASTRAL-PRO | Two-step coalescent phylogenetics from multi-labelled trees (i.e. including paralogous sequences) | Coalescent species tree with paralogous tips reconciled to species | Zhang and Mirarab (2022); https://github.com/chaoszhang/ASTER; https://github.com/chaoszhang/A-pro | |
| FastMulRFS | Two-step coalescent phylogenetics from multi-labelled trees (i.e. including paralogous sequences), using Robinson–Fould’s distances to summarise paralogous sequences | Coalescent species tree with paralogous tips reconciled to species | Molloy and Warnow (2020); https://github.com/ekmolloy/fastmulrfs | |
| SpeciesRax | Likelihood inference of species tree from gene alignments or gene family trees | Species tree and gene trees if starting with alignments | Morel et al. (2022); https://github.com/BenoitMorel/GeneRax | |
| DISCO | Performs orthology inference of each gene tree to preserve orthologous sequences and discard paralogs | Coalescent species tree with paralogous tips reconciled to species | Willson et al. (2022); https://github.com/JSdoubleL/DISCO | |
| AleRax | Likelihood inference of species tree from samples of estimated gene family trees | Species tree, reconciled and consensus gene trees, number of events | Morel et al. (2024); https://github.com/BenoitMorel/AleRax | |
| (3) Species tree inference: Phylogenetic tree reconstruction on single-sequence-per-species alignments | ||||
| IQ-TREE | Likelihood phylogenetics on concatenated data | Phylogenetic tree (and other outputs depending on analysis) | Minh et al. (2020b); http://www.iqtree.org | |
| RAxML | Likelihood phylogenetics on concatenated data | Phylogenetic tree (and other outputs depending on analysis) | Stamatakis (2014), Kozlov et al. (2019); https://github.com/stamatak/standard-RAxML; https://github.com/amkozlov/raxml-ng | |
| ASTRAL | Two-step coalescent phylogenetics | Species tree | Mirarab et al. (2014), Zhang C et al. (2018b); https://github.com/smirarab/ASTRAL | |
| SplitsTree | Implements a range of network analyses, including the popular NeighbourNet and Consensus Network algorithms | Phylogenetic network | Huson and Bryant (2006); https://uni-tuebingen.de/en/fakultaeten/mathematisch-naturwissenschaftliche-fakultaet/fachbereiche/informatik/lehrstuehle/algorithms-in-bioinformatics/software/splitstree/; https://github.com/husonlab/splitstree6 | |
| ExaBayes | Bayesian phylogenetics on concatenated data | Phylogenetic tree (and other outputs depending on analysis) | Aberer et al. (2014); https://cme.h-its.org/exelixis/web/software/exabayes/ | |
| StarBeast | Bayesian inference of gene trees and species tree under the multispecies coalescent | Posterior distribution and summary tree for species tree and gene trees | Douglas et al. (2022); https://github.com/rbouckaert/starbeast3 | |
| (3.1) Species tree inference: Gene tree assessment and phylogenomic subsampling | ||||
| GeneSortR | Sorting and subsampling phylogenomic datasets to quantify phylogenetic usefulness | Sorted alignment, partition file, gene tree file and a plot of sorted genes by estimated properties, graphical summary of metrics employed to subsample | Mongiardino Koch (2021); https://github.com/mongiardino/genesortR | |
| PhylteR | Identify outlier loci in phylogenomic datasets | Visualised output of outlier loci for removal | Comte et al. (2023); https://github.com/damiendevienne/phylter | |
| TreeShrink | Pruning long, likely erroneous branches from sets of phylogenetic trees | Pruned phylogenetic trees and corresponding alignments | Mai and Mirarab (2018); https://github.com/uym2/TreeShrink | |
| SortaDate | Phylogenomic subsampling to choose genes for phylogenetic dating | List of locus alignments of genes | Smith et al. (2018); https://github.com/FePhyFoFum/sortadate | |
| (4) Conflict assessment | ||||
| IQ-TREE | Likelihood phylogenetics on concatenated data and locus alignments or partition file | Gene concordance factors (gCFs) and site concordance factors (sCFs) on phylogeny as branch labels | Minh et al. (2020b); http://www.iqtree.org | |
| PhyParts | Identification of concordant and conflicting bipartitions | Species phylogeny with concordance and conflict as branch labels | Smith et al. (2015); https://bitbucket.org/blackrim/phyparts/src/master/ | |
| ASTRAL | Measuring concordance and discordance by percentages of supporting quartets used to produce species tree | Species phylogeny with quartet concordance and conflict as branch labels | Mirarab et al. (2014); https://github.com/smirarab/ASTRAL | |
| BUCKy | Estimating concordance factors from Bayesian MCMC trees of many loci | Species phylogeny with concordance and discordance scores as branch labels | Larget et al. (2010); https://pages.stat.wisc.edu/~ane/bucky/ | |
| Quartet Sampling | Repeated sampling of quartets to analyse branch support on molecular phylogenies | Newick tree files of various scores, a FigTree file containing all scores, and statistics files | Pease et al. (2018); https://github.com/FePhyFoFum/quartetsampling | |
| (5) Conflict investigation | ||||
| HyDe | Detects hybridisation in phylogenomic data sets | Values including identification of species and population level hybrids with ABBA-BABA tests | Blischak et al. (2018); https://github.com/pblischak/HyDe | |
| JML | Detects hybridisation on time-calibrated trees, with information about population sizes | Distances between sequences for species pairs with P-values for hybridisation according to the posterior predictive distributions | Joly et al. (2012); https://github.com/simjoly/jml | |
| Aphid | Estimating the contributions of gene flow and incomplete lineage sorting to phylogenetic conflict | Per gene tree output of conflict and the estimated cause (e.g. ILS or gene flow) in a CSV file | Galtier (2024); https://gitlab.mbb.cnrs.fr/ibonnici/aphid | |
| GRAMPA | Use homolog gene tree topologies (i.e. MULtrees) to identify placement and types of WGD | Tree and txt files detailing the estimated ploidy placement and type of polyploid | Thomas et al. (2017); https://github.com/gwct/grampa | |
| QuIBL | Uses gene tree internal branch lengths to distinguish between hybridisation and deep coalescence | For each triplet in the species tree, an estimate of the relative contribution of the locus set to ILS or gene flow | Edelman et al. (2019); https://github.com/miriammiyagi/QuIBL | |
| PhyloNet | Infer phylogenetic networks from sets of loci while accounting for both reticulation and ILS, using mostly maximum likelihood-based algorithms | Networks as Nexus files | Than et al. (2008), Wen et al. (2018); https://phylogenomics.rice.edu/html/phylonet.html | |
| PhyloNetworks | Infer phylogenetic networks from sets of loci while accounting for both reticulation and ILS, under a coalescent model | Networks as Newick files | Solís-Lemus et al. (2017); https://github.com/crsl4/PhyloNetworks.jl | |
| DiscoVista | Quantify and visualise a range of phylogenomic metrics including species tree and gene tree compatibility, branch quartet frequencies and GC content | Figures showing gene tree discordance and relative frequency of different topologies, species tree discordance and taxon occupancy | Sayyari et al. (2018); https://github.com/esayyari/DiscoVista | |
| ASTRAL | Perform a polytomy test to determine if the polyomy is ‘hard’ or ‘soft’ | Species phylogeny with significance values that indicate the presence of a hard polytomy | Mirarab et al. (2014), Sayyari and Mirarab (2018); https://github.com/smirarab/ASTRAL | |
Ultimately, the choice of extraction pipeline will depend on the performance (locus recovery), bait design, access to computational resources and research question. Some questions may require certain downstream analyses that are dependent on the output of particular locus extraction pipelines. For example, if one wants to use HybPhaser (Nauheimer et al. 2021) or ParaGone (Jackson et al. 2023) for paralogy reconciliation (see next section ‘Step 2. Paralogy reconciliation’ for more detail), the output format of HybPiper is required. As such, planning the workflow ahead and checking whether output formats are compatible for downstream analyses (or can be manually altered to be compatible with downstream analyses) can help to inform which locus extraction method should be used. In some cases, using multiple locus extraction methods could be warranted. Careful attention should also be paid to matching the most appropriate extraction method to the way the baits have been designed. All baits are designed differently; not only do these target different loci, but may also target different regions of the loci. Some of the first target-capture methods used highly conserved regions as anchors to target the surrounding highly variable regions that may or may not be protein coding (e.g. Lemmon et al. 2012). In bait kits that explicitly target protein-coding regions, a ‘locus’ might comprise a single exon, whereas in other bait kits a locus may be multiple exons, introns or the whole gene comprising exons and introns. Each locus extraction method will handle each scenario slightly differently. For example, SECAPR is designed for single-exon targets and therefore portions of the pipeline must be adapted if one wants to use this to assemble loci such as the Angiosperms353 loci that target multiple exons per locus. Owing to the differences in how assembly methods work, the methods also define and extract different regions of the targeted loci differently and care should be taken to make sure this is understood. For example, in a scenario where a multi-exon locus such as an Angiosperms353 locus is being targeted, if HybPiper is being used, when reads are mapped to the exons some reads overhang the exon edge and represent partial sequences of the introns between exons. HybPiper calls these partial intron sequences ‘flanking regions’ and there is the option of concatenating these with all other exons and the flanking regions to produce a ‘supercontig’ for the locus. However, if CAPTUS is used, the entire gene, including full introns and exons may be able to be assembled (because de novo assembly of contigs occurs before extracting the targeted sequences) and the ‘flanking regions’ refer to the DNA sequences on either side of the whole gene. Therefore, care should be taken to understand how each assembly program will handle and define the structure of the targeted loci and whether this will yield the desired output (exons v. introns v. genes). Finally, some locus extraction pipelines offer a workflow for additional steps beyond locus extraction, through to sequence alignment and even tree estimation (Fér and Schmickl 2018; Ortiz et al. 2023). Although these pipelines are user friendly, we caution against following these workflows without careful consideration and inspection of each step.
Artefactual conflict can be introduced by researchers at the locus extraction step in several ways. One cause of artefactual conflict is sequence errors introduced through misassembly; inclusion of misassembled, erroneous sequences for targeted loci (called ‘contigs’) can lead to the estimation of incorrect gene tree topologies that differ from the ‘true’ topology in other gene trees and the species tree. Therefore, misassembly issues need to be assessed and cleaned at this step of the bioinformatic pipeline to minimise artefactual phylogenetic conflict. Misassemblies can occur either through inappropriate settings for the short-read assembler program used in the locus extraction pipeline or because of poor quality data. Different assembly pipelines use different short-read assemblers; for example, HybPiper and SECAPR use SPAdes (see https://github.com/ablab/spades; Bankevich et al. 2012), whereas CAPTUS uses MEGAHIT (see https://github.com/voutcn/megahit; Li et al. 2015). Each short-read assembler performs differently depending on the pattern of coverage, the presence of highly repetitive regions, GC and AT content, and the structural variation in each dataset (Liao et al. 2019). Using a suboptimal short-read assembler through the assembly pipeline can contribute to misassemblies and errors in the resulting sequences. Poor quality data can also cause misassemblies. In cases where samples have low DNA concentration or sequencing depth, read coverage (i.e. the number of reads mapping to any given section of the reference sequence for the targeted locus) may be low or uneven. The lower the read coverage, the higher the chance of an erroneous base call in the contig by the short-read assembler due to a lack of information (Liao et al. 2019). Therefore, if loci have low coverage throughout, there is a higher chance of misassembly. Most short-read assemblers assume that there is an even coverage of reads and use an average coverage cutoff threshold for contigs (Liao et al. 2019). Although this means low coverage regions will be pruned out, the assembly of contigs with a high proportion of missing data can also result. Furthermore, loci with low complexity or many repeated regions are also notoriously difficult to correctly assemble with short-read assemblers (Liao et al. 2019). Misassemblies can be detected by stringently checking the output sequences and alignments. The quality of the assemblies can be observed by manually remapping the reads to the reference sequences and checking the coverage. Although time consuming and not always possible for all loci, this process can inform the researcher about whether there are issues with low or uneven read coverage, low complexity regions or inappropriate short-read assembler settings (e.g. pruning thresholds). Misassemblies can also be detected by manually inspecting the resulting sequence alignments for each locus, paying special attention to misaligned or gappy areas of the sequence output for each locus alignment, although the judgement of the researcher of what is ‘gappy’ or ‘well aligned’ can also introduce biases and errors. Therefore, alignment summary tools can be used to rapidly and objectively summarise the alignment quality and gappiness, and a variety of programs such as AMAS (Borowiec 2016) or SEGUL (Handika and Esselstyn 2024) can be used for this. When misassemblies have been identified through these measures, a researcher can try to ameliorate these errors by remapping the reads to identify the problem causing the misassembly (as described above), adjusting the short-read assembler settings, trying another locus assembly pipeline with a different short-read assembler, manually deleting sequences with assembly errors (although this has rarely been addressed in the literature) or automatically deleting sequences with errors. Automatic deletion of erroneous sequences can be undertaken at the alignment step and after phylogenetic trees for each locus have been generated. Deletion of erroneous sequences or erroneous parts of sequences from alignments (i.e. alignment ‘cleaning’) can be achieved using tools such as trimAl (Capella-Gutiérrez et al. 2009), ClipKIT (Steenwyk et al. 2020) and CIAlign (Tumescheit et al. 2022). Erroneous sequences can also be removed once phylogenetic trees for each locus have been generated; these sequences are likely to result in spuriously long branches, and can be detected and trimmed with tools such as TreeShrink (Mai and Mirarab 2018). Once spurious tips have been deleted, the ‘clean’ sequences should be realigned prior to estimation of final trees. Determining the threshold for parameters for all of these tools requires comprehensive data exploration and trial and error.
Missing data can also introduce artefactual conflict. Low quality samples (for example samples with low DNA or library concentrations) may yield poor coverage or biased sequence files that can result in uneven recovery across samples or loci. This can produce extremely short sequences or loci represented by very few samples that result in alignments with substantial missing data that is problematic for phylogenetic inference (Nute et al. 2018; Smirnov and Warnow 2021). Although there may be a desire to keep all samples and loci in a dataset, samples or loci with missing data can mislead phylogenetic inference through a lack of information (e.g. Smith et al. 2020), thereby resulting in discordant topologies for those samples or loci and ultimately phylogenetic conflict. There is some evidence that the impact of missing data is particularly amplified in datasets with high levels of ILS (Xi et al. 2016; Nute et al. 2018). To avoid potential biases, sample removal thresholds should be high and inspections of sequences to check for coverage (as well as the percentage of recovered length) should also be conducted. Inspection and cleaning of alignments can be achieved using the same methods for inspecting and cleaning alignments for errors as described above. Additionally, the first steps of HybPhaser (e.g. the script ‘1b_assess_dataset.R’) and CAPTUS are useful for summarising assembly quality, missingness and information content of the loci.
A third source of phylogenetic conflict at the locus extraction step is in the assembly of chimeric contigs. Chimeric contigs are sequences for a targeted locus that are formed from parts of different alleles or copies that have been stitched together (Nauheimer et al. 2021). Chimeric contigs can introduce phylogenetic conflict as these combine the phylogenetic signals of gene copies that are not homologous, and can therefore result in incorrect gene or species tree topologies that are in conflict with each other and the ‘true’ evolutionary history. Read-mapping methods such as HybPiper may be more prone to assembling chimeric contigs because reads are mapped to each exon separately and subsequently stitched together. HybPiper currently has a number of options to reduce the likelihood of assembling chimeric contigs and these should be carefully explored, particularly in cases in which paralogy is an issue and when multi-exon targets are used. Assembly first methods such as CAPTUS may do a better job at avoiding the assembly of chimeric contigs but are not necessarily immune to this problem; therefore, output should still be carefully examined. As with assessing the output of locus extraction for short-read assembler errors and missing data, chimeric contigs can be detected through inspection and cleaning of alignments, spuriously long branches in gene trees and remapping reads.
Step 2. Paralogy reconciliation
After loci have been extracted, dealing with paralogs is one of the most important parts of a phylogenomic workflow with target-capture data (Fig. 3; Smith and Hahn 2021). This is particularly important for plant phylogenomics, as gene or genome duplications are common and can produce paralogs in datasets even when using bait kits targeting ‘single-copy’ loci (De Bodt et al. 2005; Panchy et al. 2016; Ren et al. 2018; Landis et al. 2018; Almeida-Silva and Van de Peer 2023). There is an increasing number of workflows to handle paralogs in phylogenomic datasets that can be categorised into four main approaches: (1) remove paralogs (or paralogous loci), (2) mask the effects of paralogs, (3) infer orthologs, and (4) estimate species trees directly with a paralog-aware method (Fig. 3, 4). Each approach has a different philosophy and set of underlying assumptions that affects not only species tree estimation but also the downstream analyses that can be applied to investigate biological processes such as ILS, hybridisation and WGD events. The choice of approach should therefore be based on the philosophy that is most suitable for the analytical workflow the researcher wants to apply, and the biological system and questions at hand.
Schematic diagram illustrating the four main approaches used for paralog reconciliation with target-capture data in hypothetical scenarios with four samples (Samples 1–4) and four loci (Locus A–D). Loci A and C are paralogous, having multiple copies of the same locus (Copies i–iv). (a) Illustrates the removal of paralogs for a sample, whereby paralogous loci A and C are removed from the analysis and only single-copy loci B and D are retained for phylogenetic inference. This process would be repeated for each sample to obtain the final dataset for phylogenetic inference. (b) Shows paralog masking for a sample, whereby SNPs across aligned gene copies for paralogous loci A and C are coded with ambiguity characters in the consensus sequences for each locus used for phylogenetic inference. This process would be repeated for each sample to obtain the final dataset for phylogenetic inference. (c) Depicts tree-based ortholog inference, whereby all gene copies are aligned for each locus to infer homolog trees. These homolog trees are pruned (denoted by the scissors) to an ortholog subtree (denoted by the green tick) to identify orthologous sequences that are subsequently used in phylogenetic inference. In this scenario, the sequences in the clade of copies denoted by the red cross are discarded from the analysis. (d) Illustrates a scenario in which all paralogs are used in phylogenetic inference. As in (c), homolog trees are estimated for each locus using an alignment of all gene copies and these homolog trees are subsequently directly used in species tree-estimation software such as ASTRAL-Pro.
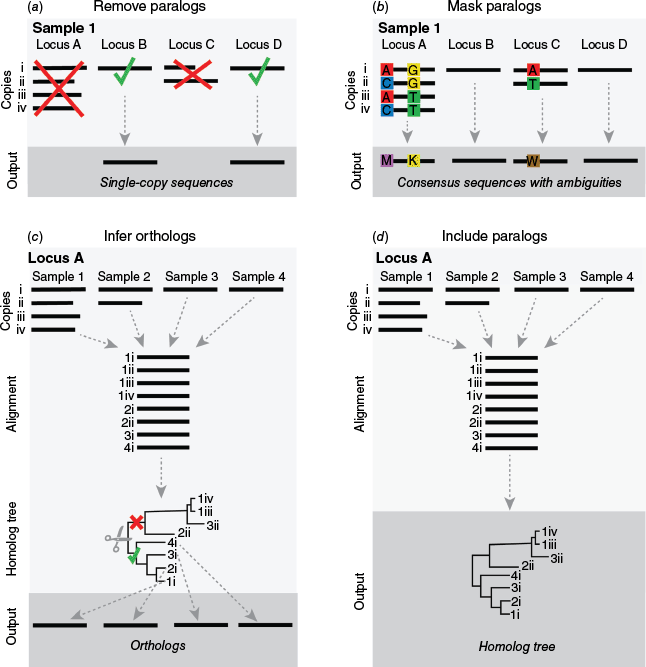
In the first approach, there are two options for removing paralogs: by filtering paralogs from paralogous loci for only one copy to be retained or excluding all paralogous loci detected (Fig. 4a). In the first option, paralogs are filtered based on a criterion such as similarity to a reference sequence, pairwise similarity, or length, for example as implemented in PPD (Zhou W et al. 2022), ParalogWizard (Ufimov et al. 2022) or the filtering steps of CAPTUS (Ortiz et al. 2023). As a result, copies of paralogous loci are removed and only the sequence that is the longest or most similar is retained for each locus. This option may be suitable for some lineages in which minimal paralogy is evident, in extremely large datasets or for handling plastid loci, where few to no paralogs are expected to be present. However, this approach clearly makes no attempt to infer orthology. As such, analysis of the remaining loci not only violates the assumptions of homology in phylogenetic inference but also runs the risk of estimating erroneous topologies and introducing artefactual conflict into phylogenetic trees because each retained copy may not share the same evolutionary history. This is especially problematic for smaller datasets and when using concatenated methods of species tree inference (Yan et al. 2022). There is some evidence to suggest that coalescent species-tree inference methods are somewhat robust to using randomly filtered paralogs for analysis in large datasets (Yan et al. 2022); however, this evidence is based on simulated data in specific biological scenarios and one empirical dataset. Moreover, this approach limits the potential for downstream investigation of conflict and gaining insight into any biological causes of conflict, as orthology is essentially ignored. Therefore, in practice for target-capture studies on plants, we advise that caution be taken if using this approach. The second option for removing paralogs (completely removing any paralogous locus; Fig. 4a) is scientifically defensible but also has limitations. Through removing paralogous loci, researchers only include single-copy loci that are more likely to be orthologous. In effect, this is an attempt to only include orthologs and can be justifiably used for tree inference. The downside of this method, however, is that the number of loci and therefore the amount of phylogenetic information can be substantially reduced (Yan et al. 2022), potentially leading to poor resolution in estimated trees in smaller bait kits (such as Angiosperms353), or in lineages that have recent WGD where all or most loci are paralogous. This approach may also remove signals of biological processes such as hybridisation and WGD events that may be in the evolutionary history of the lineage, and importantly, cannot avoid the issue that the retained single-copy loci may be hidden paralogs rather than orthologs. If identifying the presence and nature of biological processes such as WGD, reticulation, ILS and rapid radiation is of interest, a different paralog handling approach that uses the information in paralogs is likely to be more appropriate (see paralog reconciliation approaches 2–4 below).
The second approach for dealing with paralogs in target-capture datasets involves masking the paralogs with consensus sequences coded with ambiguity codes (Fig. 3, 4b). This approach, which can be implemented in pipelines such as HybPhaser (Nauheimer et al. 2021) and that of Kates et al. (2018), aims to mitigate the effects of paralogs by encoding single nucleotide polymorphisms (SNPs) from different paralogs (and alleles) as ambiguous characters. In theory, this captures the uncertainty of the base across gene copies at these sites, and therefore less weight is placed on the sites that differ across paralogs when inferring species tree topology. In doing so, this reduces the phylogenetic conflict caused by these sites across paralogs. Additionally, characterisation of the percentage of SNPs and allele divergence between paralogs through HybPhaser has been shown to be a good indication of ploidy within the phylogenetic tree (Hendriks et al. 2023) and can be used to phase paralogs and identify hybridisation events (Nauheimer et al. 2021; see section ‘Step 5. Investigating underlying causes of conflict’). One potential pitfall of this approach is that the introduction of ambiguities into the dataset may eliminate potentially important phylogenetic signal at those sites that can theoretically decrease the resolution of the tree. Potts et al. (2014) found this to be true for a series of short (<~1100 bp) single-gene datasets but Kates et al. (2018) did not find the same for target-capture data; further research is needed to test the effects of this approach on phylogenetic informativeness.
The third approach involves inferring orthologous sequences from all sequences based on gene tree topologies (Fig. 3, 4c). This approach, summarised by Yang and Smith (2014), takes gene trees with all gene copies (herein referred to as ‘homolog trees’) and identifies subtrees that only contain nodes representing speciation events (herein referred to as ‘ortholog subtrees’), rather than nodes that may be a result of gene duplication. These subtrees include sequences for each gene that is orthologous (i.e. share a common ancestor), that can subsequently be extracted from the original dataset, aligned and used for species tree estimation. There are several algorithms for pruning homolog trees to ortholog subtrees. Choice of algorithm depends on the availability and quality of outgroup sequences (that the Maximum Inclusion, MI, algorithm does not require) and the trade-off between retrieving few ortholog subtrees with good sampling (Monophyletic Outgroups, MO, algorithm) v. many ortholog subtrees with many missing samples (Rooted subTrees, RT, algorithm) (Yang and Smith 2014). For reliable ortholog identification, carefully cleaning the initial, raw homolog trees by removing any spurious sequences (e.g. by pruning especially long branches) and reducing any monophyletic tips of the same species (that could represent alleles or neopolyploids) to one representative sequence to produce clean homolog trees is important. Orthology inference (and other downstream analyses such as WGD mapping, GRAMPA and ASTRAL-Pro – see the section ‘Step 5. Investigating underlying causes of conflict’) can subsequently be performed on the clean homolog trees (also often referred to as ‘multi-labelled trees’). Tree-based ortholog inference can be implemented through the scripts developed by Morales-Briones et al. (2021), or through the software ParaGone (Jackson et al. 2023). Through identifying orthologous sequences, the underlying assumption of homology in evolutionary models is maintained and the conflicting signal of paralogs is eliminated, resulting in robust phylogenomic inferences. This approach of generating homolog trees and ortholog subtrees also has the advantage of facilitating many options in downstream analyses for meaningful conflict investigation and inferring the underlying biological processes.
The fourth approach to dealing with paralogs entails the estimation of species trees using methods explicitly designed to accommodate paralogs (Fig. 3, 4d), as summarised in Smith and Hahn (2021). Rather than building homolog trees only to prune out all paralogs and only use orthologs for species tree inference (as in the previous orthology inference approach), paralog-aware methods use all gene copies from the homolog trees to infer the species tree topology. Instead of assuming a single gene tree topology across all loci, paralog-aware methods explicitly model and account for gene duplication and loss events in the estimation of species trees, though the method of doing so varies between programs. Programs such as ASTRAL-Pro (Zhang C et al. 2020; Zhang and Mirarab 2022), FastMulRFS (Molloy and Warnow 2020), SpeciesRax (Morel et al. 2022) and iGTP (Chaudhary et al. 2010) use homolog trees and model gene evolution, duplication and loss events using two-step coalescent or parsimony-style approaches. Decomposition methods such as DISCO (Willson et al. 2022) apply a tree-pruning algorithm to the homolog trees (similar to the third paralog-reconciliation approach), splitting homolog trees into ortholog subtrees (also known as ‘orthogroups’) prior to species tree estimation, usually under the coalescent. As with the orthology-inference approach to paralogs, optimal implementation of these methods is dependent on the use of clean homolog trees, rather than the raw homolog trees. Through integrating information across paralogous loci while accommodating gene tree discordance, these methods offer a sound option for accurate species tree estimation in complex evolutionary scenarios. However, two-step coalescent-based methods such as ASTRAL-Pro come with some drawbacks, such as treating gene tree topology as fixed even where nodes may be poorly supported because of short individual gene alignments. This potentially misleads species tree inference and results in undefined branch lengths on the phylogeny (Mirarab et al. 2016; Simmons and Gatesy 2021). These issues may be mitigated with the development of new paralog-aware species tree estimation methods such as AleRax (Morel et al. 2024) that uses the distribution of homolog tree topologies to inform species tree inference; however, this method is yet to be applied in a plant phylogenetic context. Nevertheless, resolving paralogy before phylogenetic analysis gives the researcher more methodological and software options for investigating evolutionary processes.
Each of these approaches is based on a distinct philosophy and set of underlying assumptions, influencing not only species tree estimation but also the possibilities for downstream analyses, and the associated interpretability and robustness. In some cases, taking multiple complementary approaches to dealing with paralogs may yield additional insight into any conflict present in the dataset and help to answer research questions pertaining to underlying biological sources of conflict.
Step 3. Phylogenomic reconstruction of gene trees and species trees
Following the first three paralogy reconciliation approaches (i.e. once a sequence copy from each locus has been chosen), there are multiple methods available for species tree inference (Fig. 3). Tree-inference methods have been comprehensively reviewed (see e.g. Leaché and Rannala 2011; Simmons and Gatesy 2015; Mirarab et al. 2016), therefore we will not review these in-depth in this paper. Briefly, the two main methods currently used to infer species trees from target-capture data are Maximum Likelihood analyses conducted on concatenated sequence alignments (such as with IQ-TREE, Nguyen et al. 2015; Minh et al. 2020b; and RAxML, Stamatakis 2014; Kozlov et al. 2019) and two-step coalescent approaches based on gene tree topologies (such as with ASTRAL, Mirarab et al. 2014). Bayesian analysis of phylogenomic datasets can also be performed using ExaBayes (Aberer et al. 2014) and for smaller datasets of fewer than 100 terminals, Bayesian inference under the multispecies coalescent (e.g. with StarBEAST, Douglas et al. 2022) is another computationally feasible option. Each method comes with a uniqueset of assumptions that may be more or less suitable depending on the scale of taxonomic sampling, size of study group and lineage. Using methods that rely on a concatenated sequence alignment, it is assumed that the evolutionary history of all loci is congruent with the evolutionary history of the species; however, because this is not true, phylogenetic conflict can manifest as poorly supported nodes or erroneous yet strongly supported relationships if the strongest signal is incongruent with the species history. Using a two-step approach enables the estimation of independent evolutionary histories of each gene, but is dependent on the use of loci with adequate phylogenetic information to produce well-supported gene trees. Furthermore, the degree of gene-tree topology error and ILS present in a dataset can also influence the choice of tree-inference method, as conflict can increase the computational effort required (e.g. Tea et al. 2022). We recommend using multiple tree-estimation methods for target-capture datasets, especially because any conflict may provide insight into artefactual issues or biological processes (see section ‘Step 5. Investigating underlying causes of conflict’).
Artefactual conflict can arise during phylogenetic tree reconstruction through inappropriate choice of evolutionary models (such as substitution model) and gene tree estimation error (Cai et al. 2021). As such, it is good practice to conduct phylogenetic tree reconstruction with multiple approaches, carefully considering the assumptions of all choices made in tree estimation models, and inspecting gene trees for signs of error. Error in gene tree topologies can be caused by a number of factors, such as the inclusion of erroneous sequences, uninformative loci (due to slow mutation rates or short loci) or loci (or sites within loci) with extremely high rates of mutation prone to saturation and homoplasy. Filtering gene trees, or phylogenomic subsampling, can reduce artefactual conflict by selecting a subset of genes that are considered reliable. Tools such as GeneSortR (Mongiardino Koch 2021) and PhylteR (Comte et al. 2023) perform comparative analyses to identify a set of gene trees that have higher phylogenetic utility and accuracy, and remove potential outlier gene trees. GeneSortR is particularly extensive in comparisons, calculating average pairwise distance, compositional heterogeneity, level of saturation, root-to-tip variance, Robinson–Foulds distance to a reference topology, average bootstrap support, and proportion of variable sites. This also has the added benefit of producing easy-to-interpret and publication-ready images of the summarised outputs. TreeShrink (Mai and Mirarab 2018) is another useful tool to reduce artefactual gene tree conflict by identifying and pruning outlier long branches, thereby removing spurious samples. In combination with locus assembly and alignment assessment, gene tree assessment and phylogenomic subsampling can reduce the impact of non-biological conflict in the dataset and allow for clearer inferences of the true biological cause of conflict (see section ‘Investigating patterns and underlying causes of tree conflict’).
Should a researcher want to continue with producing a dated phylogeny with node-dating, special considerations need to be made to deal with target-capture datasets. Bayesian approaches to obtaining a dated phylogeny (e.g. BEAST, see https://www.beast2.org/; Bouckaert et al. 2014; MCMCTree in PAML, see http://abacus.gene.ucl.ac.uk/software/paml.html; Yang 2007) are computationally demanding and become intractable with large datasets (Barba-Montoya et al. 2021). This can be solved by subsampling genes to choose the most clocklike and similar to the species tree, as implemented in SortaDate (Smith et al. 2018) or using more computationally efficient phylogenetic dating methods such as penalised likelihood (Sanderson 2002), as implemented in treePL (see https://github.com/blackrim/treePL; Smith and O’Meara 2012), the R package ape (chronos) (see https://cran.r-project.org/package=ape; Paradis 2013; Paradis and Schliep 2019) and r8s (see https://sourceforge.net/projects/r8s/; Sanderson 2003) or the relative rate framework (RRF) as implemented in RelTime (Tamura et al. 2012, 2018). However, in cases in which extensive phylogenetic conflict is present and caused by biological processes such as reticulation, ILS or simultaneous speciation, extreme caution should be used in dating analyses. Currently, there is no method that can date a phylogeny that deviates from a bifurcating tree and as such, trying to apply a molecular clock that assumes a bifurcating birth–death process of species evolution to such a phylogenetic tree could lead to erroneous results (see section ‘Conclusions and future perspectives’).
Step 4. Conflict assessment
Before being able to identify the cause of phylogenetic conflict, one must first be able to pinpoint where the conflict occurs and the degree of conflict. Conflict may manifest as discordance between the topologies of species trees estimated with different methods or different data types or between gene trees. Conflict in topology across species trees inferred with different data types or methods (e.g. discrepancies in the topologies of plastid and nuclear phylogenies or discordance between coalescent and concatenated phylogenies) is usually identified visually and qualitatively described (Fig. 3). Conflict between the topologies of gene trees can be quantified on the resulting species tree in three main ways: through support values, concordance vectors (sensu Lanfear and Hahn 2024) and internode certainty (IC) (Fig. 3).
Support values, such as bootstrap values or posterior probabilities, are statistical measures of confidence for the existence of any given branch, akin to standard errors (Lanfear and Hahn 2024). Although they are are important measures, the increasing amount of data from high-throughput sequencing datasets means that support values tend towards the maximum, often giving inflated measures of confidence (Kumar et al. 2012; Thomson and Brown 2022). Concordance vectors, on the other hand, are statistical measures of the variation in the relationships of any given branch, analogous to standard deviation. Unlike support values, these are more robust to the effects of larger datasets, giving an informative summary of the variation in the topology of the taxa or clades at each node independent of the size of the dataset. Concordance vectors can be calculated in three ways: as gene concordance factors, as quartet concordance factors and as site concordance factors. These are reviewed in depth in Lanfear and Hahn (2024) and here we provide only a brief summary of the major differences between the three measures.
In short, gene concordance factors (gCFs) compare the topology for each node of each gene tree to the topology of the species tree and summarise the proportion of gene trees that have a topology concordant with the species tree (Ané et al. 2007; Baum 2007; Smith et al. 2015; Lanfear and Hahn 2024). gCFs can be calculated in several ways and the exact measures of concordance differ slightly depending on the method used. The most computationally feasible and popular methods for large datasets are implemented in IQ-TREE2 (see https://github.com/iqtree/iqtree2; Minh et al. 2020a), BUCKy (Larget et al. 2010) and PhyParts (Smith et al. 2015) that can also calculate concordance based on homolog trees (i.e. can account for duplications).
Quartet concordance factors (qCFs) are estimated by subsampling all (or many) sets of four taxa for each locus (‘quartets’), estimating the unrooted topology for each quartet and subsequently counting the proportion of quartets that are congruent with the species tree. Tools available to calculate qCFs include the program ASTRAL and the subsequent versions (e.g. Mirarab et al. 2014; Sayyari and Mirarab 2016) and Quartet Sampling (Pease et al. 2018).
Site concordance factors (sCFs) sample quartets of taxa for each node of the species tree and use parsimony or maximum likelihood to count the number of informative sites (of a single locus or concatenated loci) that support each of three possible topologies for these taxa (Minh et al. 2020a). Currently, this method is only implemented in IQ-TREE2 (Minh et al. 2020a; Mo et al. 2023); however, sCFs are more susceptible to the effects of homoplasy than other concordance vectors and may therefore overestimate discordance (Kück et al. 2022).
Another way to measure conflict within a species tree is by calculating internode certainty that can be seen as a summary of the aforementioned concordance vectors that compares the support for a given branch to the support for the best-supported alternative resolution of that branch (Salichos and Rokas 2013; Zhou X et al. 2020). Internode certainty can also be compared to branch length to gain an indication of potential factors that may be causing conflict. Visualising these quantified conflicts and the relative frequencies of different topological combinations can also be conducted through DiscoVista (Sayyari et al. 2018). Each measure of conflict has nuanced meaning, interpretation and pitfalls (Lanfear and Hahn 2024), therefore characterising conflict through a number of methods is always good practice.
Step 5. Investigating underlying causes of conflict
If care has been taken to reduce artefactual conflict, the remaining conflict measured in phylogenetic trees is likely largely due to biological sources of conflict. As described above, the four main biological sources of conflict are (1) paralogy, (2) reticulation, (3) deep coalescence and ILS, and (4) simultaneous speciation or rapid radiation (Fig. 2). Given ‘ideal’ data, differentiating between these patterns and reliably inferring the underlying evolutionary process in each case would be possible; however, sufficiently clear evidence may be unavailable with reduced-representation sequencing methods such as target-capture sequencing due to hidden paralogy or failure to capture or assemble all existing copies. A further complicating matter is that there is no one method that can satisfactorily model and test for paralogy, reticulation and ILS simultaneously. Therefore carefully selecting a suite of methods to test for and tease apart the effect of each of these processes if conflict is detected is important.
As previously explained, paralogy is caused either by gene duplication or WGD followed by lineage diversification. WGD events can be identified through target enrichment data in several ways. Locus extraction software such as HybPiper and CAPTUS may be able to infer the presence and number of paralogs for each locus (Johnson et al. 2016; Ortiz et al. 2023) depending on data quality and sequencing depth. These are useful for extracting all sequence copies and for gaining an indication of the amount of paralogy present in a dataset; however, these detected ‘paralogs’ are also likely to comprise divergent alleles and contigs with sequencing errors, therefore further processing is required to identify paralogs that are the result of gene or genome duplication events. Another method to characterise the degree of paralogy is through HybPhaser (Nauheimer et al. 2021). HybPhaser enables the user to define the threshold of heterozygosity that most likely represents true paralogs (rather than sequencing errors or alleles) for these to be quantified. Furthermore, heterozygosity has been shown to be correlated with ploidy level and can therefore be used to characterise lineages that have a history of genome duplication (Hendriks et al. 2023). Alternatively, the paralog output of locus extraction software can be processed by initially building clean homolog trees from all paralogs, extracting orthogroups from each homolog tree and mapping these to the species tree to count the number of gene duplication events that occurred at each node (e.g. Yang et al. 2018; Morales-Briones et al. 2021). Although these gene duplication mapping approaches have been shown to be useful for large (transcriptomic and genomic) datasets, the application in smaller target-capture datasets (particularly Angiosperms353 datasets) has not been extensively tested. Homolog trees can also be reconciled with species trees using programs such as GRAMPA (Thomas et al. 2017). GRAMPA uses a modified duplication-loss (DL) reconciliation algorithm (e.g. Goodman et al. 1979; Page 1994) to determine whether hypothesised genome duplication events are best explained by allo- or autopolyploidisation events. However, as with any DL-based method, GRAMPA does not account for ILS and can only investigate genome duplication at one node at a time, therefore use of such methods requires careful consideration and interpretation of results. The development of new reconciliation algorithms that can account for the coalescent process is an important area of future research to disentangle WGD and ILS in phylogenomic datasets (Boussau and Scornavacca 2020; Mishra et al. 2023). When possible, any of these analyses can be combined with additional sources of evidence, such as Ks plots from transcriptomic and genomic data or karyological data, to pinpoint WGD events in a species tree (e.g. Yang et al. 2018).
Dealing with hidden paralogy remains an area in need of further research in phylogenomics. The extent of hidden paralogy in plant evolution is yet to be quantified and as yet there are no formal methods available to manage hidden paralogs. Use of appropriate and well-designed bait kits may minimise this issue, and careful inspection of sequence alignments and homolog trees may enable a researcher to infer loci where hidden paralogy is an issue. However, at this point, hidden paralogy continues to be an unavoidable source of phylogenetic conflict.
Several approaches are available for testing the presence of reticulation, and several comprehensive reviews already widely address the issue of hybridisation and introgression in phylogenomic datasets (e.g. Hibbins and Hahn 2022; Steenwyk et al. 2023; Stull et al. 2023). We focus on methods that are applicable to target-capture data.
Introgression can be difficult to separate from ILS due to the similar signature that these leave in the data. Many methods for the detection of introgression work by comparing the depth of coalescence between estimated gene trees and the species tree, and infer introgression if the coalescence of gene lineages is too recent to be plausibly explained by deep coalescence (e.g. Joly et al. 2009; see ‘Deep coalescence and ILS’ below). Programs such as JML (Joly 2012), QuIBL (Edelman et al. 2019) and Aphid (Galtier 2024) compare branch lengths of taxon triplets from ortholog gene trees and by examining differences in branch lengths – shorter for gene flow and longer for deep coalescence – provide estimates of speciation times and ancestral population sizes, and quantify the impact of each process on phylogenetic conflict.
In cases of allopolyploid speciation, reticulation can be difficult to distinguish from autopolyploidisation because phylogenetic conflict is caused by both a non-bifurcating pattern of gene inheritance due to the crossing of two species and paralogy. In these cases, WGD mapping approaches such as GRAMPA (Thomas et al. 2017) are useful for inferring allopolyploid events from autopolyploid events (see previous section ‘Paralogy’) and inferring putative parental lineages.
Further approaches available for detection of reticulation (with and without polyploidisation) in target-capture datasets include phasing methods, whereby reads of putative hybrids are phased into subgenomes and placed separately into the species tree to identify putative parental lineages. This is commonly achieved in target-capture data through HybPhaser (Nauheimer et al. 2021) and has been shown to be highly effective in cases of neoallopolyploidy (e.g. Bloesch et al. 2022; Bradican et al. 2023); however, the method requires careful selection of the presence of diploid references for putative parental clades, and is often unsuitable for groups with complex or ancient reticulation (e.g. McLay et al. 2023). The need for a diploid reference is overcome through the Bayesian implementation of phasing in homologiser (Freyman et al. 2023) but may require subsampling of target-capture datasets to reduce computational demands.
Other available methods are derived from population genetics ABBA-BABA (or ‘D-statistic’) tests, whereby any deviation in site pattern probabilities from what would be expected in a bifurcating tree indicates reticulation or ILS. Such tests can be implemented in programs such as HyDe (Blischak et al. 2018). However, the site pattern probabilities expected in ABBA-BABA methods are calculated under a suite of assumptions, including symmetrical gene-flow between populations and constant substitution rate across lineages and genes, that may be unrealistic and could lead to inaccurate results (Frankel and Ané 2023a, 2023b). Therefore, these methods should be applied and interpreted with care.
Finally, network-based methods can be used to explore and depict reticulate evolutionary relationships but these methods are still in infancy and present a much-needed area for development. Distance-based methods such as Neighbour-Net (Bryant and Moulton 2004) and split decomposition methods such as SplitsTree (Huson 1998) are computationally feasible for phylogenomic datasets, but do not explicitly incorporate models of evolution, nor account for biological processes such as ILS. Other phylogenetic network packages can implement more complex models, including the likelihood methods used in PhyloNet (Than et al. 2008) and more recently developed Bayesian and coalescent methods (e.g. Yu and Nakhleh 2015; Solís-Lemus and Ané 2016; Wen et al. 2016; Zhang C et al. 2018a), such as those applied in PhyloNetworks (Solís-Lemus et al. 2017). These can provide robust estimations of phylogenetic networks but remain computationally intensive (often prohibitively so), restricting analysis to datasets of very few terminals. As phylogenetic network methods develop, these will be powerful tools to model and understand evolution in the presence of reticulation. However, networks are models that are more parameter-rich than bifurcating trees, therefore complex, reticulate scenarios will tend to be more statistically probable, even when these may not be true (Blair and Ané 2020). Therefore, results of phylogenetic network analyses should be evaluated critically, and are usually most useful as a complement to bifurcating trees rather than as a replacement.
Despite the expected incongruence between gene trees due to deep coalescence, the underlying species tree can still generally be reliably inferred under the assumption that ILS is the process causing the incongruence, i.e. under the multispecies coalescent. Although there are multiple methods for estimating the species tree, the most relevant to target-capture data that accounts for deep coalescence are summary approaches such as ASTRAL that take gene trees as input. If deep coalescence is the main cause of conflict in the data, target-capture data are particularly promising for resolving the species tree because individual loci may be long enough to produce relatively resolved gene trees and there are many loci. However, depending on the biological system and bait set, if most loci have little phylogenetic signal, this can mislead methods such as ASTRAL and make estimating the species tree more difficult (Molloy and Warnow 2018), underscoring the importance of evaluating phylogenetic signal and gene trees earlier.
Given that deep coalescence can leave a genetic signature similar to that of reticulation, most tests of deep coalescence also test for reticulation to differentiate the effect of these processes. Such tests include ABBA-BABA tests and branch-length based methods such as Aphid (Galtier 2024) and QuIBL (Edelman et al. 2019).
As simultaneous speciation is best represented as a polytomy in a phylogenetic tree, most methods available to test for simultaneous speciation and rapid radiations are statistical ‘polytomy tests’. These tests treat a polytomy as the null hypothesis (whereby the branch length is zero) and reject the null hypothesis based on data (Swofford et al. 1996; Anisimova and Gascuel 2006). Some versions also incorporate a power test to facilitate the differentiation of soft and hard polytomies (Walsh et al. 1999). Alternative Bayesian approaches such as that described by Lewis et al. (2005) are also available. However, these methods can only be applied to single-locus data. As such, the most popular method currently used for phylogenomic data is the polytomy test available through ASTRAL (Sayyari and Mirarab 2018). This method is also based on the concept of rejecting the null hypothesis of zero-branch-length polytomies in the tree, can test across multiple gene trees and also accounts for ILS but by nature is sensitive to errors in gene tree topology (Sayyari and Mirarab 2018). Regardless of the analysis conducted, conclusively inferring simultaneous speciation and differentiating it from a rapid radiation or an effect of a deficit of data is usually very difficult, unless the study is conducted with large quantities of phylogenetic data at a shallow phylogenetic scale, and in conjunction with a great deal of ecological and biological knowledge for the group in question. Realistically, the exact mode of evolution in most cases of rapid radiation and simultaneous speciation is therefore impossible to know.
Conclusions and future perspectives
Given the recent and rapid advancement of target-capture data for plant phylogenetic studies, there are many areas of the bioinformatic workflow that need improvement and research to further reduce artefactual conflict. One important area is locus extraction and assembly; the accurate and complete detection of paralogs resulting from gene duplication remains a challenge for plant phylogenomics, especially in groups currently without reference genomes. Aside from the unavoidable issue of hidden paralogy (see ‘Paralogy’ in the ‘Biological sources of conflict’ section) and misassembly issues that can arise from short-read assembler errors (see section ‘Step 1. Locus extraction’), there are indications that current assembly approaches may be underestimating real paralogy in Angiosperms353 datasets based on comparisons with reference genomes (Theodore Allnut, pers. comm.). Although the extents of such issues are currently unknown, further refinement of locus extraction and assembly programs, along with more affordable reference genomes and target-capture sequence from those same references, will greatly assist future studies. Furthermore, a better understanding of hidden paralogy in plants, and how different bait sets and different targeted loci (e.g. introns v. exons) perform is needed to further develop best practices for reducing artefactual noise and investigating the causes of conflict.
Even when phylogenomic analysis is carefully conducted in such a manner as to minimise data artefacts, phylogenetic conflict is often inevitable and expected given underlying biological processes. Although this conflict can often be carefully investigated to identify processes such as reticulation, paralogy, deep coalescence or rapid or simultaneous radiation as the cause, many methods to detect these processes are still being developed. The inability of many conflict interrogation analyses to account for more than two processes at once can make differentiating the effects on phylogenetic conflict and the influence on evolution difficult. Phylogenetic network methods also have great need for improvement for these to be applied meaningfully to large datasets. Currently, one of the main hindrances to the further development of these analyses is modelling the complex interactions between ILS, reticulation, paralogy and polytomies in such large datasets. Machine learning will likely play a significant role in overcoming these obstacles in future, although this would also come with a unique set of caveats and limitations (Mo et al. 2024). In the meantime, the limitations of the data and assumptions of models used should always be acknowledged and considered while these methods develop. The biological conclusions we make need to carefully consider and include the inherent (and real) uncertainty in our study systems.
Although identification of conflict and the underlying biological processes offers interesting insights into the mode of plant evolution, this also presents challenges for downstream evolutionary analyses such as dating, diversification analyses, ancestral area reconstruction and ancestral trait reconstruction. In cases in which conflict is caused by noise from paralogs or deep coalescence, researchers may opt for downstream analyses that can account for the topological uncertainty at nodes with high degrees of conflict. In cases of reticulation and simultaneous speciation, however, reasonably conducting these analyses is more difficult, as most cannot yet account for evolution that is not modelled by a bifurcating tree. In these cases, we encourage researchers to be realistic about which analyses can be justifiably conducted, and to be transparent about assumption violations and uncertainty in results when they are conducted. Creative solutions can possibly be found in these scenarios by, for example, conducting analyses on subsets of taxa or trees. However, overall development of dating, diversification and ancestral state reconstruction models that can account for these processes is another area where a great deal of research is needed, especially for plant evolutionary research.
We have long known that plant evolution is complex, with reticulation, WGD events, ILS and rapid radiations commonly reported and therefore phylogenetic conflict being inherent within many plant lineages should be unsurprising. Although target capture has made conflict more distinct, in some cases this can also provide unprecedented capacity to empirically test for the underlying biological processes that are the cause, providing new insights into the extraordinary complexities of plant evolution. For the Australian flora, this has meant shedding light on long-standing questions previously unanswerable due to a ‘lack of resolution’ intractable from previously available technologies. To date, target-capture studies have greatly enhanced our understanding of the timing and tempo of radiations (Joyce et al. 2023; Nge et al. 2024), the role of hybridisation and introgression in evolution (Nge et al. 2021a; Bloesch et al. 2022; McLay et al. 2023 – Adenanthos), polyploidy and WGD events (Nge et al. 2021b; Schmidt-Lebuhn et al. 2024a), and the evolution of diverse, ecologically important groups in Australia (Peakall et al. 2021; Schmidt-Lebuhn and Bovill 2021; McLay et al. 2023; Crisp et al. 2024). These have also shed light on biogeography within Australia (Nge et al. 2021a, 2021b – Calytrix and Pomaderris) and Australia’s biotic connection with other land-masses (Nge et al. 2021a; Joyce et al. 2023 – Pomaderris and Sapindales), demonstrating Australia’s role as both a source and sink of global plant diversity (Pillon et al. 2021; Van Dijk et al. 2023). Furthermore, these have aided in taxonomic classification and the description of new species (Simpson et al. 2022; Cooper et al. 2023; Crisp et al. 2024; Schmidt-Lebuhn and Grealy 2024). These studies are only scratching the surface but have clearly been an extraordinary advancement for our understanding of the Australian flora. We envisage that greater adoption of target-capture approaches through collective (GAP and the Australian Angiosperm Tree of Life – Schmidt-Lebuhn et al., in prep.) group-specific studies (e.g. Stage 2 GAP phylogenomics – https://www.genomicsforaustralianplants.com/phylogenomics/, accessed May 2024) will spearhead research on the evolution and systematics of the Australian flora, and highlight this on a global stage.
These Australian examples show that although much methodological development is needed, the advancement of target-capture data has nonetheless facilitated a step-change in plant phylogenomic research. The difficulties of dealing with conflict within datasets and the vast array of methods involved in analysing this type of data offer new challenges to overcome and complexity to decipher. However, surmounting these challenges will ultimately provide a more comprehensive understanding, and more realistic and accurate evolutionary reconstruction of plants in Australia and worldwide.
Declaration of funding
E. M. Joyce is supported by funding from the Prinzessin Therese von Bayern Stiftung.
Acknowledgements
The authors sincerely thank S. Bellot for her comments that greatly improved the manuscript.
References
Aberer AJ, Kobert K, Stamatakis A (2014) ExaBayes: massively parallel bayesian tree inference for the whole-genome era. Molecular Biology and Evolution 31, 2553-2556.
| Crossref | Google Scholar | PubMed |
Aïnouche M, Wendel J (2014) Polyploid speciation and genome evolution: lessons from recent allopolyploids. In ‘Evolutionary Biology: Genome Evolution, Speciation, Coevolution and Origin of Life’. (Ed. P Pontarotti) pp. 87–113. (Springer: Cham, Switzerland) 10.1007/978-3-319-07623-2_5
Alix K, Gérard PR, Schwarzacher T, Heslop-Harrison J (2017) Polyploidy and interspecific hybridization: partners for adaptation, speciation and evolution in plants. Annals of Botany 120, 183-194.
| Crossref | Google Scholar | PubMed |
Almeida-Silva F, Van de Peer Y (2023) Whole-genome duplications and the long-term evolution of gene regulatory networks in angiosperms. Molecular Biology and Evolution 40, msad141.
| Crossref | Google Scholar | PubMed |
Andermann T, Cano , Zizka A, Bacon C, Antonelli A (2018) SECAPR—a bioinformatics pipeline for the rapid and user-friendly processing of targeted enriched Illumina sequences, from raw reads to alignments. PeerJ 6, e5175.
| Crossref | Google Scholar |
Andermann T, Torres Jiménez MF, Matos-Maraví P, Batista R, Blanco-Pastor JL, Gustafsson ALS, Kistler L, Liberal IM, Oxelman B, Bacon CD, Antonelli A (2020) A guide to carrying out a phylogenomic target sequence capture project. Frontiers in Genetics 10, 1407.
| Crossref | Google Scholar | PubMed |
Ané C, Larget B, Baum DA, Smith SD, Rokas A (2007) Bayesian estimation of concordance among gene trees. Molecular Biology and Evolution 24, 412-426.
| Crossref | Google Scholar | PubMed |
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Systematic Biology 55, 539-552.
| Crossref | Google Scholar | PubMed |
Baker WJ, Dodsworth S, Forest F, Graham SW, Johnson MG, McDonnell A, Pokorny L, Tate JA, Wicke S, Wickett NJ (2021) Exploring Angiosperms353: an open, community toolkit for collaborative phylogenomic research on flowering plants. American Journal of Botany 108, 1059-1065.
| Crossref | Google Scholar | PubMed |
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology: A Journal of Computational Molecular Cell Biology 19, 455-477.
| Crossref | Google Scholar | PubMed |
Barba-Montoya J, Tao Q, Kumar S (2021) Assessing rapid relaxed-clock methods for phylogenomic dating. Genome Biology and Evolution 13, evab251.
| Crossref | Google Scholar | PubMed |
Baum DA (2007) Concordance trees, concordance factors, and the exploration of reticulate genealogy. TAXON 56, 417-426.
| Crossref | Google Scholar |
Blair C, Ané C (2020) Phylogenetic trees and networks can serve as powerful and complementary approaches for analysis of genomic data. Systematic Biology 69, 593-601.
| Crossref | Google Scholar | PubMed |
Blischak PD, Chifman J, Wolfe AD, Kubatko LS (2018) HyDe: a Python package for genome-scale hybridization detection. Systematic Biology 67, 821-829.
| Crossref | Google Scholar | PubMed |
Bloesch Z, Nauheimer L, Elias Almeida T, Crayn D, Field AR (2022) HybPhaser identifies hybrid evolution in Australian Thelypteridaceae. Molecular Phylogenetics and Evolution 173, 107526.
| Crossref | Google Scholar | PubMed |
Bolnick DI (2006) Multi-species outcomes in a common model of sympatric speciation. Journal of Theoretical Biology 241, 734-744.
| Crossref | Google Scholar | PubMed |
Bomblies K (2020) When everything changes at once: finding a new normal after genome duplication. Proceedings of the Royal Society of London – B. Biological Sciences 287, 20202154.
| Crossref | Google Scholar | PubMed |
Borowiec ML (2016) AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4, e1660.
| Crossref | Google Scholar | PubMed |
Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Computational Biology 10, e1003537.
| Crossref | Google Scholar | PubMed |
Boussau B, Scornavacca C (2020) Reconciling gene trees with species trees. In ‘Phylogenetics in the Genomic Era’. (Eds C Scornavacca, F Delsuc, N Galtier) pp. 3.2:1–3.2:23. (Published by the authors, CC BY-NC-ND) Available at https://hal.science/hal-02535529
Bradican JP, Tomasello S, Boscutti F, Karbstein K, Hörandl E (2023) Phylogenomics of southern European taxa in the Ranunculus auricomus species complex: the apple doesn’t fall far from the tree. Plants 12, 3664.
| Crossref | Google Scholar | PubMed |
Breinholt JW, Carey SB, Tiley GP, Davis EC, Endara L, McDaniel SF, Neves LG, Sessa EB, Von Konrat M, Chantanaorrapint S, Fawcett S, Ickert-Bond SM, Labiak PH, Larraín J, Lehnert M, Lewis LR, Nagalingum NS, Patel N, Rensing SA, Testo W, Vasco A, Villarreal JC, Williams EW, Burleigh JG (2021) A target enrichment probe set for resolving the flagellate land plant tree of life. Applications in Plant Sciences 9, e11406.
| Crossref | Google Scholar | PubMed |
Bryant D, Moulton V (2004) Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Molecular Biology and Evolution 21, 255-265.
| Crossref | Google Scholar | PubMed |
Cai L, Xi Z, Lemmon EM, Lemmon AR, Mast A, Buddenhagen CE, Liu L, Davis CC (2021) The perfect storm: gene tree estimation error, incomplete lineage sorting, and ancient gene flow explain the most recalcitrant ancient angiosperm clade, Malpighiales. Systematic Biology 70, 491-507.
| Crossref | Google Scholar | PubMed |
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972-1973.
| Crossref | Google Scholar | PubMed |
Cardillo M, Weston PH, Reynolds ZKM, Olde PM, Mast AR, Lemmon EM, Lemmon AR, Bromham L (2017) The phylogeny and biogeography of Hakea (Proteaceae) reveals the role of biome shifts in a continental plant radiation. Evolution 71, 1928-1943.
| Crossref | Google Scholar | PubMed |
Chaudhary R, Bansal MS, Wehe A, Fernández-Baca D, Eulenstein O (2010) iGTP: a software package for large-scale gene tree parsimony analysis. BMC Bioinformatics 11, 574.
| Crossref | Google Scholar | PubMed |
Clark JW, Donoghue PCJ (2018) Whole-genome duplication and plant macroevolution. Trends in Plant Science 23, 933-945.
| Crossref | Google Scholar | PubMed |
Comte A, Tricou T, Tannier E, Joseph J, Siberchicot A, Penel S, Allio R, Delsuc F, Dray S, De Vienne DM (2023) PhylteR: efficient identification of outlier sequences in phylogenomic datasets. Molecular Biology and Evolution 40, msad234.
| Crossref | Google Scholar | PubMed |
Cooper WE, Crayn DM, Joyce EM (2023) Aglaia fellii W.E.Cooper & Joyce (Meliaceae), a new species for Cape York Peninsula. Australian Journal of Taxonomy 16, 1-9.
| Crossref | Google Scholar |
Crisp MD, Minh BQ, Choi B, Edwards RD, Hereward J, Kulheim C, Lin YP, Meusemann K, Thornhill AH, Toon A, Cook LG (2024) Perianth evolution and implications for generic delimitation in the eucalypts (Myrtaceae), including the description of the new genus, Blakella. Journal of Systematics and Evolution jse 62, 942-962.
| Crossref | Google Scholar |
De Bodt S, Maere S, Van de Peer Y (2005) Genome duplication and the origin of angiosperms. Trends in Ecology & Evolution 20, 591-597.
| Crossref | Google Scholar | PubMed |
del Pozo JC, Ramirez-Parra E (2015) Whole genome duplications in plants: an overview from Arabidopsis. Journal of Experimental Botany 66, 6991-7003.
| Crossref | Google Scholar | PubMed |
DeSalle R, Absher R, Amato G (1994) Speciation and phylogenetic resolution. Trends in Ecology & Evolution 9, 297-298.
| Crossref | Google Scholar | PubMed |
Dillenberger MS, Kadereit JW (2017) Simultaneous speciation in the European high mountain flowering plant genus Facchinia (Minuartia sl, Caryophyllaceae) revealed by genotyping-by-sequencing. Molecular Phylogenetics and Evolution 112, 23-35.
| Crossref | Google Scholar | PubMed |
Douglas J, Jiménez-Silva CL, Bouckaert R (2022) StarBeast3: adaptive parallelized Bayesian inference under the multispecies coalescent. Systematic Biology 71, 901-916.
| Crossref | Google Scholar | PubMed |
Edelman NB, Mallet J (2021) Prevalence and adaptive impact of introgression. Annual Review of Genetics 55, 265-283.
| Crossref | Google Scholar | PubMed |
Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow RB, García-Accinelli G, Van Belleghem SM, Patterson N, Neafsey DE, Challis R, Kumar S, Moreira GRP, Salazar C, Chouteau M, Counterman BA, Papa R, Blaxter M, Reed RD, Dasmahapatra KK, Kronforst M, Joron M, Jiggins CD, McMillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, Mallet J (2019) Genomic architecture and introgression shape a butterfly radiation. Science 366, 594-599.
| Crossref | Google Scholar | PubMed |
Faircloth BC (2016) PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics 32, 786-788.
| Crossref | Google Scholar | PubMed |
Fér T, Schmickl RE (2018) HybPhyloMaker: target enrichment data analysis from raw reads to species trees. Evolutionary Bioinformatics Online 14, 1176934317742613.
| Crossref | Google Scholar | PubMed |
Flagel LE, Wendel JF (2009) Gene duplication and evolutionary novelty in plants. The New Phytologist 183, 557-564.
| Crossref | Google Scholar | PubMed |
Fowler RM, McLay TGB, Schuster TM, Buirchell BJ, Murphy DJ, Bayly MJ (2020) Plastid phylogenomic analysis of tribe Myoporeae (Scrophulariaceae). Plant Systematics and Evolution 306, 52.
| Crossref | Google Scholar |
Frankel LE, Ané C (2023a) Summary tests of introgression are highly sensitive to rate variation across lineages. bioRxiv 2023, 2023.01.26.525396 [Preprint, published 26 January 2023].
| Crossref | Google Scholar |
Frankel LE, Ané C (2023b) Summary tests of introgression are highly sensitive to rate variation across lineages. Systematic Biology 72(6), 1357-1369.
| Crossref | Google Scholar | PubMed |
Freyman WA, Johnson MG, Rothfels CJ (2023) homologizer: phylogenetic phasing of gene copies into polyploid subgenomes. Methods in Ecology and Evolution 14, 1230-1244.
| Crossref | Google Scholar |
Frost LA, Bedoya AM, Lagomarsino LP (2024) Artifactual orthologs and the need for diligent data exploration in complex phylogenomic datasets: a museomic case study from the Andean Flora. Systematic Biology 73, 308-322.
| Crossref | Google Scholar | PubMed |
Galtier N (2024) An approximate likelihood method reveals ancient gene flow between human, chimpanzee and gorilla. Peer Community Journal 4, e3.
| Crossref | Google Scholar |
Goodman M, Czelusniak J, Moore GW, Romero-Herrera AE, Matsuda G (1979) Fitting the gene lineage into its species lineage, a parsimony strategy illustrated by cladograms constructed from globin sequences. Systematic Biology 28, 132-163.
| Crossref | Google Scholar |
Gunn BF, Murphy DJ, Walsh NG, Conran JG, Pires JC, Macfarlane TD, Birch JL (2020) Evolution of Lomandroideae: multiple origins of polyploidy and biome occupancy in Australia. Molecular Phylogenetics and Evolution 149, 106836.
| Crossref | Google Scholar | PubMed |
Gunn BF, Murphy DJ, Walsh NG, Conran JG, Pires JC, Macfarlane TD, Crisp MD, Cook LG, Birch JL (2024) Genomic data resolve phylogenetic relationships of Australian mat-rushes, Lomandra (Asparagaceae: Lomandroideae). Botanical Journal of the Linnean Society 204, 1-22.
| Crossref | Google Scholar |
Hammer TA, Biffin E, van Dijk K, Thiele KR, Waycott M (2025) A framework phylogeny of the diverse guinea-flowers (Hibbertia, Dilleniaceae) using high-throughput sequence data. Australian Systematic Botany 38(2), SB24009.
| Crossref | Google Scholar |
Hancock LP, Obbens F, Moore AJ, Thiele K, De Vos JM, West J, Holtum JAM, Edwards EJ (2018) Phylogeny, evolution, and biogeographic history of Calandrinia (Montiaceae). American Journal of Botany 105, 1021-1034.
| Crossref | Google Scholar | PubMed |
Handika H, Esselstyn JA (2024) SEGUL: ultrafast, memory‐efficient and mobile‐friendly software for manipulating and summarizing phylogenomic datasets. Molecular Ecology Resources 24(7), e13964.
| Crossref | Google Scholar |
Hart ML, Forrest LL, Nicholls JA, Kidner CA (2016) Retrieval of hundreds of nuclear loci from herbarium specimens. TAXON 65, 1081-1092.
| Crossref | Google Scholar |
Hendriks KP, Kiefer C, Al-Shehbaz IA, Bailey CD, Hooft van Huysduynen A, Nikolov LA, Nauheimer L, Zuntini AR, German DA, Franzke A, Koch MA, Lysak MA, Toro-Núñez Ó, Özüdoğru B, Invernón VR, Walden N, Maurin O, Hay NM, Shushkov P, Mandáková T, Schranz ME, Thulin M, Windham MD, Rešetnik I, Španiel S, Ly E, Pires JC, Harkess A, Neuffer B, Vogt R, Bräuchler C, Rainer H, Janssens SB, Schmull M, Forrest A, Guggisberg A, Zmarzty S, Lepschi BJ, Scarlett N, Stauffer FW, Schönberger I, Heenan P, Baker WJ, Forest F, Mummenhoff K, Lens F (2023) Global Brassicaceae phylogeny based on filtering of 1,000-gene dataset. Current Biology 33, 4052-4068.
| Crossref | Google Scholar | PubMed |
Hibbins MS, Hahn MW (2022) Phylogenomic approaches to detecting and characterizing introgression. Genetics 220, iyab173.
| Crossref | Google Scholar | PubMed |
Hoelzer GA, Meinick DJ (1994) Patterns of speciation and limits to phylogenetic resolution. Trends in Ecology & Evolution 9, 104-107.
| Crossref | Google Scholar | PubMed |
Huson DH (1998) SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14, 68-73.
| Crossref | Google Scholar | PubMed |
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23(2), 254-267.
| Crossref | Google Scholar |
Jackson C, McLay T, Schmidt-Lebuhn AN (2023) hybpiper-nf and paragone-nf: containerization and additional options for target capture assembly and paralog resolution. Applications in Plant Sciences 11, e11532.
| Crossref | Google Scholar | PubMed |
Johnson MG, Gardner EM, Liu Y, Medina R, Goffinet B, Shaw AJ, Zerega NJ, Wickett NJ (2016) HybPiper: extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Applications in Plant Sciences 4, 1600016.
| Crossref | Google Scholar | PubMed |
Johnson MG, Pokorny L, Dodsworth S, Botigué LR, Cowan RS, Devault A, Eiserhardt WL, Epitawalage N, Forest F, Kim JT, Leebens-Mack JH, Leitch IJ, Maurin O, Soltis DE, Soltis PS, Wong GK, Baker WJ, Wickett NJ (2019) A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Systematic Biology 68, 594-606.
| Crossref | Google Scholar | PubMed |
Joly S (2012) JML: testing hybridization from species trees. Molecular Ecology Resources 12, 179-184.
| Crossref | Google Scholar | PubMed |
Joly S, McLenachan PA, Lockhart PJ (2009) A statistical approach for distinguishing hybridization and incomplete lineage sorting. The American Naturalist 174, 54-70.
| Crossref | Google Scholar | PubMed |
Joyce EM, Appelhans MS, Buerki S, Cheek M, de Vos JM, Pirani JR, Zuntini AR, Bachelier JB, Bayly MJ, Callmander MW, Devecchi MF, Pell SK, Groppo M, Lowry PP, Mitchell J, Siniscalchi CM, Munzinger J, Orel HK, Pannell CM, Nauheimer L, Sauquet H, Weeks A, Muellner-Riehl AN, Leitch IJ, Maurin O, Forest F, Nargar K, Thiele KR, Baker WJ, Crayn DM (2023) Phylogenomic analyses of Sapindales support new family relationships, rapid Mid-Cretaceous Hothouse diversification, and heterogeneous histories of gene duplication. Frontiers in Plant Science 14, 1063174.
| Crossref | Google Scholar | PubMed |
Joyce EM, Schmidt-Lebuhn AN, Orel HK, Nge FJ, Anderson BM, Hammer TA, McLay TGB (2024) Navigating phylogenetic conflict and evolutionary inference in plants with target capture data. EcoEvoRxiv 2024, Version 2 [Preprint, published 27 May 2024, updated 7 March 2025].
| Crossref | Google Scholar |
Karimi N, Grover CE, Gallagher JP, Wendel JF, Ané C, Baum DA (2020) Reticulate evolution helps explain apparent homoplasy in floral biology and pollination in baobabs (Adansonia; Bombacoideae; Malvaceae). Systematic Biology 69, 462-478.
| Crossref | Google Scholar | PubMed |
Kates HR, Johnson MG, Gardner EM, Zerega NJC, Wickett NJ (2018) Allele phasing has minimal impact on phylogenetic reconstruction from targeted nuclear gene sequences in a case study of Artocarpus. American Journal of Botany 105, 404-416.
| Crossref | Google Scholar | PubMed |
Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A (2019) RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453-4455.
| Crossref | Google Scholar | PubMed |
Kück P, Romahn J, Meusemann K (2022) Pitfalls of the site-concordance factor (sCF) as measure of phylogenetic branch support. NAR Genomics and Bioinformatics 4, lqac064.
| Crossref | Google Scholar | PubMed |
Kumar S, Filipski AJ, Battistuzzi FU, Kosakovsky Pond SL, Tamura K (2012) Statistics and truth in phylogenomics. Molecular Biology and Evolution 29, 457-472.
| Crossref | Google Scholar | PubMed |
Landis JB, Soltis DE, Li Z, Marx HE, Barker MS, Tank DC, Soltis PS (2018) Impact of whole‐genome duplication events on diversification rates in angiosperms. American Journal of Botany 105, 348-363.
| Crossref | Google Scholar | PubMed |
Lanfear R, Hahn MW (2024) The meaning and measure of concordance factors in phylogenomics. Molecular Biology and Evolution 41, msae214.
| Crossref | Google Scholar | PubMed |
Larget BR, Kotha SK, Dewey CN, Ané C (2010) BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26, 2910-2911.
| Crossref | Google Scholar | PubMed |
Larridon I, Zuntini AR, Barrett RL, Wilson KL, Bruhl JJ, Goetghebeur P, Baker WJ, Brewer GE, Epitawalage N, Fairlie I, Forest F, Sabino Kikuchi IAB, Pokorny L, Semmouri I, Spalink D, Simpson DA, Muasya AM, Roalson EH (2021) Resolving generic limits in Cyperaceae tribe Abildgaardieae using targeted sequencing. Botanical Journal of the Linnean Society 196, 163-187.
| Crossref | Google Scholar |
Leaché AD, Rannala B (2011) The accuracy of species tree estimation under simulation: a comparison of methods. Systematic Biology 60, 126-137.
| Crossref | Google Scholar | PubMed |
Lemmon AR, Emme SA, Lemmon EM (2012) Anchored hybrid enrichment for massively high-throughput phylogenomics. Systematic Biology 61(5), 727-744.
| Crossref | Google Scholar | PubMed |
Lewis PO, Holder MT, Holsinger KE (2005) Polytomies and Bayesian phylogenetic inference. Systematic Biology 54, 241-253.
| Crossref | Google Scholar | PubMed |
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W (2015) MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674-1676.
| Crossref | Google Scholar | PubMed |
Liao X, Li M, Zou Y, Wu F, Yi‐Pan , Wang J (2019) Current challenges and solutions of de novo assembly. Quantitative Biology 7, 90-109.
| Crossref | Google Scholar |
McKinnon GE, Steane DA, Potts BM, Vaillancourt RE (1999) Incongruence between chloroplast and species phylogenies in Eucalyptus subgenus Monocalyptus (Myrtaceae). American Journal of Botany 86, 1038-1046.
| Crossref | Google Scholar | PubMed |
McLay TGB, Birch JL, Gunn BF, Ning W, Tate JA, Nauheimer L, Joyce EM, Simpson L, Schmidt-Lebuhn AN, Baker WJ, Forest F, Jackson CJ (2021) New targets acquired: improving locus recovery from the Angiosperms353 probe set. Applications in Plant Sciences 9, e11420.
| Crossref | Google Scholar | PubMed |
McLay TGB, Fowler RM, Fahey PS, Murphy DJ, Udovicic F, Cantrill DJ, Bayly MJ (2023) Phylogenomics reveals extreme gene tree discordance in a lineage of dominant trees: hybridization, introgression, and incomplete lineage sorting blur deep evolutionary relationships despite clear species groupings in Eucalyptus subgenus Eudesmia. Molecular Phylogenetics and Evolution 187, 107869.
| Crossref | Google Scholar | PubMed |
Maddison W (1989) Reconstructing character evolution on polytomous cladograms. Cladistics 5, 365-377.
| Crossref | Google Scholar | PubMed |
Maddison WP (1997) Gene trees in species trees. Systematic Biology 46, 523-536.
| Crossref | Google Scholar |
Mai U, Mirarab S (2018) TreeShrink: fast and accurate detection of outlier long branches in collections of phylogenetic trees. BMC Genomics 19, 272.
| Crossref | Google Scholar | PubMed |
Marques DA, Meier JI, Seehausen O (2019) A combinatorial view on speciation and adaptive radiation. Trends in Ecology & Evolution 34, 531-544.
| Crossref | Google Scholar | PubMed |
Mason AS, Wendel JF (2020) Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Frontiers in Genetics 11, 1014.
| Crossref | Google Scholar | PubMed |
Matsubayashi KW, Yamaguchi R (2022) The speciation view: disentangling multiple causes of adaptive and non-adaptive radiation in terms of speciation. Population Ecology 64, 95-107.
| Crossref | Google Scholar |
Minh BQ, Hahn MW, Lanfear R (2020a) New methods to calculate concordance factors for phylogenomic datasets. Molecular Biology and Evolution 37, 2727-2733.
| Crossref | Google Scholar | PubMed |
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R (2020b) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Molecular Biology and Evolution 37, 1530-1534.
| Crossref | Google Scholar | PubMed |
Mirarab S, Reaz R, Bayzid MdS, Zimmermann T, Swenson MS, Warnow T (2014) ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, 541-8.
| Crossref | Google Scholar | PubMed |
Mirarab S, Bayzid MS, Warnow T (2016) Evaluating summary methods for multilocus species tree estimation in the presence of incomplete lineage sorting. Systematic Biology 65, 366-380.
| Crossref | Google Scholar | PubMed |
Mishra S, Smith ML, Hahn MW (2023) reconcILS: a gene tree-species tree reconciliation algorithm that allows for incomplete lineage sorting. bioRxiv 2023, 2023.11.03.565544 [Preprint, published 13 October 2024].
| Crossref | Google Scholar |
Mo YK, Lanfear R, Hahn MW, Minh BQ (2023) Updated site concordance factors minimize effects of homoplasy and taxon sampling. Bioinformatics 39, btac741.
| Crossref | Google Scholar | PubMed |
Mo YK, Hahn MW, Smith ML (2024) Applications of machine learning in phylogenetics. Molecular Phylogenetics and Evolution 196, 108066.
| Crossref | Google Scholar | PubMed |
Molloy EK, Warnow T (2018) To include or not to include: the impact of gene filtering on species tree estimation methods. Systematic Biology 67, 285-303.
| Crossref | Google Scholar | PubMed |
Molloy EK, Warnow T (2020) FastMulRFS: fast and accurate species tree estimation under generic gene duplication and loss models. Bioinformatics 36, i57-i65.
| Crossref | Google Scholar | PubMed |
Mongiardino Koch N (2021) Phylogenomic subsampling and the search for phylogenetically reliable loci. Molecular Biology and Evolution 38, 4025-4038.
| Crossref | Google Scholar | PubMed |
Morales-Briones DF, Gehrke B, Huang C-H, Liston A, Ma H, Marx HE, Tank DC, Yang Y (2021) Analysis of paralogs in target enrichment data pinpoints multiple ancient polyploidy events in Alchemilla s.l. (Rosaceae). Systematic Biology 71, 190-207.
| Crossref | Google Scholar | PubMed |
Morel B, Schade P, Lutteropp S, Williams TA, Szöllősi GJ, Stamatakis A (2022) SpeciesRax: a tool for maximum likelihood species tree inference from gene family trees under duplication, transfer, and loss. Molecular Biology and Evolution 39, msab365.
| Crossref | Google Scholar | PubMed |
Morel B, Williams TA, Stamatakis A, Szöllősi GJ (2024) AleRax: a tool for gene and species tree co-estimation and reconciliation under a probabilistic model of gene duplication, transfer, and loss. Bioinformatics 40, btae162.
| Crossref | Google Scholar | PubMed |
Nauheimer L, Cui L, Clarke C, Crayn DM, Bourke G, Nargar K (2019) Genome skimming provides well resolved plastid and nuclear phylogenies, showing patterns of deep reticulate evolution in the tropical carnivorous plant genus Nepenthes (Caryophyllales). Australian Systematic Botany 32, 243-254.
| Crossref | Google Scholar |
Nauheimer L, Weigner N, Joyce E, Crayn D, Clarke C, Nargar K (2021) HybPhaser: a workflow for the detection and phasing of hybrids in target capture data sets. Applications in Plant Sciences 9, e11441.
| Crossref | Google Scholar | PubMed |
Nevill PG, Després T, Bayly MJ, Bossinger G, Ades PK (2014) Shared phylogeographic patterns and widespread chloroplast haplotype sharing in Eucalyptus species with different ecological tolerances. Tree Genetics & Genomes 10, 1079-1092.
| Crossref | Google Scholar |
Nge FJ, Biffin E, Thiele KR, Waycott M (2021a) Reticulate evolution, ancient chloroplast haplotypes, and rapid radiation of the Australian plant genus Adenanthos (Proteaceae). Frontiers in Ecology and Evolution 8, 616741.
| Crossref | Google Scholar |
Nge FJ, Kellermann J, Biffin E, Waycott M, Thiele KR (2021b) Historical biogeography of Pomaderris (Rhamnaceae): continental vicariance in Australia and repeated independent dispersals to New Zealand. Molecular Phylogenetics and Evolution 158, 107085.
| Crossref | Google Scholar | PubMed |
Nge FJ, Biffin E, Waycott M, Thiele KR (2022) Phylogenomics and continental biogeographic disjunctions: insight from the Australian starflowers (Calytrix). American Journal of Botany 109, 291-308.
| Crossref | Google Scholar | PubMed |
Nge FJ, Kellermann J, Biffin E, Thiele KR, Waycott M (2024) Rise and fall of a continental mesic radiation in Australia: spine evolution, biogeography, and diversification of Cryptandra (Rhamnaceae: Pomaderreae). Botanical Journal of the Linnean Society 204, 327-342.
| Crossref | Google Scholar |
Nge FJ, Biffin E, Rye BL, Wilson PG, van Dijk K, Thiele KR, Waycott M, Barrett MD (2025) Australian biogeography, climate-dependent diversification and phylogenomics of the spectacular Chamelaucieae tribe (Myrtaceae). Australian Systematic Botany 38(1), SB24014.
| Crossref | Google Scholar |
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution 32, 268-274.
| Crossref | Google Scholar | PubMed |
Nicol DA, Saldivia P, Summerfield TC, Heads M, Lord JM, Khaing EP, Larcombe MJ (2024) Phylogenomics and morphology of Celmisiinae (Asteraceae: Astereae): taxonomic and evolutionary implications. Molecular Phylogenetics and Evolution 195, 108064.
| Crossref | Google Scholar | PubMed |
Nute M, Chou J, Molloy EK, Warnow T (2018) The performance of coalescent-based species tree estimation methods under models of missing data. BMC Genomics 19, 286.
| Crossref | Google Scholar | PubMed |
Orel HK, McLay TGB, Neal WC, Forster PI, Bayly MJ (2023) Plastid phylogenomics of the Eriostemon group (Rutaceae; Zanthoxyloideae): support for major clades and investigation of a backbone polytomy. Australian Systematic Botany 36, 355-385.
| Crossref | Google Scholar |
Orel HK, McLay TG, Guja LK, Duretto MF, Bayly MJ (2024) Genomic data inform taxonomy and conservation of critically endangered shrubs: a case study of Zieria (Rutaceae) species from eastern Australia. Botanical Journal of the Linnean Society 205, 292-312.
| Crossref | Google Scholar |
Orel HK, McLay TGB, Forster PI, Bayly MJ (2025) Target capture sequencing clarifies key relationships in the Eriostemon group (Rutaceae: Zanthoxyloideae) and supports a reclassification of Philotheca, including the recognition of two new genera. TAXON. [Published online early 6 February 2025].
| Crossref | Google Scholar |
Ortiz EM, Höwener A, Shigita G, Raza M, Maurin O, Zuntini A, Forest F, Baker WJ, Schaefer H (2023) A novel phylogenomics pipeline reveals complex pattern of reticulate evolution in Cucurbitales. bioRxiv 2023, 2023.10.27.564367 [Preprint, published 26 December 2024].
| Crossref | Google Scholar |
Ostevik KL, Andrew RL, Otto SP, Rieseberg LH (2016) Multiple reproductive barriers separate recently diverged sunflower ecotypes. Evolution 70, 2322-2335.
| Crossref | Google Scholar | PubMed |
Page RD (1994) Maps between trees and cladistic analysis of historical associations among genes, organisms, and areas. Systematic Biology 43, 58-77.
| Crossref | Google Scholar |
Pamilo P, Nei M (1988) Relationships between gene trees and species trees. Molecular Biology and Evolution 5, 568-583.
| Crossref | Google Scholar | PubMed |
Panchy N, Lehti-Shiu M, Shiu S-H (2016) Evolution of gene duplication in plants. Plant Physiology 171, 2294-2316.
| Crossref | Google Scholar | PubMed |
Paradis E (2013) Molecular dating of phylogenies by likelihood methods: a comparison of models and a new information criterion. Molecular Phylogenetics and Evolution 67, 436-444.
| Crossref | Google Scholar | PubMed |
Paradis E, Schliep K (2019) ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526-528.
| Crossref | Google Scholar | PubMed |
Peakall R, Wong DCJ, Phillips RD, Ruibal M, Eyles R, Rodriguez-Delgado C, Linde CC (2021) A multitiered sequence capture strategy spanning broad evolutionary scales: application for phylogenetic and phylogeographic studies of orchids. Molecular Ecology Resources 21, 1118-1140.
| Crossref | Google Scholar | PubMed |
Pease JB, Brown JW, Walker JF, Hinchliff CE, Smith SA (2018) Quartet Sampling distinguishes lack of support from conflicting support in the green plant tree of life. American Journal of Botany 105, 385-403.
| Crossref | Google Scholar | PubMed |
Pillon Y, Hopkins HCF, Maurin O, Epitawalage N, Bradford J, Rogers ZS, Baker WJ, Forest F (2021) Phylogenomics and biogeography of Cunoniaceae (Oxalidales) with complete generic sampling and taxonomic realignments. American Journal of Botany 108, 1181-1200.
| Crossref | Google Scholar | PubMed |
Potts AJ, Hedderson TA, Grimm GW (2014) Constructing phylogenies in the presence of intra-individual site polymorphisms (2ISPs) with a focus on the nuclear ribosomal cistron. Systematic Biology 63, 1-16.
| Crossref | Google Scholar | PubMed |
Rannala B, Edwards VS, Leaché A, Yang Z (2020) The multi-species coalescent model and species tree inference. In ‘Phylogenetics in the Genomic Era)’. pp. 3.3:1–3.3:21. (Published by the authors, CC BY-NC-ND) Available at https://hal.science/hal-02535622v1
Raza M, Ortiz EM, Schwung L, Shigita G, Schaefer H (2023) Resolving the phylogeny of Thladiantha (Cucurbitaceae) with three different target capture pipelines. BMC Ecology and Evolution 23, 75.
| Crossref | Google Scholar | PubMed |
Ren R, Wang H, Guo C, Zhang N, Zeng L, Chen Y, Ma H, Qi J (2018) Widespread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Molecular Plant 11, 414-428.
| Crossref | Google Scholar | PubMed |
Salichos L, Rokas A (2013) Inferring ancient divergences requires genes with strong phylogenetic signals. Nature 497, 327-331.
| Crossref | Google Scholar | PubMed |
Sanderson MJ (2002) Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Molecular Biology and Evolution 19, 101-109.
| Crossref | Google Scholar | PubMed |
Sanderson MJ (2003) r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19, 301-302.
| Crossref | Google Scholar | PubMed |
Sayyari E, Mirarab S (2016) Fast coalescent-based computation of local branch support from quartet frequencies. Molecular Biology and Evolution 33, 1654-1668.
| Crossref | Google Scholar | PubMed |
Sayyari E, Mirarab S (2018) Testing for polytomies in phylogenetic species trees using quartet frequencies. Genes 9, 132.
| Crossref | Google Scholar | PubMed |
Sayyari E, Whitfield JB, Mirarab S (2018) DiscoVista: interpretable visualizations of gene tree discordance. Molecular Phylogenetics and Evolution 122, 110-115.
| Crossref | Google Scholar | PubMed |
Scannell DR, Byrne KP, Gordon JL, Wong S, Wolfe KH (2006) Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 440, 341-345.
| Crossref | Google Scholar | PubMed |
Schmidt-Lebuhn AN (2022) Sequence capture data support the taxonomy of Pogonolepis (Asteraceae: Gnaphalieae) and show unexpected genetic structure. Australian Systematic Botany 35(4), 317-325.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Bovill J (2021) Phylogenomic data reveal four major clades of Australian Gnaphalieae (Asteraceae). TAXON 70, 1020-1034.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Grealy A (2024) Transfer of Cotula alpina to the genus Leptinella (Asteraceae: Anthemideae). Australian Systematic Botany 37, SB23012.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Egli D, Grealy A, Nicholls JA, Zwick A, Dymock JJ, Gooden B (2024a) Genetic data confirm the presence of Senecio madagascariensis in New Zealand. New Zealand Journal of Botany 62, 1-13.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Chen SH, Grealy A (2024b) Elachanthus, Isoetopsis and Kippistia are nested in the genus Minuria (Asteraceae: Astereae). Australian Systematic Botany 37(4), SB23028.
| Crossref | Google Scholar |
Shee ZQ, Frodin DG, Cámara-Leret R, Pokorny L (2020) Reconstructing the complex evolutionary history of the Papuasian Schefflera radiation through herbariomics. Frontiers in Plant Science 11, 258.
| Crossref | Google Scholar | PubMed |
Simmons MP, Gatesy J (2015) Coalescence vs. concatenation: sophisticated analyses vs. first principles applied to rooting the angiosperms. Molecular Phylogenetics and Evolution 91, 98-122.
| Crossref | Google Scholar | PubMed |
Simmons MP, Gatesy J (2021) Collapsing dubiously resolved gene-tree branches in phylogenomic coalescent analyses. Molecular Phylogenetics and Evolution 158, 107092.
| Crossref | Google Scholar | PubMed |
Simpson J, Conran JG, Biffin E, Van Dijk K, Waycott M (2022) The Crinum flaccidum (Amaryllidaceae) species complex in Australia. Australian Systematic Botany 35, 395-402.
| Crossref | Google Scholar |
Siniscalchi CM, Hidalgo O, Palazzesi L, Pellicer J, Pokorny L, Maurin O, Leitch IJ, Forest F, Baker WJ, Mandel JR (2021) Lineage‐specific vs. universal: a comparison of the Compositae1061 and Angiosperms353 enrichment panels in the sunflower family. Applications in Plant Sciences 9, e11422.
| Crossref | Google Scholar |
Smirnov V, Warnow T (2021) Phylogeny estimation given sequence length heterogeneity. Systematic Biology 70(2), 268-282.
| Crossref | Google Scholar |
Smith ML, Hahn MW (2021) New approaches for inferring phylogenies in the presence of paralogs. Trends in Genetics 37, 174-187.
| Crossref | Google Scholar | PubMed |
Smith ML, Hahn MW (2022) The frequency and topology of pseudoorthologs. Systematic Biology 71, 649-659.
| Crossref | Google Scholar | PubMed |
Smith SA, O’Meara BC (2012) treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 28, 2689-2690.
| Crossref | Google Scholar | PubMed |
Smith SA, Moore MJ, Brown JW, Yang Y (2015) Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evolutionary Biology 15, 150.
| Crossref | Google Scholar | PubMed |
Smith SA, Brown JW, Walker JF (2018) So many genes, so little time: a practical approach to divergence-time estimation in the genomic era. PLoS ONE 13, e0197433.
| Crossref | Google Scholar | PubMed |
Smith BT, Mauck WM, Benz BW, Andersen MJ (2020) Uneven missing data skew phylogenomic relationships within the lories and lorikeets. Genome Biology and Evolution 12, 1131-1147.
| Crossref | Google Scholar | PubMed |
Solís-Lemus C, Ané C (2016) Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genetics 12, e1005896.
| Crossref | Google Scholar | PubMed |
Solís-Lemus C, Bastide P, Ané C (2017) PhyloNetworks: a package for phylogenetic networks. Molecular Biology and Evolution 34(12), 3292-3298.
| Crossref | Google Scholar |
Soltis DE, Albert VA, Leebens-Mack J, Bell CD, Paterson AH, Zheng C, Sankoff D, de Pamphilis CW, Wall PK, Soltis PS (2009) Polyploidy and angiosperm diversification. American Journal of Botany 96, 336-348.
| Crossref | Google Scholar | PubMed |
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312-1313.
| Crossref | Google Scholar | PubMed |
Steenwyk JL, Buida TJ, Li Y, Shen X-X, Rokas A (2020) ClipKIT: a multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biology 18, e3001007.
| Crossref | Google Scholar | PubMed |
Steenwyk JL, Li Y, Zhou X, Shen X-X, Rokas A (2023) Incongruence in the phylogenomics era. Nature Reviews Genetics 24, 834-850.
| Crossref | Google Scholar | PubMed |
Stegemann S, Keuthe M, Greiner S, Bock R (2012) Horizontal transfer of chloroplast genomes between plant species. Proceedings of the National Academy of Sciences of the United States of America 109, 2434-2438.
| Crossref | Google Scholar | PubMed |
Strobel V (2018) Pold87/academic-keyword-occurrence: first release. Zenodo 2018, v1.0.0 [Dataset, published 14 April 2014].
| Crossref | Google Scholar |
Struck TH (2013) The impact of paralogy on phylogenomic studies – a case study on annelid relationships. PLoS ONE 8, e62892.
| Crossref | Google Scholar | PubMed |
Stull GW, Pham KK, Soltis PS, Soltis DE (2023) Deep reticulation: the long legacy of hybridization in vascular plant evolution. The Plant Journal 114, 743-766.
| Crossref | Google Scholar | PubMed |
Suarez-Gonzalez A, Lexer C, Cronk QCB (2018) Adaptive introgression: a plant perspective. Biology Letters 14, 20170688.
| Crossref | Google Scholar | PubMed |
Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S (2012) Estimating divergence times in large molecular phylogenies. Proceedings of the National Academy of Sciences of the United States of America 109, 19333-19338.
| Crossref | Google Scholar | PubMed |
Tamura K, Tao Q, Kumar S (2018) Theoretical foundation of the RelTime method for estimating divergence times from variable evolutionary rates. Molecular Biology and Evolution 35, 1770-1782.
| Crossref | Google Scholar | PubMed |
Tea Y-K, Xu X, DiBattista JD, Lo N, Cowman PF, Ho SYW (2022) Phylogenomic analysis of concatenated ultraconserved elements reveals the recent evolutionary radiation of the fairy wrasses (Teleostei: Labridae: Cirrhilabrus). Systematic Biology 71, 1-12.
| Crossref | Google Scholar | PubMed |
Than C, Ruths D, Nakhleh L (2008) PhyloNet: a software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinformatics 9, 322.
| Crossref | Google Scholar | PubMed |
Thomson RC, Brown JM (2022) On the need for new measures of phylogenomic support. Systematic Biology 71, 917-920.
| Crossref | Google Scholar | PubMed |
Thomas GWC, Ather SH, Hahn MW (2017) Gene-tree reconciliation with MUL-trees to resolve polyploidy events. Systematic Biology 66, 1007-1018.
| Crossref | Google Scholar | PubMed |
Tumescheit C, Firth AE, Brown K (2022) CIAlign: a highly customisable command line tool to clean, interpret and visualise multiple sequence alignments. PeerJ 10, e12983.
| Crossref | Google Scholar | PubMed |
Ufimov R, Gorospe JM, Fér T, Kandziora M, Salomon L, van Loo M, Schmickl R (2022) Utilizing paralogues for phylogenetic reconstruction has the potential to increase species tree support and reduce gene tree discordance in target enrichment data. Molecular Ecology Resources 22, 3018-3034.
| Crossref | Google Scholar | PubMed |
Van Dijk K, Waycott M, Biffin E, Creed JC, Albertazzi FJ, Samper-Villarreal J (2023) Phylogenomic insights into the phylogeography of Halophila baillonii Asch. Diversity 15, 111.
| Crossref | Google Scholar |
Vatanparast M, Powell A, Doyle JJ, Egan AN (2018) Targeting legume loci: a comparison of three methods for target enrichment bait design in Leguminosae phylogenomics. Applications in Plant Sciences 6, e1036.
| Crossref | Google Scholar | PubMed |
Walsh HE, Kidd MG, Moum T, Friesen VL (1999) Polytomies and the power of phylogenetic inference. Evolution 53, 932-937.
| Crossref | Google Scholar | PubMed |
Waycott M, Van Dijk K, Biffin E (2021) A hybrid capture RNA bait set for resolving genetic and evolutionary relationships in angiosperms from deep phylogeny to intraspecific lineage hybridization. bioRxiv 2021, 2021.09.06.456727 [Preprint, published 7 September 2021].
| Crossref | Google Scholar |
Wen D, Yu Y, Nakhleh L (2016) Bayesian inference of reticulate phylogenies under the multispecies network coalescent. PLoS Genetics 12, e1006006.
| Crossref | Google Scholar | PubMed |
Wen D, Yu Y, Zhu J, Nakhleh L (2018) Inferring phylogenetic networks using PhyloNet. Systematic Biology 67(4), 735-740.
| Crossref | Google Scholar |
Whitfield JB, Lockhart PJ (2007) Deciphering ancient rapid radiations. Trends in Ecology & Evolution 22, 258-265.
| Crossref | Google Scholar | PubMed |
Williamson L, Biffin E, Hammer T, van Dijk K, Conran J, Waycott M (2025) Phylogenomics of Australian sundews (Drosera: Droseraceae). Australian Systematic Botany 38, SB24016.
| Crossref | Google Scholar |
Willson J, Roddur MS, Liu B, Zaharias P, Warnow T (2022) DISCO: species tree inference using multicopy gene family tree decomposition. Systematic Biology 71, 610-629.
| Crossref | Google Scholar | PubMed |
Xi Z, Liu L, Davis CC (2016) The impact of missing data on species tree estimation. Molecular Biology and Evolution 33, 838-860.
| Crossref | Google Scholar | PubMed |
Xiong H, Wang D, Shao C, Yang X, Yang J, Ma T, Davis CC, Liu L, Xi Z (2022) Species tree estimation and the impact of gene loss following whole-genome duplication. Systematic Biology 71, 1348-1361.
| Crossref | Google Scholar | PubMed |
Yan Z, Smith ML, Du P, Hahn MW, Nakhleh L (2022) Species tree inference methods intended to deal with incomplete lineage sorting are robust to the presence of paralogs. Systematic Biology 71, 367-381.
| Crossref | Google Scholar | PubMed |
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24, 1586-1591.
| Crossref | Google Scholar | PubMed |
Yang Y, Smith SA (2014) Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution 31, 3081-3092.
| Crossref | Google Scholar | PubMed |
Yang Y, Moore MJ, Brockington SF, Mikenas J, Olivieri J, Walker JF, Smith SA (2018) Improved transcriptome sampling pinpoints 26 ancient and more recent polyploidy events in Caryophyllales, including two allopolyploidy events. New Phytologist 217, 855-870.
| Crossref | Google Scholar | PubMed |
Yu Y, Nakhleh L (2015) A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 16, S10.
| Crossref | Google Scholar | PubMed |
Zhang C, Mirarab S (2022) ASTRAL-Pro 2: ultrafast species tree reconstruction from multi-copy gene family trees. Bioinformatics 38, 4949-4950.
| Crossref | Google Scholar | PubMed |
Zhang C, Ogilvie HA, Drummond AJ, Stadler T (2018a) Bayesian inference of species networks from multilocus sequence data. Molecular Biology and Evolution 35, 504-517.
| Crossref | Google Scholar |
Zhang C, Rabiee M, Sayyari E, Mirarab S (2018b) ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 153.
| Crossref | Google Scholar |
Zhang C, Scornavacca C, Molloy EK, Mirarab S (2020) ASTRAL-Pro: quartet-based species-tree inference despite paralogy. Molecular Biology and Evolution 37, 3292-3307.
| Crossref | Google Scholar | PubMed |
Zhang C, Zhao Y, Braun EL, Mirarab S (2021) TAPER: pinpointing errors in multiple sequence alignments despite varying rates of evolution. Methods in Ecology and Evolution 12(11), 2145-2158.
| Crossref | Google Scholar |
Zhang Q, Folk RA, Mo Z-Q, Ye H, Zhang Z-Y, Peng H, Zhao J-L, Yang S-X, Yu X-Q (2023) Phylotranscriptomic analyses reveal deep gene tree discordance in Camellia (Theaceae). Molecular Phylogenetics and Evolution 188, 107912.
| Crossref | Google Scholar | PubMed |
Zhang Z, Xie P, Guo Y, Zhou W, Liu E, Yu Y (2022) Easy353: a tool to get Angiosperms353 genes for phylogenomic research. Molecular Biology and Evolution 39, msac261.
| Crossref | Google Scholar | PubMed |
Zhou W, Soghigian J, Xiang QJ (2022) A new pipeline for removing paralogs in target enrichment data. Systematic Biology 71, 410-425.
| Crossref | Google Scholar | PubMed |
Zhou X, Lutteropp S, Czech L, Stamatakis A, Looz MV, Rokas A (2020) Quartet-based computations of internode certainty provide robust measures of phylogenetic incongruence. Systematic Biology 69, 308-324.
| Crossref | Google Scholar | PubMed |
Zuntini A, Carruthers T, Maurin O, Bailey PC, Leempoel K, Brewer GE, Epitawalage N, Françoso E, Gallego-Paramo B, McGinnie C, Negrão R, Roy SR, Simpson L, Toledo Romero E, Barber VMA, Botigué L, Clarkson JJ, Cowan RS, Dodsworth S, Johnson MG, Kim JT, Pokorny L, Wickett NJ, Antar GM, DeBolt L, Gutierrez K, Hendriks KP, Hoewener A, Hu A-Q, Joyce EM, Kikuchi IABS, Larridon I, Larson DA, de Lírio EJ, Liu J-X, Malakasi P, Przelomska NAS, Shah T, Viruel J, Allnutt TR, Ameka GK, Andrew RL, Appelhans MS, Arista M, Ariza MJ, Arroyo J, Arthan W, Bachelier JB, Bailey CD, Barnes HF, Barrett MD, Barrett RL, Bayer RJ, Bayly MJ, Biffin E, Biggs N, Birch JL, Bogarín D, Borosova R, Bowles AMC, Boyce PC, Bramley GLC, Briggs M, Broadhurst L, Brown GK, Bruhl JJ, Bruneau A, Buerki S, Burns E, Byrne M, Cable S, Calladine A, Callmander MW, Cano Á, Cantrill DJ, Cardinal-McTeague WM, Carlsen MM, Carruthers AJA, de Castro Mateo A, Chase MW, Chatrou LW, Cheek M, Chen S, Christenhusz MJM, Christin P-A, Clements MA, Coffey SC, Conran JG, Cornejo X, Couvreur TLP, Cowie ID, Csiba L, Darbyshire I, Davidse G, Davies NMJ, Davis AP, van Dijk K, Downie SR, Duretto MF, Duvall MR, Edwards SL, Eggli U, Erkens RHJ, Escudero M, de la Estrella M, Fabriani F, Fay MF, Ferreira Pde L, Ficinski SZ, Fowler RM, Frisby S, Fu L, Fulcher T, Galbany-Casals M, Gardner EM, German DA, Giaretta A, Gibernau M, Gillespie LJ, González CC, Goyder DJ, Graham SW, Grall A, Green L, Gunn BF, Gutiérrez DG, Hackel J, Haevermans T, Haigh A, Hall JC, Hall T, Harrison MJ, Hatt SA, Hidalgo O, Hodkinson TR, Holmes GD, Hopkins HCF, Jackson CJ, James SA, Jobson RW, Kadereit G, Kahandawala IM, Kainulainen K, Kato M, Kellogg EA, King GJ, Klejevskaja B, Klitgaard BB, Klopper RR, Knapp S, Koch MA, Leebens-Mack JH, Lens F, Leon CJ, Léveillé-Bourret É, Lewis GP, Li D-Z, Li L, Liede-Schumann S, Livshultz T, Lorence D, Lu M, Lu-Irving P, Luber J, Lucas EJ, Luján M, Lum M, Macfarlane TD, Magdalena C, Mansano VF, Masters LE, Mayo SJ, McColl K, McDonnell AJ, McDougall AE, McLay TGB, McPherson H, Meneses RI, Merckx VSFT, Michelangeli FA, Mitchell JD, Monro AK, Moore MJ, Mueller TL, Mummenhoff K, Munzinger J, Muriel P, Murphy DJ, Nargar K, Nauheimer L, Nge FJ, Nyffeler R, Orejuela A, Ortiz EM, Palazzesi L, Peixoto AL, Pell SK, Pellicer J, Penneys DS, Perez-Escobar OA, Persson C, Pignal M, Pillon Y, Pirani JR, Plunkett GM, Powell RF, Prance GT, Puglisi C, Qin M, Rabeler RK, Rees PEJ, Renner M, Roalson EH, Rodda M, Rogers ZS, Rokni S, Rutishauser R, de Salas MF, Schaefer H, Schley RJ, Schmidt-Lebuhn A, Shapcott A, Al-Shehbaz I, Shepherd KA, Simmons MP, Simões AO, Simões A, Rita G, Siros M, Smidt EC, Smith JF, Snow N, Soltis DE, Soltis PS, Soreng RJ, Sothers CA, Starr JR, Stevens PF, Straub SCK, Struwe L, Taylor JM, Telford IRH, Thornhill AH, Tooth I, Trias-Blasi A, Udovicic F, Utteridge TMA, Del Valle JC, Verboom GA, Vonow HP, Vorontsova MS, de Vos JM, Al-Wattar N, Waycott M, Welker CAD, White AJ, Wieringa JJ, Williamson LT, Wilson TC, Wong SY, Woods LA, Woods R, Worboys S, Xanthos M, Yang Y, Zhang Y-X, Zhou M-Y, Zmarzty S, Zuloaga FO, Antonelli A, Bellot S, Crayn DM, Grace OM, Kersey PJ, Leitch IJ, Sauquet H, Smith SA, Eiserhardt WL, Forest F, Baker WJ (2024) Phylogenomics and the rise of the angiosperms. Nature 629, 843-850.
| Crossref | Google Scholar | PubMed |