

Genomic selection in French dairy cattle
D. Boichard A F , F. Guillaume A B , A. Baur C , P. Croiseau A , M. N. Rossignol D , M. Y. Boscher D , T. Druet E , L. Genestout D , J. J. Colleau A , L. Journaux C , V. Ducrocq A and S. Fritz CA INRA, UMR1313 Gabi, 78350 Jouy-en-Josas, France.
B Institut de l’Elevage, 78350 Jouy-en-Josas, France.
C UNCEIA, 149 Rue de Bercy, 75595 Paris, France.
D Labogena, 78350 Jouy-en-Josas, France.
E Liège University, Belgium.
F Corresponding author. Email: didier.boichard@jouy.inra.fr
Animal Production Science 52(3) 115-120 https://doi.org/10.1071/AN11119
Submitted: 21 June 2011 Accepted: 27 November 2011 Published: 6 March 2012
Journal Compilation © CSIRO Publishing 2012 Open Access CC BY-NC-ND
Abstract
Genomic selection is implemented in French Holstein, Montbéliarde, and Normande breeds (70%, 16% and 12% of French dairy cows). A characteristic of the model for genomic evaluation is the use of haplotypes instead of single-nucleotide polymorphisms (SNPs), so as to maximise linkage disequilibrium between markers and quantitative trait loci (QTLs). For each trait, a QTL-BLUP model (i.e. a best linear unbiased prediction model including QTL random effects) includes 300–700 trait-dependent chromosomal regions selected either by linkage disequilibrium and linkage analysis or by elastic net. This model requires an important effort to phase genotypes, detect QTLs, select SNPs, but was found to be the most efficient one among all tested ones. QTLs are defined within breed and many of them were found to be breed specific. Reference populations include 1800 and 1400 bulls in Montbéliarde and Normande breeds. In Holstein, the very large reference population of 18 300 bulls originates from the EuroGenomics consortium. Since 2008, ~65 000 animals have been genotyped for selection by Labogena with the 50k chip. Bulls genomic estimated breeding values (GEBVs) were made official in June 2009. In 2010, the market share of the young bulls reached 30% and is expected to increase rapidly. Advertising actions have been undertaken to recommend a time-restricted use of young bulls with a limited number of doses. In January 2011, genomic selection was opened to all farmers for females. Current developments focus on the extension of the method to a multi-breed context, to use all reference populations simultaneously in genomic evaluation.
Introduction
In 2000, a large-scale program of marker-assisted selection (MAS) was implemented in the three main French dairy cattle breeds (Holstein, Normande and Montbéliarde) (Boichard et al. 2002, 2006). It was a first generation program with 14 chromosome regions traced by 45 microsatellites markers. No population-wide linkage disequilibrium was assumed and only within-family information was used. After 7 years of activity and more than 70 000 animals genotyped, the efficiency of this program was shown to be close to its expectation (Guillaume et al. 2008a, 2008b), i.e. rather limited but large enough to be profitable through a reduction by 15% of the number of bulls entering progeny test without any loss of genetic gain. But since 2005, it was anticipated that high-throughput SNP chips would rapidly open the way to MAS based on linkage disequilibrium or to genomic selection (Gautier et al. 2007). When the BovineSNP50™ beadchip (Illumina Inc, San Diego, USA) became available in late 2007, this 50k chip was immediately and intensively used to upgrade the MAS program and boost its efficiency. However, the experience gained from the first program deeply influenced the implementation and the practical use of genomic selection. Indeed, on one hand, some initial technical choices appeared to be very important for a good and sustainable efficiency and were maintained in the new system. On the other hand, the industry had been already deeply involved in MAS for a long time and this resulted in a very good acceptability of the new product and its immediate use in selection, with drastic changes in the management of breeding programs. In the current paper, we present the ideas, the implementation and the application of genomic selection in French dairy cattle.
Strategy for genomic selection implementation
The main features of the strategy implemented for genomic selection can be summarised as follows. A first major characteristic is the use of haplotypes, instead of single SNPs, to maximise linkage disequilibrium between markers and QTLs. Hayes et al. (2007) proposed this solution, as well as Meuwissen et al. (2001) in the initial paper on genomic selection. Indeed, it is believed that individual SNPs are unlikely to be systematically close to the QTLs as well as in complete disequilibrium with them. Many reasons could be advocated to justify this statement, including the limited density of the marker SNPs, their extremely low probability to be the causative variants, their low informativity, with only two alleles and unbalanced frequencies, or their selection based on informativity. Even with a much higher marker density, Yang et al. (2010) showed that a part of the ‘missing heritability’ could be attributed to a lack of linkage disequilibrium between markers and causative variants. Combining neighbouring SNPs into haplotypes is a simple way to increase informativity and is likely to generate a more complete linkage disequilibrium, at the expense of some over-parameterisation. Because these haplotypes are limited in size compared with conserved segments within breeds, their association with the QTLs is likely to be well conserved in the population and over several generations. Another way to express this idea is as follows: two random copies in the population of the same allele of a highly informative haplotype are more likely to be identical by descent than for a SNP and, therefore, to be associated with the same QTL allele.
An important potential drawback of using haplotypes is the large number of effects to estimate and, consequently, the loss of accuracy because of the number of effects to estimate. Therefore, it is very important to target the most important regions of the genome and to assume that most markers have null effects. This explains the superiority of the BayesB (Meuwissen et al. 2001) approaches or its different approximations (BayesCΠ, BayesR), particularly when the marker density increases, but the same arguments could be used with haplotypes to reduce the number of effects to estimate.
Therefore, only several targeted regions were included in the model. However, they should be numerous enough to account for a proportion of the genetic variability as high as possible and the strategy implies a trade-off in the number of regions accounted for. In practice, 300–700 small regions were targeted, corresponding to an estimated 60–70% of the total genetic variance explained, depending on the traits.
Two strategies were used to define these QTL regions. On the one hand, 20–40 large QTLs were detected in a conventional QTL detection approach, with a linkage equilibrium and linkage analysis procedure (Meuwissen and Goddard 2000). These large QTLs were well characterised by their location and their variance component, and they were traced by haplotypes of four or five SNPs, so as to define 10–15 haplotypes, thus ensuring a good probability of linkage disequilibrium. However, apart from a few exceptions such as DGAT1 (Grisart et al. 2002) for milk production and composition, most of these supposedly large QTLs explained only 1–2% of the genetic variance each and were not sufficient for an accurate breeding value prediction.
On the second hand, to maximise the part of variance explained by markers, the model also included several hundred trait-dependent chromosomal regions selected on the basis of an elastic-net approach. Elastic net is a selection procedure combining least absolute shrinkage and selection operator (LASSO) and ridge regression. Main characteristics and results are presented in Croiseau et al. (2011).
As for the larger QTLs, these regions are traced by haplotypes. Haplotypes were defined by grouping all selected SNPs in the same centimorgan. When a marker was alone, the two flanking SNPs were selected to define a haplotype of at least three markers. These small QTLs were given an arbitrary, small and identical variance component.
Although this approach is targeted on QTLs, it should be noted that it also accounts for relationships between animals and long-range linkage disequilibrium. Indeed, one or two dozens of haplotypes per chromosome already provide a good coverage of the genome.
The model used (Eqn 1) for the evaluation was a simple QTL–BLUP with one effect per haplotype allele and a residual polygenic effect, accounting for 30–40% of the genetic variance.

with y being the vector of records, µ a mean, u the vector of residual polygenic effects assumed to be normally distributed N(0, Aσu2), qj the vector of QTL alleles for haplotype j N(0, Iσqj2), nq the number of QTLs, and e the vector of residuals assumed to be normally distributed N(0, Rσe2).
For computational reasons, to avoid a large dense matrix and also to avoid redundancies with the molecular information, the covariance structure A of the polygenic part was based on pedigree and not on markers. On average, ~7000 haplotype effects were estimated per trait. Phenotypes were derived from the polygenic conventional evaluation procedures. In the initial implementation, both males and females had records, with the major advantage of no need for post-processing, all information being already accounted for. Records of males were daughter yield deviations (or deregressed proofs for foreign bulls) with appropriate weights, whereas genotyped females were characterised by their yield deviation. However, records have been restricted to males since 2010. This decision was motivated by the very peculiar situation of genotyped females with likely overestimated performances at least for some traits. It is anticipated that this situation could evolve, particularly when the number of genotyped females increases and when they become more representative of the overall population.
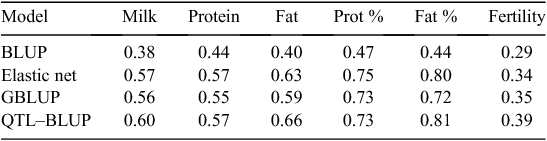
This procedure based on a QTL–BLUP is quite unusual compared with those used in other countries. Nevertheless, it was applied because it was found to be the most efficient one among all tested methods. Table 1 presents a comparison of the correlation between genomic evaluations and daughter yield deviations in the Holstein validation population. It can be observed that correlations were higher for all traits with the QTL–BLUP than those with Genomic BLUP (GBLUP), usually considered as a reference. The same conclusion was also true in Montbéliarde and Normande breeds, characterised by more limited reference populations. It should be noticed that for the validation of the approach, QTL detection and SNP selection were based only on the training population without the validation set. Admittedly, the QTL–BLUP approach requires a large initial tuning effort to select SNPs and haplotypes, to be repeated for each new trait and each breed. It also requires to phase genotypes on a routine basis, a task which could be very demanding in the future if hundreds of thousands animals are genotyped and evaluated yearly, but this effort will be necessary anyway to impute genotypes from low-density chips.
|
The overall evaluation pipeline includes (1) a step of genotype checking, pedigree verification and genotype reconstruction based on relatives and (2) a phasing step. This step uses several approaches based on pedigree and population information (initially LinkPhase and DualPhase, then DagPhase (Druet and Georges 2010). Beagle (Browning and Browning 2007) has also been used and, surprisingly, in spite of the complex existing family structure, was found to be as efficient as DagPhase, whereas the latter is expected to better account for relationships. Moreover, the directed acyclic graph could be computed once and stored, saving a lot of time for subsequent analyses. Consequently, the final choice across methods is still subject to evolving. At the end of this step, complete and phased genotypes are stored, ready for any kind of analyses. (3) The third step is a conventional QTL–BLUP. Equations are limited to genotyped animals with data (i.e. presently sires with daughter yield deviations or deregressed proofs) and their ancestors. Nearly every bull progeny-tested in the past 10–15 years (according to the breed) was genotyped, limiting the bias due to selection in such a strategy. Polygenic values of the parents and haplotype effect estimates are then combined to predict the breeding values of progeny without data. Reliabilities are computed by combining elements of the inverse of the coefficient matrix. To compute reliabilities of candidates, all necessary terms of the matrix should be taken care of, including covariance between haplotypes present in candidates and polygenic values of the parents. According to traits and animals, estimated reliabilities ranged from 0.50 to 0.60 in Normande and Montbeliarde breeds, and from 0.60 to 0.70 in Holstein, these latter values resulting from the much larger reference population.
Genomic selection is implemented in Holstein, Montbéliarde and Normande breeds, which represent 70, 16 and 12% of the French dairy population (4 millions cows). QTLs are defined within breed and a majority of them were found to be breed-specific. Reference populations have been gradually built since 2008 and included 1800 and 1400 progeny-tested bulls in Montbéliarde and Normande breeds in February 2011. In Holstein, we started with a population of 4000 French bulls available in 2009. In fall 2009, EuroGenomics, a European consortium gathering the Dutch, Nordic, German and French artificial insemination industries and several research organisations was formed. In this framework, reference populations of the four partners were shared, leading to the very large common reference population of 18 300 bulls (Lund et al. 2011).
Practical use of genomic selection
The practical implementation of genomic selection was extremely fast. In April 2008, the genotypes of the first 3200 bulls were obtained, allowing the first tests with real data. In September 2008, a service was opened by Labogena to genotype the candidates. In October 2008, the first genomic evaluations were released to the industry as non-official indicators. In June 2009, genomic evaluations were made official for males, allowing the use of young bulls in artificial insemination in the French population. In Fall 2009, the Eurogenomics consortium was created, with important consequences on reference population sharing, but also additional collaboration on scientific fields such as comparison of methods (Lund et al. 2011), imputation (Dassonneville et al. 2011), use of high-density chip, or sequencing. In January 2011, genomic evaluation was opened as a commercial service for females.
Since September 2008, ~2000–2500 animals were genotyped each month with the BovineSNP50™ beadchip, 40–50% of them being females for the selection of bull dams. The evaluation is run nine times a year. Since June 2009, three evaluations per year have an official status for males. Until now, all effects have been re-estimated at every run but the system is now well stabilised and it is going to be simplified with a re-estimation of QTL effects only at the official evaluations. Genomic evaluation is run as follows: because Institut National de la Recherche Agronomique (INRA) is responsible for the official evaluation in France, INRA is also responsible for genomic evaluation. Since April 2010, the newly created Valogene company is licenced to sell the service for females. It contracts with different genotyping laboratories (3 presently) and buys the chips from Illumina so as to decrease the genotyping cost. Presently, the service is provided only with 50k genotypes, to both maximise evaluation quality and maximise the 50k volume and get a low price for the chip. Valogene customers could be individual farmers or, preferentially, retailers such as artificial insemination centres, breed associations, milk-recording organisations, or any structure providing services to farmers. Female GEBVs are made official either immediately or at latest at 18 months of age, on the customer’s request.
As soon as June 2009, strong recommendations accompanied the GEBV released. These recommendations, supported by the comparison of various scenarios simulated by Colleau et al. (2009) on the basis on the Montbeliarde breed, were three-fold:
-
Use young bulls without progeny test. If progeny test is continued, the interest of genomic selection is limited as genetic trend and inbreeding trend are hardly modified. In contrast, if only young bulls are used, the theoretical genetic trend could be nearly doubled, mainly because of a reduced generation interval but also because of an increase in accuracy for females and a potentially high selection intensity in the first step of the breeding scheme, when selection is rather cheap (selection of bull dams and of young male calves). This result is in agreement with Schaeffer (2006) who first demonstrated the potential of genomic selection for genetic trend and other authors (Colleau et al. 2009; Sørensen and Sørensen 2009; Pryce et al. 2010). Of course, stopping progeny test before using bulls does not mean that evaluations based on progeny no longer exist. In fact, because these young bulls may have hundreds of daughters in herds with performance recording, they will receive a more accurate progeny evaluation than after conventional progeny test. Therefore, they will efficiently contribute to the reference population upgrading.
-
Many young bulls should be used and each of them should contribute only a few thousand inseminations, over only a short period of time before being replaced. Limiting the use of each bull decreases generation interval and also limits the potentially unpopular effects of the somewhat lower accuracy of genomic evaluations than with conventional progeny test. Because genomic selection could be twice as fast as conventional selection, inbreeding trend could be also doubled. The key is to use a much higher number of bulls and of bull sires (Colleau et al. 2009; Sørensen and Sørensen 2009) to counterbalance the effect of generation interval on inbreeding, without affecting genetic trends. In fact, using each marketed young bull also as a bull sire could be optimal, with a rule similar to poultry breeding where each sire is replaced by one of his sons. Such a strategy is favourable to genetic trend but also to inbreeding and to the robustness of the results. Indeed, this strategy leads to a 20% lower inbreeding trend than after conventional progeny test. As genomic selection puts less emphasis on family information (Daetwyler et al. 2007) and because the early selection steps are cheap, more diversity could be used by the breeders with limited hazard and strong potential benefits on long-term variability.
-
An appealing scenario for breeding companies would have been to use former young bulls for a second crop of daughters after they receive their evaluation based on progeny information. For them, this scenario presents strong advantages, such as a better use of the best bulls, a more limited change in comparison with the past, a better fit to breeders’ demand, a better profitability for the company. This scenario, however, is not recommended. In such a scenario with 50% insemination by young bulls and 50% by the best bulls returning to service, Colleau et al. (2009) found that the genetic trend was also nearly doubled but the inbreeding trend was strongly increased to an unaffordable level, namely twice the level of a conventional scheme which is already very high. Moreover, these older bulls returned to service would be in competition with their own sons and even their grand-sons, which are likely to be better.
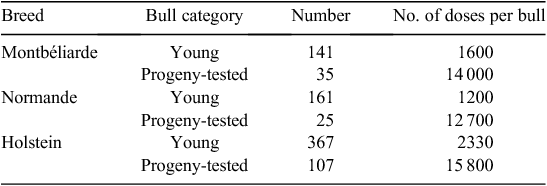
A first evaluation of the practices based on the 2010 inseminations was made after the first year of use of the young bulls. This period, of course, was not representative of what could happen after several years of genomic selection. A large excess of bulls was available, with the coexistence of progeny-tested bulls, ‘true’ young bulls and bulls waiting for their progeny-test results. In 2010, 669 young bulls were marketed and represented 30% of the market share over the whole country and more than 40% in some regions (Table 2). It is anticipated that this proportion would increase in the next years to rapidly represent the largest share of the market. The average number of inseminations per young bull was quite low, 1200–2300 according to breeds. These surprisingly low figures could be attributed to the large number of available top bulls and are expected to increase up to ~5000–7000 in the next years.
|
Selection intensity was quite high. These 669 bulls were selected of more than 20 000 genotyped bulls. Table 3 presents the average EBV of French young bulls and conventional progeny-tested bulls used in 2010. In Normande and Montbeliarde breeds, both categories were rather similar, with a superiority of 1.5 and 1.8 genetic standard deviations for total merit index. Compared with conventional bulls, some additional selection intensity was applied on longevity. In Holstein breed, a stronger selection of young bulls was applied on type and longevity, and to a lower extent, fertility, resulting in a very high total merit index (+2.7) and a large superiority (+0.5) over conventional bulls. The additional effort on type and longevity was not expected, because recommendations were targeted on fertility and somatic cell counts.

|
Perspectives
Genomic selection is only in its early steps and will benefit from important evolutions in the near future.
-
Massive extension to females. Females have been involved in genomic selection since the beginning with ~40% of the genotyped animals, as potential bull dams. In 2011, a commercial service is proposed and available to any farmer. Presently, it is proposed on the basis of the 50k chip. With an anticipated large market, a reasonable price could be proposed for a high quality service. The 3k chip was not proposed because the quality of its imputation on the 50k chip was not high enough for a reliable evaluation in any of the three breeds, and even rather poor in Montbeliarde and Normande. We participated in the Illumina 7k low-density chip project and will propose this solution when it is on the market. It is anticipated that a large proportion of the French heifers could be genotyped if the corresponding cost is low enough. The interest of heifer genotyping is enhanced when within-herd selection pressure is increased, for instance when sexed semen is used (Sørensen et al. 2011).
-
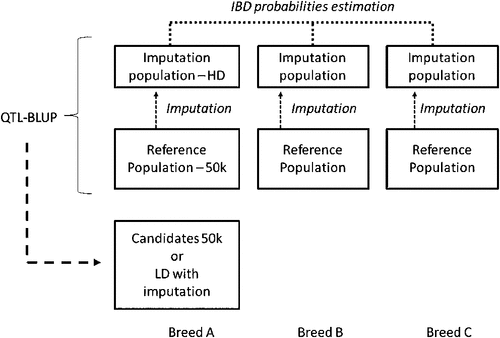
Across-breed evaluation. Across-breed evaluation is a necessary evolution in genomic selection. Only widespread breeds can afford the very large reference population required for an efficient genomic selection. Across-breed evaluations offer new opportunities for nearly all breeds other than Holstein, because their reference populations are under the optimal size. Furthermore, it becomes a necessity for every breed for new traits difficult or expensive to record; sharing data across breeds would then be the most convenient way to build a sufficiently large reference population. Finally, it is a good pilot project for other production systems (particularly beef) and other species that cannot afford the investments made in dairy. In 2010, a high-density (HD) chip with 777k SNPs was made available by Illumina. This tool is currently used to genotype several hundreds of bulls within each breed, for a total of 5000 dairy and beef animals. The resolution (1 SNP/4 kb) should be high enough to find identical-by-descent chromosomal segments across breeds and, therefore, estimate the effects of conserved haplotypes. Therefore, we are confident that the method used in French genomic selection could be extended to the across-breed situation. Of course, the complete reference populations should not be genotyped with the HD chip because 50k genotypes could be efficiently imputed to the HD. Figure 1 illustrates the ideal situation we would like to implement in 2012.
-
Selection for new traits. With respect to new traits, genomic selection opens new opportunities provided that reference populations could be generated. Indeed, genomic selection disconnects the measurement of the phenotype from selection, and a trait previously very difficult to select for could be selected if it is recorded on several tens of thousands of animals. In contrast to a classical reference population based on conventional large-scale performance recording and a large number of progeny tested bulls, these new reference populations will be composed of females with their own performances.
|
Several traits are very good candidates for such a selection. A first category is related to fine-milk composition. New methods are presently under development to predict milk composition in fatty acids and protein through mid-infrared spectrometry. Based on the same approach as the current prediction of overall fat and protein content by milk analysis laboratory, these predictions could be implemented at a marginal cost. Although the proportion of saturated fat is considered to be too high, it is still unclear, however, what should be the breeding objective and what would be the incentive for farmers to produce a less saturated milk fat.
A second category involves various health traits. Many health traits could be easily recorded but little effort was made in France in the past to do this, because these traits have a very low heritability. Hoof pathology could be scored at trimming and the Dutch experience shows it is valuable information (Stoop et al. 2010). More generally, all sanitary events could be recorded by recovering information from the sanitary notebook, recording of which is mandatory in Europe. This should give access to useful information relative to metabolic diseases and metritis. Many other phenotypes are possible to collect, relative to feeding, behaviour, health or reproduction, particularly with the development of precision farming and various electronic devices.
Acknowledgements
The Cartofine and Amasgen projects were funded by the National Research Agency (ANR) and ApisGene. The authors thank A. Legarra, C. Robert-Granié and C. Colombani for numerous discussions and their contribution to the comparison between methods within the Amasgen project.
References
Boichard D, Fritz S, Rossignol MN, Boscher MY, Malafosse A, Colleau JJ (2002) Implementation of marker-assisted selection in dairy cattle. In ‘Proceedings of the 7th world congress of genetics applied to livestock production, Montpellier, France’. Communication no. 22-03.Boichard D, Fritz S, Rossignol MN, Guillaume F, Colleau JJ, Druet T (2006) Implementation of marker-assisted selection: practical lessons from dairy cattle. In ‘Proceedings of the 8th world congress of genetics applied to livestock production, Belo Horizonte, Brazil’. Communication no. 22-11.
Browning SR, Browning BL (2007) Rapid and accurate haplotype phasing and missing data inference for whole genome association studies using localized haplotype clustering. American Journal of Human Genetics 81, 1084–1097.
| Rapid and accurate haplotype phasing and missing data inference for whole genome association studies using localized haplotype clustering.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1KmsL3M&md5=334109ceaf9982afbf5138f23f9b26d5CAS |
Colleau JJ, Fritz S, Guillaume F, Baur A, Dupassieux D, Boscher MY, Journaux L, Eggen A, Boichard D (2009) Simulating the potential of genomic selection in dairy cattle breeding. Rencontres Recherches Ruminants 16, 419 [in French].
Croiseau P, Legarra A, Guillaume F, Fritz S, Baur A, Colombani C, Robert-Granié C, Boichard D, Ducrocq V (2011) Fine tuning genomic evaluations in dairy cattle through SNP pre-selection with the Elastic-Net algorithm. Genetics Research 93, 409–417.
| Fine tuning genomic evaluations in dairy cattle through SNP pre-selection with the Elastic-Net algorithm.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1KrsbfL&md5=3da12e73ba3bcd6b1fdf7bb71bc9d232CAS |
Daetwyler HD, Villanueva B, Bijma P, Woolliams JA (2007) Inbreeding in genome-wide selection. Journal of Animal Breeding and Genetics 124, 369–376.
| Inbreeding in genome-wide selection.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD2sjjvFyhsQ%3D%3D&md5=563aee84967b25051aca608a0dd290a9CAS |
Dassonneville R, Brøndum RF, Druet T, Fritz S, Guillaume F, Guldbrandtsen B, Lund MS, Ducrocq V, Su G (2011) Impact of imputing markers from a low density chip on the reliability of genomic breeding values in Holstein populations. Journal of Dairy Science 94, 3679–3686.
| Impact of imputing markers from a low density chip on the reliability of genomic breeding values in Holstein populations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvFahtrw%3D&md5=96a7a47100085d4d8458f33f210348f8CAS |
Druet T, Georges M (2010) A hidden Markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping. Genetics 184, 789–798.
| A hidden Markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFOnurvN&md5=18bb77ec25c91292a97809f342a86c64CAS |
Gautier M, Faraut T, Moazami-Goudarzi K, Navratil V, Foglio M, Grohs C, Boland A, Garnier JG, Boichard D, Lathrop GM, Gut IG, Eggen A (2007) Genetic and haplotypic structure in 14 European and African cattle breeds. Genetics 177, 1059–1070.
| Genetic and haplotypic structure in 14 European and African cattle breeds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlyrs7rK&md5=d95b8fc7c596048e8a57c4c16a26a004CAS |
Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, Spelman R, Georges M, Snell R (2002) Positional candidate cloning of a QTL in dairy cattle: identication of a missense mutation in the bovine DGAT1 gene with a major effect on milk yield and composition. Genome Research 12, 222–231.
| Positional candidate cloning of a QTL in dairy cattle: identication of a missense mutation in the bovine DGAT1 gene with a major effect on milk yield and composition.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhtlKksrs%3D&md5=101173298a98503d2e18b1ec8d71a0feCAS |
Guillaume F, Fritz S, Boichard D, Druet T (2008a) Estimation by simulation of the efficiency of the French marker-assisted selection program in dairy cattle. Genetics, Selection, Evolution 40, 91–102.
| Estimation by simulation of the efficiency of the French marker-assisted selection program in dairy cattle.Crossref | GoogleScholarGoogle Scholar |
Guillaume F, Fritz S, Boichard D, Druet T (2008b) Correlations of marker-assisted breeding values with progeny-test breeding values for eight hundred ninety-nine French Holstein bulls. Journal of Dairy Science 91, 2520–2522.
| Correlations of marker-assisted breeding values with progeny-test breeding values for eight hundred ninety-nine French Holstein bulls.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmsVWmurY%3D&md5=80cca5f9064f9b60516035ced93f2b31CAS |
Hayes BJ, Chamberlain AJ, McPartlan H, MacLeod I, Sethuraman L, Goddard ME (2007) Accuracy of marker-assisted selection with single markers and marker haplotypes in cattle. Genetical Research 89, 215–220.
| Accuracy of marker-assisted selection with single markers and marker haplotypes in cattle.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovV2nsQ%3D%3D&md5=40a7173cb6eda32b2092f5ecce7abc0cCAS |
Lund MS, de Roos APW, de Vries AG, Druet T, Ducrocq V, Fritz S, Guillaume F, Guldbrandtsen B, Liu Z, Reents R, Schrooten C, Seefried M, Su G (2011) Common reference of four European Holstein populations increases reliability of genomic predictions. Genetics, Selection, Evolution 43, 43
| Common reference of four European Holstein populations increases reliability of genomic predictions.Crossref | GoogleScholarGoogle Scholar |
Meuwissen THE, Goddard ME (2000) Fine mapping of quantitative trait loci using linkage disequilibria with closely linked marker loci. Genetics 155, 421–430.
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829.
Pryce JE, Goddard ME, Raadsma HW, Hayes BJ (2010) Deterministic models of breeding scheme designs that incorporate genomic selection. Journal of Dairy Science 93, 5455–5466.
| Deterministic models of breeding scheme designs that incorporate genomic selection.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvVSjtQ%3D%3D&md5=4324442e41a5cdb6838940b03b371fdaCAS |
Schaeffer LR (2006) Strategy for applying genome-wide selection in dairy cattle. Journal of Animal Breeding and Genetics 123, 218–223.
| Strategy for applying genome-wide selection in dairy cattle.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD28vlvFCitA%3D%3D&md5=f2323721cc79f7ab1ccb3d6057ddefe7CAS |
Sørensen MK, Sørensen AC (2009) Inbreeding rates in breeding programs with different strategies for using genomic selection. In ‘Interbull meeting, Barcelona, Spain’, 21–24 August 2009. Available at http://www-interbull.slu.se/bulletins/bulletin40/Sorensen.pdf [Verified 8 December 2011]
Sørensen MK, Voergaard J, Pedersen LD, Berg P, Sørensen AC (2011) Genetic gain in dairy cattle populations is increased using sexed semen in commercial herds. Journal of Animal Breeding and Genetics 128, 267–275.
| Genetic gain in dairy cattle populations is increased using sexed semen in commercial herds.Crossref | GoogleScholarGoogle Scholar |
Stoop M, de Jong G, van Pelt M, van der Linde C (2010) Implementation of a claw health index in The Netherlands. In ‘Interbull meeting, Riga, Latvia’, 31 May – 4 June 2010. Available at http://www.interbull.org/images/stories/Stoop_Claw_20100601.pdf [Verified 8 December 2011]
Yang JA, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, Goddard ME, Visscher PM (2010) Common SNPs explain a large proportion of the heritability for human height. Nature Genetics 42, 565–569.
| Common SNPs explain a large proportion of the heritability for human height.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXns1GisL8%3D&md5=c9db73a48ec56a558783f98f92da10efCAS |