

An integrated approach in gene-expression landscape profiling to identify housekeeping and tissue-specific genes in cattle
Peng Li A , Yun Zhu A , Xiaolong Kang A , Xingang Dan A , Yun Ma A and Yuangang Shi A B
A , Xiaolong Kang A , Xingang Dan A , Yun Ma A and Yuangang Shi A B
A College of Agriculture, Ningxia University, Helan Mountain West Road 489, 750021, Yinchuan, Ningxia Hui Autonomous Region, China.
B Corresponding author. Email: shi_yg@nxu.edu.cn
Animal Production Science 61(16) 1643-1651 https://doi.org/10.1071/AN20689
Submitted: 25 December 2020 Accepted: 7 July 2021 Published: 13 September 2021
Journal Compilation © CSIRO 2021 Open Access CC BY-NC-ND
Abstract
Context: High-throughput transcriptome sequencing (RNA-Seq) has been widely applied in cattle studies. Public databases such as the National Center for Biotechnology Information (NCBI) contain large collections of gene expression data from various cattle tissues that can be used in gene expression analysis research
Aims: This study was conducted to investigate patterns of transcriptome variation across tissues of cattle through large-scale identification of housekeeping genes (i.e. those crucial to maintaining basic cellular activity) and tissue-specific genes in cattle tissues.
Methods: Using data available in the NCBI Sequence Read Archive database, we analysed 1377 transcriptome data sequences from 60 bovine tissue types, identified tissue-specific and housekeeping genes, and set up a web-based bovine gene expression analysis tool.
Key results: We found 101 genes widely expressed in almost all tissue and screened out five housekeeping genes: RPL35A, eIF4A2, GAPDH, IPO5 and PAK2. Focusing on 12 major organs, we found 861 genes specifically expressing in these tissues. Furthermore, 187 significantly differentially expressed genes were found among six types of muscle tissues. All expression data were made available at our new website http://cattleExp.org, which can be freely accessed for future gene expression analyses.
Conclusions: The housekeeping genes and tissue-specific genes identified will provide more information for researchers studying gene expression in cattle.
Implications: The web-based cattle gene expression analysis tool will make it easy for researchers to access large public datasets. Users can easily access all publicly available RNA data and upload their own RNA-Seq data.
Key words: beef cattle, bioinformatics, differentially expressed genes, gene regulation, genomics, muscle tissue, public database, reverse genetics, RNA-Seq.
Introduction
Two major types genes are housekeeping and tissue-specific genes. Housekeeping genes are crucial to maintaining basic cellular activity necessary for organ function and thus life (Thellin et al. 1999; Eisenberg and Levanon 2013; Curina et al. 2017). Housekeeping genes can also be used as internal reference genes when correcting and quantifying gene expression levels. Many diagnostic and research quantification techniques use housekeeping gene expression levels as a baseline to standardise values and determine differential gene expression (DGE) (Thellin et al. 1999; Rubie et al. 2005; Curina et al. 2017). Additionally, systematic mining of tissue-specific genes can improve our understanding of particular tissue morphology and biological behaviour of organisms, as well as providing in-depth research information, especially on animal genetics and breeding (Ardlie et al. 2015; Curina et al. 2017; Ramírez-González et al. 2018). The screening of tissue-specific genes through reverse genetics on a large scale is a fast way to obtain important genes.
Analysis using RNA sequencing (RNA-Seq) has become a powerful tool for transcriptomics research (Sultan et al. 2008; Wang et al. 2008b; Robinson and Oshlack 2010). It can effectively sequence millions of nucleotides and is the most accurate method for genome-wide gene expression research (Wang et al. 2008b). Cattle researchers conducting RNA-Seq transcriptomics have discovered many candidate genes and altered functions related to beef quality, tissues and feed efficiency (Fang et al. 2017; Khansefid et al. 2017), such as the morphology and formation of rumen epithelial tissue and the liver response of cattle with high feed efficiency (Mukiibi et al. 2019).
Large collections of the cattle RNA-Seq data are publicly available and shared on the National Center for Biotechnology Information (NCBI) and European Bioinformatics Institute websites. However, these are raw sequencing data obtained from different research projects of different laboratories; thus, they are not analysed in a unified way and are inconvenient to use directly. There have been many studies on gene expression in various tissues of cattle, through large-scale data analysis, especially of tissue-specific genes and housekeeping genes. In this study, we analysed 1377 RNA-Seq data sequences annotated with 60 tissue types. Through tissue group comparisons, we identified tissue-specific genes and housekeeping genes among those tissue types. Finally, we set up a web-based tool for cattle gene expression analysis, where users can easily access all public RNA data and upload their own sequencing data.
Data and methods
In total, 1377 RNA-Seq records from 97 studies were downloaded from the NCBI SRA (Sequence Read Archive) database. Of these records, 1128 samples annotated to 65 tissues (Table S1), and the most abundant tissues were blood, liver, muscle and mammary gland (Fig. 1a, Table S2). All raw sample data were filtered to remove low-quality reads. The bovine genome (ARS-UCD1.2) obtained from Ensembl (https://www.ensembl.org/index.html) was used as a reference. Kallisto (Weijers et al. 2012) was used to analyse clean reads and determine coding genes, non-coding genes (small and long non-coding genes) and pseudogenes. Well-annotated samples were selected to identify housekeeping and tissue-specific genes. Principal component analysis (PCA) was applied for profiling the similarities of all samples based on gene transcripts per million (TPM) reads.

|
We analysed 354 samples from tissues of 12 major organs to identify tissue-specific genes. Each tissue group was compared with each of the other 11 groups. A tissue-specific gene was defined as a DGE overlap among the 11 comparison groups.
The general framework of the application of cattle RNA-Seq data from the NCBI SRA database is shown in Fig. 2. Public RNA-Seq SRA data of cattle were downloaded from NCBI and converted to the FASTQ file format using the NCBI sratoolkit (version 2.9.6). Low-quality reads and adapters were removed using the Fastp program version 0.12.4 (Chen et al. 2018). Kallisto version 0.45.0 (Weijers et al. 2012) was used to compute gene expression TPM. Ensembl genome bos_taurus.ARS-UCD1.2 and gene annotation version 100 were used as references genes. KOBAS 3.0 (Wu et al. 2006; Xie et al. 2011) was applied for Gene Ontology (GO; Ashburner et al. 2000) and Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto 2000) pathway enrichment analyses. PCA was performed with the R basic function ‘prcomp’ and the gene expression heatmap was generated by R package ‘pheatmap’ version 1.0.12 (The R Foundation, Vienna). Housekeeping genes were defined as those having expression in each sample (ln(TPM + 1) >0.1), and the most stably expressed housekeeping gene had a ln(TPM + 1) >1 in every sample. A comparison of any two tissue differentially expressed genes was calculated by edgeR v3.11 (DOI: 10.18129/B9.bioc.edgeR) with false discovery rate (FDR) <0.05. Tissues-specific genes were defined as genes differentially up-expressed in 11 tissue group comparisons. The cattleExp website was built with Vue.js (https://vuejs.org) as the front-end framework, and node (Express, http://expressjs.com) as the back-end framework.
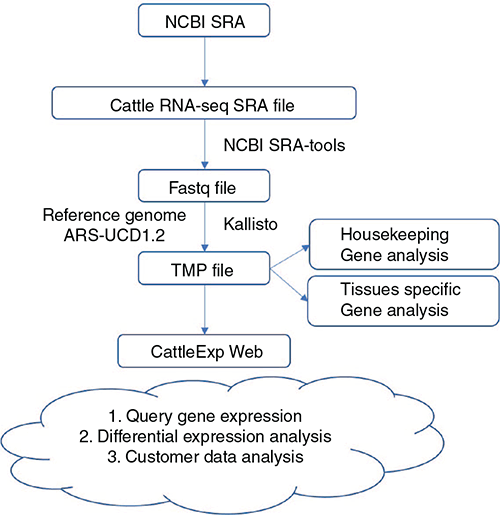
|
Results
The PCA of all samples showed the basic outlines of the relationship of different tissues and samples. The annotations of muscle tissues include ‘skeletal muscle’, ‘longissimus dorsi’, ‘heart’, ‘intercostal muscle’, ‘latissimus dorsi muscle’, ‘gluteus’, ‘scapula muscle’ and ‘muscle’. PCA clearly discriminated longissimus dorsi samples from skeletal muscle samples (Fig. 1b). Most of the samples annotated as ‘blood’ were very similar in gene expression and well repeated from different studies (orange colour in the upper left corner of Fig. 1b), whereas no other tissue type exhibited such a similar distribution pattern. In the annotated data, the most abundant tissues were ‘blood’, ‘liver’, ‘muscle’ and ‘breast’ (Table S2), and 27 607 genes from 1377 different tissue samples of cattle were quantitatively analysed. Among them, there were 21 880 coding genes, 5235 non-coding genes and 492 pseudogenes; 27 539 genes were expressed in at least one sample. In total, 1220 samples of tissue information were used to identify housekeeping and tissue-specific genes.
By analysing the expression levels of genes in all tissues, 101 housekeeping genes were screened out, and the expression characteristics of these genes were analysed by composing a heatmap as shown in Fig. 3; the housekeeping genes with yellow lines sustained and stable high expression, and those with blue lines were consistently expressed but expressed at relatively low levels. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), ribosomal protein L35A (RPL35A), eukaryotic translation initiation factor 4A2 (eIF4A2), importin 5 (IPO5), and p21 (RAC1) activated kinase 2 (PAK2) had the most stable expression in all tissues (minimum variance of expression volume between samples).

|
Constitutive housekeeping gene function was examined through GO enrichment analysis of 101 housekeeping genes (Table 2), revealing that most genes were involved in basic activities of living cells, such as cell replication and energy metabolism. Most of the enrichment results indicate that these genes are related to transcription and translation, peptide biosynthesis, peptide metabolism, amide biosynthesis, cellular amide metabolism, ribosomal large subunit biogenesis, cytosolic large ribosomal subunit, protein-containing complex, large ribosomal subunit, cytosolic ribosome and cellular macromolecule biosynthesis.
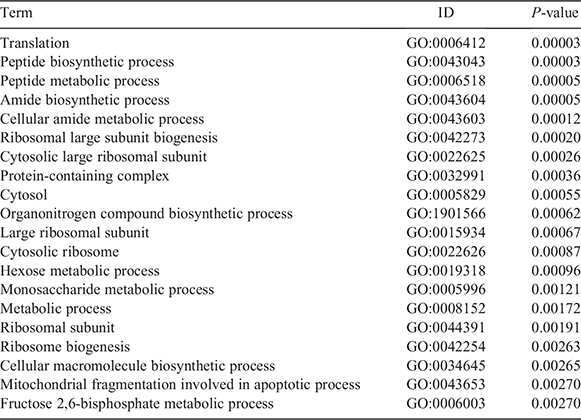
|
Six muscle types (heart, latissimus dorsi muscle, longissimus dorsi, semimembranosus muscle, shoulder muscle, skeletal muscle) and fat tissue were compared with each other to find genes specifically expressing in different tissue types. SLC51B (solute carrier family 51 β subunit), HSPB2 (heat shock protein family B [small] member 2), CKMT2 (muscle creatine kinase, mitochondrial 2) and MYOZ2 (myozenin 2) were identified as muscle-specific genes because they maintained high levels of expression in muscle tissue only. In total, 187 significantly differentially expressed genes were found among six types of muscle tissues.
In the analysis of 354 samples from tissues of 12 major organs, where each tissue group was compared with other groups and a tissue-specific gene was defined as a DGE overlap among the 11 comparison groups, we determined 861 genes from the 12 tissues (Table 1, Table S3). We compared the sample repeat number (i.e. no. of samples of the same tissue) and tissue-specific gene number (i.e. no. of genes from each tissue) and found that the tissue-specific gene number depended on the tissue itself and did not correlate with the sample repeat number. The heatmap with TPM of tissue-specific genes (Fig. 3) shows that samples were well clustered and that each tissue had several highly expressed genes (highlighted blocks; the highest expression levels were observed in the liver and rumen (red lines in Fig. 3).
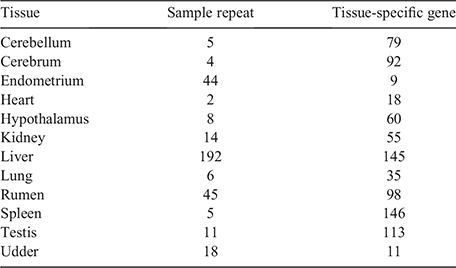
|
All data in the study have been integrated into the cattleExp database (http://cattleExp.org). Most of the samples contain extensive background material such as information on tissue sources, growth conditions, and sampling period. Users can query gene expression data with one gene ID or multiple gene IDs of selected samples (Fig. 4a), then an interactive graphical interface will follow the query submission. This web database is also a tool for differential gene calculation. Users can select any sample group from the database or upload their own sample as a TMP file (temporary backup) for gene differential analysis. Within minutes, users can submit a differential analysis request and the website will return a page with a gene expression volcano plot and gene TPM bar plot (Fig. 4b). All results of differential analysis can be download into a CSV file.

|
Discussion
The housekeeping gene is used as reference gene in many experiments. Relatively highly expressed and stable housekeeping genes have been considered as internal reference genes. However, increasing numbers of studies have shown that the gene expression levels of commonly used housekeeping genes such as β-actin (ACTB), GAPDH, and ribosomal RNA (rRNA) are unstable under different experimental conditions. Therefore, the determination of reference genes requires verification of the stability of specific tissues (i.e. housekeeping genes of cell lines). The choice of housekeeping genes depends on the circumstances. For a broader study, the variability in expression levels of commonly used housekeeping genes means that there is no ‘one size fits all’ gene that can be used to standardise gene expression data. Vandesompele et al. (2002) proposed the use of several housekeeping genes simultaneously when normalising gene expression levels. It is desirable to use between three and five housekeeping genes, and gene expression analysis should be quantified in conjunction with several housekeeping genes. GAPDH and RPL35A were the most widely and highest expressed genes of all genes (Fig. 5). GAPDH is a well-known housekeeping gene in humans and mice and is reported as expressed in 72 human tissues (Barber et al. 2005). RPL35A is a protein coding gene. Diseases associated with RPL35A include Diamond-Blackfan anaemia-5. Among its related pathways are rRNA processing in the nucleus and cytosol and viral mRNA translation. RPL35A and its coding protein are very highly conserved during the process of evolution and can be used in the study of molecular evolution. GO annotations related to this gene include tRNA binding. PAK2 showed lower expression than GAPDH and RPL35A, but it was still very constitutively expressed. It is activated by proteolytic enzymes during caspase-mediated apoptosis; therefore, it may play a role in regulating apoptosis of cells (Edlinger et al. 2017; Lee et al. 2019). With GO enrichment analysis, we found that those were the most stably expressed genes and can be applied as an internal control gene for relative quantification of genes. Other important biological processes include cellular energy conversion and metabolism, such as monosaccharide metabolism, fructose 2,6-bisphosphate metabolism and mitochondrial fragmentation involved in apoptotic process. 18S RNA is another a commonly used housekeeping gene, but it does not have a poly-A RNA-Seq library prepared with oligo(dT). In addition, it is an immature mRNA. So, it cannot used as housekeeping gene for most RNA-Seq data.

|
Muscle tissue is an important economic trait in beef cattle breeding; therefore, we investigated potential relationships in gene expression between different muscle tissues. Six muscle types and fat tissue was compared in order to find genes specifically expressing in particular tissue. SLC51B, HSPB2, CKMT2 and MYOZ2 were identified as muscle-specific genes because they maintained high levels of expression in muscle tissue only. Genes with low expression levels occurring in non-muscle tissue such as fat, liver, rumen and colon also had high and stable expression in muscle tissue. Therefore, we speculate that those are new muscle-specific genes that have yet to be reported in cattle. Another gene, CRYAB (crystallin α B), expressed in many types of tissues and showed very high expression levels in muscles, and especially the heart. This gene is not known to be a muscle-specific expressing gene, but it is important for muscle tissues (Ruan et al. 2020; Zhu et al. 2020). In total, we found three heart-specific, 12 longissimus dorsi-specific, and six skeletal muscle-specific genes compared with the group (Table S4). Our next endeavour is to understand how those muscle-specific genes control tissue development and function.
A gene expression webtool (cattleExp: http://www.cattleExp.org) was developed as part of the study. The database also has upload facilities, designed for comparison with public datasets. The normalised expression values we store in the database support all of these analysis functions. After data processing is standardised, all of these values are placed on the same level for further analysis. The analysis component consists of three independent functional parts. By using these functions, researchers can obtain expression information from three aspects: single gene expression level, and tissue or development gene differential analysis. We hope that this webtool can inspire researchers in the following ways. First, scientists can understand the stage-specific expression of genes in certain species. Second, the database compares the dynamic expression of multiple genes in specific functional pathways. Third, the tool allows researchers to understand the overall expression level of genes in different tissues and development stages. Through the above information, researchers can deepen their understanding of specific genes or functions. Hypergeometric tests performed on each set of compared data draw out the important biological functions of organisms in specific pathways. The elements of the webtool combine biology, genes and functions to enable full understanding of the cattle gene expression.
Conclusions
Based on the analysis of 1377 transcriptome data sequences annotated by 60 tissue types, five bovine housekeeper genes were identified: RPI35A, eIF4A2, GAPDH, IPO5 and PAK2. We have described a large dataset of multi-tissue cattle gene expression. Identification of specific genes in the organisation of genes, especially identification of muscle-related genes, can aid understanding of the growth and development of cattle muscles via gene expression. Muscle-expressed genes can be used as molecular markers to predict meat quality at a given time. More housekeeping genes have been defined, and so we have more choices when needing internal references. In some extreme cases, more internal controls can be avoided. European researchers used L13, EF1a, Tubb2, GADPH and β-actin as reference genes in the analysis. They found that L13 and EF 1A were the most suitable reference genes for European perch, while Tubb2, GADPH and β-actin 1 were the least suitable reference genes. Therefore, internal reference should be selected first rather than directly used, and the final result of this study can only provide a reference. Finally, researchers can query and compare gene expression more easily by using the webtool (cattleExp: http://www.cattleExp.org) that was developed.
Data availability
The data used to support the findings of this study are included within the article and supplementary files.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
Research was supported by Ningxia Hui Autonomous Region’s Key Research and Development program (2017BY078).
Acknowledgements
We are grateful to Ningxia Hui Autonomous Region for its Key Research and Development Program, Ningxia University for the continuous instruction we received for this study, and our mentor for his constant guidance in finishing this paper.
References
Ardlie KG, DeLuca DS, Segrè AV, Sullivan TJ, Young TR, Gelfand ET, et al (2015) The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660.| The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans.Crossref | GoogleScholarGoogle Scholar |
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene Ontology: tool for the unification of biology. Nature Genetics 25, 25
| Gene Ontology: tool for the unification of biology.Crossref | GoogleScholarGoogle Scholar | 10802651PubMed |
Barber RD, Harmer DW, Coleman RA, Clark BJ (2005) GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiological Genomics 21, 389–395.
| GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues.Crossref | GoogleScholarGoogle Scholar | 15769908PubMed |
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890.
| Fastp: an ultra-fast all-in-one FASTQ preprocessor.Crossref | GoogleScholarGoogle Scholar | 30423086PubMed |
Curina A, Termanini A, Barozzi I, Prosperini E, Simonatto M, Polletti S, Silvola A, Soldi M, Austenaa L, Bonaldi T, Ghisletti S, Natoli G (2017) High constitutive activity of a broad panel of housekeeping and tissue-specific cis-regulatory elements depends on a subset of ETS proteins. Genes & Development 31, 399–412.
| High constitutive activity of a broad panel of housekeeping and tissue-specific cis-regulatory elements depends on a subset of ETS proteins.Crossref | GoogleScholarGoogle Scholar |
Edlinger L, Berger-Becvar A, Menzl I, Hoermann G, Greiner G, Grundschober E, Bago-Horvath Z, Al-Zoughbi W, Hoefler G, Brostjan C, Gille L, Moriggl R, Spittler A, Sexl V, Hoelbl-Kovacic A (2017) Expansion of BCR/ABL1+ cells requires PAK2 but not PAK1. British Journal of Haematology 179, 229–241.
| Expansion of BCR/ABL1+ cells requires PAK2 but not PAK1.Crossref | GoogleScholarGoogle Scholar | 28707321PubMed |
Eisenberg E, Levanon EY (2013) Human housekeeping genes, revisited. Trends in Genetics 29, 569–574.
| Human housekeeping genes, revisited.Crossref | GoogleScholarGoogle Scholar | 23810203PubMed |
Fang L, Sahana G, Su G, Yu Y, Zhang S, Lund MS, Sørensen P (2017) Integrating sequence-based GWAS and RNA-seq provides novel insights into the genetic basis of mastitis and milk production in dairy cattle. Scientific Reports 7, 45560
| Integrating sequence-based GWAS and RNA-seq provides novel insights into the genetic basis of mastitis and milk production in dairy cattle.Crossref | GoogleScholarGoogle Scholar | 28358110PubMed |
Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28, 27–30.
| KEGG: Kyoto encyclopedia of genes and genomes.Crossref | GoogleScholarGoogle Scholar | 10592173PubMed |
Khansefid M, Millen CA, Chen Y, Pryce JE, Chamberlain AJ, Vander Jagt CJ, Gondro C, Goddard ME (2017) Gene expression analysis of blood, liver, and muscle in cattle divergently selected for high and low residual feed intake. Journal of Animal Science 95, 4764–4775.
| Gene expression analysis of blood, liver, and muscle in cattle divergently selected for high and low residual feed intake.Crossref | GoogleScholarGoogle Scholar | 29293712PubMed |
Lee JS, Mo Y, Gan H, Burgess RJ, Baker DJ, van Deursen JM, Zhang Z (2019) Pak2 kinase promotes cellular senescence and organismal aging. Proceedings of the National Academy of Sciences of the United States of America 116, 13311–13319.
| Pak2 kinase promotes cellular senescence and organismal aging.Crossref | GoogleScholarGoogle Scholar | 31209047PubMed |
Mukiibi R, Vinsky M, Keogh K, Fitzsimmons C, Stothard P, Waters SM, Li C (2019) Liver transcriptome profiling of beef steers with divergent growth rate, feed intake, or metabolic body weight phenotypes. Journal of Animal Science 97, 4386–4404.
| Liver transcriptome profiling of beef steers with divergent growth rate, feed intake, or metabolic body weight phenotypes.Crossref | GoogleScholarGoogle Scholar | 31583405PubMed |
Ramírez-González RH, Borrill P, Lang D, Harrington SA, Brinton J, Venturini L, Davey M, Jacobs J, Van Ex F, Pasha A, Khedikar Y, Robinson SJ, Cory AT, Florio T, Concia L, Juery C, Schoonbeek H, Steuernagel B, Xiang D, Ridout CJ, Chalhoub B, Mayer KFX, Benhamed M, Latrasse D, Bendahmane A, Wulff BBH, Appels R, Tiwari V, Datla R, Choulet F, Pozniak CJ, Provart NJ, Sharpe AG, Paux E, Spannagl M, Bräutigam A, Uauy C (2018) The transcriptional landscape of polyploid wheat. Science 361, eaar6089
| The transcriptional landscape of polyploid wheat.Crossref | GoogleScholarGoogle Scholar | 30115783PubMed |
Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology 11, R25
| A scaling normalization method for differential expression analysis of RNA-seq data.Crossref | GoogleScholarGoogle Scholar | 20196867PubMed |
Ruan H, Li Y, Wang X, Sun B, Fang W, Jiang S, Liang C (2020) CRYAB inhibits migration and invasion of bladder cancer cells through the PI3K/AKT and ERK pathways. Japanese Journal of Clinical Oncology 50, 254–260.
| CRYAB inhibits migration and invasion of bladder cancer cells through the PI3K/AKT and ERK pathways.Crossref | GoogleScholarGoogle Scholar | 31829429PubMed |
Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, Brittner B, Ludwig B, Schilling M (2005) Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Molecular and Cellular Probes 19, 101–109.
| Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues.Crossref | GoogleScholarGoogle Scholar | 15680211PubMed |
Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O’Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML (2008) A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321, 956–960.
| A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome.Crossref | GoogleScholarGoogle Scholar | 18599741PubMed |
Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennan G, Grisar T, Igout A, Heinen E (1999) Housekeeping genes as internal standards: use and. Journal of Biotechnology 75, 291–295.
| Housekeeping genes as internal standards: use and.Crossref | GoogleScholarGoogle Scholar | 10617337PubMed |
Vandesompele J, Preter KD, Patty NF, Poppe B, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 3, research0034.1
| Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes.Crossref | GoogleScholarGoogle Scholar | 12184808PubMed |
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB (2008a) Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
| Alternative isoform regulation in human tissue transcriptomes.Crossref | GoogleScholarGoogle Scholar | 18978772PubMed |
Wang X, Sun Q, McGrath SD, Mardis ER, Soloway PD, Clark AG (2008b) Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One 3, e3839
| Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain.Crossref | GoogleScholarGoogle Scholar | 19066630PubMed |
Weijers SR, De Jonge J, Van Zanten O, Benedetti L, Langeveld J, Menkveld HW, Van Nieuwenhuijzen AF (2012) KALLISTO: cost effective and integrated optimization of the urban wastewater system Eindhoven. Water Practice and Technology 7, wpt2012036
| KALLISTO: cost effective and integrated optimization of the urban wastewater system Eindhoven.Crossref | GoogleScholarGoogle Scholar |
Wu J, Mao X, Cai T, Luo J, Wei L (2006) KOBAS server: a web-based platform for automated annotation and pathway identification. Nucleic Acids Research 34, W720–W724.
| KOBAS server: a web-based platform for automated annotation and pathway identification.Crossref | GoogleScholarGoogle Scholar | 16845106PubMed |
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li C-Y, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research 39, W316–W322.
| KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases.Crossref | GoogleScholarGoogle Scholar | 21715386PubMed |
Zhu B, Xue F, Li G, Zhang C (2020) CRYAB promotes osteogenic differentiation of human bone marrow stem cells via stabilizing β-catenin and promoting the Wnt signalling. Cell Proliferation 53, 1–12.
| CRYAB promotes osteogenic differentiation of human bone marrow stem cells via stabilizing β-catenin and promoting the Wnt signalling.Crossref | GoogleScholarGoogle Scholar |
* These authors contributed equally to this work.