

Molecular Metal Oxide Cluster-Surface Modified Titanium(iv) Dioxide Photocatalysts
Michael Nolan A C , Anna Iwaszuk A and Hiroaki Tada B CA Tyndall National Institute, Lee Maltings, University College Cork, Cork, Ireland.
B Department of Applied Chemistry, School of Science and Engineering, Kinki University, 3-4-1, Kowakae, Higashi-Osaka, Osaka 577-8502, Japan.
C Corresponding authors. Email: h-tada@apch.kindai.ac.jp or michael.nolan@tyndall.ie

Michael Nolan received a BSc in Chemistry with German in 1997 and a Masters and PhD in Microelectronic Engineering from University College Cork in 1999 and 2004. He was a postdoctoral fellow at Trinity College Dublin with Professor Graeme Watson and has been at Tyndall National Institute since 2005, and a staff researcher since 2009. |

Anna Iwaszuk was born in Poland. She obtained a BSc in Analytical Chemistry from Hogeschool Zeeland in the Netherlands and MSc in Chemical and Process Engineering from Technical University of Szczecin, Poland. She is currently a PhD student studying metal oxide interfaces with Dr Nolan at the Tyndall National Institute. |

Hiroaki Tada received his BS and MS in Engineering from Kyoto University. He received his Dr degree in engineering from Kyoto University in 1991. He joined the research group of Professor A. T. Bell at University of California, Berkeley as an invited scholar in 2002. He is currently a full professor of the School of Science and Engineering at Kinki University. |
Australian Journal of Chemistry 65(6) 624-632 https://doi.org/10.1071/CH11451
Submitted: 30 November 2011 Accepted: 30 January 2012 Published: 24 April 2012
Abstract
The surface modification of TiO2 with molecular sized metal oxide clusters has recently been shown to be a promising approach for providing TiO2 with visible-light activity and/or improved UV activity. This short review summarizes the effects of the surface modification of TiO2 with the oxides of iron and tin selected from d- and p-blocks, respectively, on the photocatalytic activity. Fe(acac)3 and [Sn(acac)2]Cl2 chemisorption on the TiO2 surface occurs by ligand-exchange and ion-exchange, respectively. Taking advantage of the strong adsorption, we formed extremely small metal oxide clusters on TiO2 by the chemisorption–calcination cycle (CCC) technique with their loading amount strictly controlled. The iron oxide surface modification of P-25 (anatase/rutile = 4 : 1, w/w, Degussa) gives rise to a high level of visible-light activity and a concomitant increase in the UV-light activity for the degradation of model organic pollutants. On the other hand, only the UV-light activity is increased by the tin oxide surface modification of ST-01 (anatase, Ishihara Sangyo). This striking difference can be rationalized on the basis of the material characterization and DFT calculations, which show that FeOx surface modification of rutile leads to visible-light activity, while SnO2-modified anatase enhances only the UV-light activity. We propose the mechanisms behind the FeOx and SnO2 surface modification, where the surface-to-bulk and bulk-to-surface interfacial electron transfer are taken into account in the former and the latter, respectively.
Introduction
TiO2 has attracted much attention as an ‘eco-catalyst’ for the purification of polluted water and air owing to its UV-light induced powerful oxidation ability, physicochemical stability, and abundance in nature.[1,2] In general, the photocatalytic activity of TiO2 strongly depends on the crystal form and the exposed crystal plane.[3] For the oxidation of organics, anatase usually exhibits a higher UV-light activity than rutile,[4] which can be improved by coupling with rutile.[5] The present major challenge in TiO2 photocatalysis is providing it with visible-light activity at the same time as increasing its UV-light activity. For this purpose, doping of metal ions and anions into TiO2 has been intensively studied.[6–12] However, the defects introduced into TiO2 lead to a decrease in its crystallinity reducing the carrier mobility and increasing the recombination probability. Consequently, if visible-light activity is obtained, the UV-light activity decreases in most cases. On the other hand, bulk- or nano-coupling of appropriate metal oxides with TiO2 enables the increase of its UV-light activity. For example, the SnO2-coupling significantly increases the UV-light activity of TiO2 because of the long-range charge separation assisted by the high electric conductivity of SnO2.[13–15] Conversely, Fe2O3-[16] or WO3-coupling[17] reduces the UV-light activity, giving no visible-light activity. As an alternative, Kisch and coworkers have devised the photosensitization of TiO2 by its surface modification with PtIV chloride.[18] This approach is attractive in that a visible-light response can be induced by a simple procedure without the introduction of impurity/vacancy levels. The research groups of Ohno and Hashimoto have reported that the surface modification of rutile TiO2 with Fe3+,[19,20] Cr,[21] and Cu[22] by the impregnation method leads to high visible-light activities for the decomposition of model organic pollutants. However, the effect is small for anatase TiO2. Libera et al. reported the synthesis of iron(iii)-oxo clusters adsorbed on TiO2 which showed visible-light absorption and photocatalytic activity for methylene blue degradation.[23] The interface between the two oxides can result in electron and hole separation upon photoexcitation which is one of the key factors in efficient photocatalysis. Heterostructures of TiO2 with metals,[24,25] semiconductors[26,27] and heterostructures of two different metal oxides[28–30] have been shown to induce visible light absorption and improved photocatalytic activity when compared with the isolated materials. The formation of a heterostructure of two metal oxides could provide a promising approach for developing materials with improved photocatalytic activity.
Recently, we have shown that the surface modification of anatase and anatase/rutile TiO2, having the highest level of UV-light activity among the commercial ones, with iron oxide[31] and nickel oxide clusters[32] by the chemisorption–calcination cycle (CCC) technique yields a high level of visible-light activity and a concomitant increase in the UV-light activity. In contrast, the surface modification by molecular SnO2 affords no visible-light activity, while the UV-light activity does increase.[33] Thus, it is of great importance for the design of this new type of photocatalyst to clarify the action mechanism of the surface modification, which is quite different from those of the usual metal oxide–TiO2 bulk- and nano-coupling systems.
This short review summarizes our recent experimental and theoretical studies on the iron oxide and tin oxide surface modified anatase/rutile and anatase TiO2 photocatalysts, respectively. Following the introduction, the experimental results are described, including the metal oxide cluster formation on the TiO2 surface by the CCC technique (MO/TiO2), the optical property, photocatalytic activity, and band energy of MO/TiO2, and the electron transfer from MO/TiO2 to O2. Results of simulations using first principles density functional theory (DFT) calculations for FeOx and SnO2-modified TiO2 are then presented. Finally the action mechanisms of the FeOx and SnO2 surface modifications on the photocatalytic activity of TiO2 are discussed.
Experimental Results
Molecular Metal Oxide Formation on TiO2 by the CCC Technique
We developed the CCC technique, where metal complexes such as metal acetylacetonates are adsorbed by chemical bonds, and the organic part is oxidized by post-heating, for preparing metal oxide clusters at a molecular scale (Scheme 1).[34] For FeOx-modified TiO2, Fe(acac)3 is chemisorbed on the TiO2 surface by ligand-exchange between the acac-ligand and the surface Ti–OH group (Eqn 1).

|
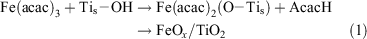
where the subscript s denotes the surface adsorbed species.
On the other hand, [Sn(acac)2]Cl2 is chemisorbed onto the TiO2 surface by ion-exchange between H+ and the [Sn(acac)2]2+ ion (Eqn 2).[35]
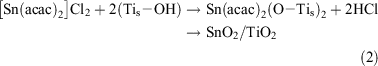
X-Ray photoelectron spectroscopy (XPS) of the iron oxide and tin oxide surface modified TiO2 confirmed that the valence states of Fe and Sn are +3/+2 (mixed valency) and +4, respectively. Post-heating the complex-adsorbed TiO2 at 773 K in air completely oxidizes the residual acac ligands. The Fe (or Sn) on the TiO2 surface was dissolved by treatment with 35 % HCl, and the solid was completely dissolved into 96 % H2SO4 at 353 K. The Fe (or Sn) amounts in both the solutions were almost equal. Also, no particles were observed on the TiO2 surfaces of the iron oxide and tin oxide surface modified samples by high resolution-transmission electron microscopy. Thus, FeOx (or SnO2) species can be formed on the TiO2 surface in a highly dispersed state at a molecular level by the CCC technique. The use of hydrolysis-resistant Fe(acac)3 (or [Sn(acac)2]Cl2) as a precursor and the post-heating after its strong chemisorption on the TiO2 surface are considered to contribute to the formation of the unique FeOx (or SnO2) species. The Fe (or Sn) loading amount is expressed by the number of Fe (or Sn) ions per unit of TiO2 surface area (Γ/ions nm–2). The sample with a given loading, Γ, is designated as MO(Γ)/TiO2 below.
Optical Properties
The optical properties of MO/TiO2 structures are of primary importance in determining the photocatalytic activity. Fig. 1A shows UV-vis absorption spectra of FeOx(Γ)/TiO2. TiO2 only shows strong absorption, because of the interband transition, in the UV-light region. As a result of the surface modification of TiO2 with FeOx, bandgap narrowing takes place, while the intensity of the d–d transition band is very weak at Γ < 0.36. A similar spectroscopic feature was observed for TiO2 doped with Cr and N prepared by physical methods such as ion implantation and magnetron sputtering.[10,35] In contrast, on chemically doping Cr and N ions into TiO2, fairly strong shoulders by the d–d transition appear in the visible region with the absorption edge almost invariant.[12]
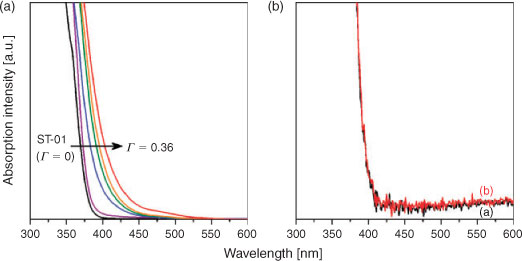
|
Fig. 1B shows UV-vis absorption spectra of TiO2 and SnO2(0.024)/TiO2. No change in the absorption spectrum is observed after the surface modification, which is true for the other samples with varying Γ. In this manner, the surface modification of TiO2 with FeOx extends light absorption into the visible region, whereas no spectroscopic change is induced by the surface modification with SnO2.
Photocatalytic Activities
P-25 (anatase/rutile = 4 : 1, w/w) and ST-01 (anatase), having the highest level of UV-light activities among commercial TiO2, were used as the standard TiO2 photocatalyst. To evaluate the relative photocatalytic activities of FeOx/TiO2 (k) with respect to that of pristine TiO2 (k0), the rate constants were determined under the same irradiation conditions with the same amount of photocatalysts. As a liquid-phase test reaction, the photocatalytic degradation of 2-naphthol (2-NAP) was carried out under illumination of visible light (λ > 400 nm) and UV light (330 < λ < 400 nm). 2-NAP, which is the starting material of azo-dyes and transparent at λ > 330 nm, was used as a model water pollutant. The 2-NAP degradation apparently follows first-order kinetics. Fig. 2a shows the relative first-order pseudo-rate constants for the FeOx/P-25 photocatalyzed degradation of 2-NAP under illumination of visible-light (kvis/kvis0) and UV-light (kUV/kUV0) as a function of Γ. A high level of visible-light activity is induced by the FeOx surface modification. However, the kvis value at Γ ≈ 0.5 (0.7 h–1) is below half the kUV value at Γ = 0 (1.6 h–1). This fact points out the importance of the compatibility of the visible-light with the UV-light activities. A marked increase in the UV-light activity with the surface modification is observed at the same time. In each case, the plot exhibits a volcano-type curve with a maximum at Γ ≈ 0.5 ions nm–2. Under the optimum conditions, the kvis and kUV values increase as compared with those for TiO2 by factors of 7.9 and 4.2, respectively. As a gas-phase test reaction, the photocatalytic degradation of acetaldehyde (CH3CHO) was carried out under illumination of visible-light and UV-light. CH3CHO, which is a volatile organic compound responsible for sick-house syndrome, was used as a model air pollutant. Fig. 2b shows the plots of kvis/kvis0 and kUV/kUV0 vs. Γ. Drastic increases in the visible-light and UV-light activities with the FeOx surface modification are observed. Remarkable enhancing effects by the FeOx surface modification were also obtained for ST-01.[31] Evidently, the FeOx surface modification gives rise to a noticeable visible-light activity and a concomitant significant increase in the UV-light activity.
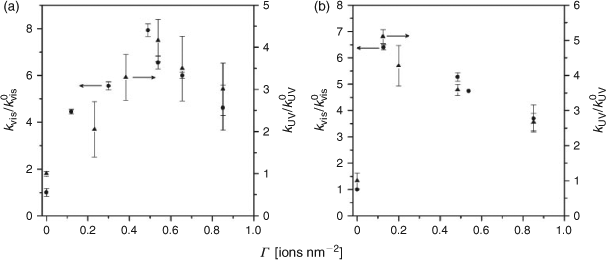
|
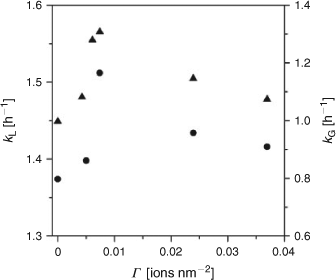
Highly active ST-01 was modified by the SnO2 species by the CCC technique. As predicted from the absorption spectrum, SnO2/TiO2 was inactive for the degradations of 2-NAP and CH3CHO under illumination of visible light. On the other hand, UV-light irradiation led to the degradations of 2-NAP and CH3CHO apparently with first-order kinetics followed. Fig. 3 shows the first-order pseudo-rate constants for the SnO2/ST-01 photocatalzyed degradation of 2-NAP (kL/kL0) and CH3CHO (kG/kG0) under UV-light irradiation as a function of Γ. The plots of both the kL and kG exhibit parallel volcano-shaped dependency on Γ with a maximum at Γ ≈ 0.0074 ions nm–2 and the activities further decreased at Γ > 0.04. The SnO2 surface modification scarcely affected the adsorptivity for 2-NAP in the dark at Γ < 0.04. Certainly, the SnO2 surface modification has a positive effect on its UV-light activity for both the liquid-phase and gas-phase reactions, although the effects are much smaller than those by the FeOx surface modification. Also, this optimum Γ value in the SnO2/TiO2 system is smaller as compared with that in the FeOx/TiO2 system by a factor of more than one-order of magnitude. This result indicates that the optimum loading amount of the metal oxide clusters strongly depends on their type. Interestingly, it has recently been shown that the SnOx surface modification of ZnGa2O4 by an impregnation method remarkably increases not only the UV-light activity but also the visible-light activity.[36]
|
TiO2 Band Energy Modification
XPS measurements were performed to gain information about the filled energy levels of FeOx/TiO2. Fig. 4a shows the valence band (VB)-XPS spectra for FeOx/TiO2. The emission from the O2p-VB extends from 3 to 9 eV. Closer inspection of the VB-top (inset) indicates an upwards shift of 0.4 eV with an increase in Γ, which is comparable with the decrease in the bandgap (Eg) with the FeOx surface modification. The effective mixing between the surface Fe3+ levels and O2p as a result of the Tis–O–Fe interfacial bond is considered to yield a surface d-sub-band dispersing around the energy level to overlap with the VB(TiO2).[37] This interpretation explains the net decrease in the Eg of TiO2. From the Eg for FeOx(Γ ≈ 0.5)/TiO2 (~2.9 eV), the top of the surface d-sub-band is estimated to be situated at +2.4 V vs. a standard hydrogen electrode (SHE).[38] In addition, Fig. 4b shows VB-XPS spectra for SnO2(Γ)/TiO2. In contrast to the FeOx/TiO2 system, the VB-top is not changed by the SnOx surface modification, irrespective of Γ.

|
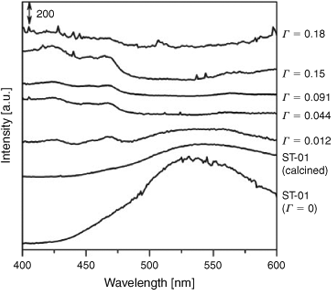
Information about empty levels can be obtained by photoluminescence (PL) spectroscopy. Fig. 5 shows PL spectra of FeOx(Γ)/TiO2 at 77 K: excitation wavelength = 320 nm. TiO2 (ST-01) has a broad emission band centered at 538 nm (E1). Heating it at 773 K for 1 h in air remarkably weakens the E1 signal intensity. This PL band is assignable to the emission from the surface oxygen vacancy levels of anatase TiO2.[39] On modifying TiO2 with the FeOx species, the intensity further decreases to disappear at Γ > 0.044 ions nm–2, while two new emissions appear at 423 (E2) and 468 nm (E3). The E2 and E3 signals can be attributed to the emissions from extrinsic levels resulting from the surface FeOx species. These findings suggest that the excited electrons in the conduction band (CB) of TiO2 are transferred to the empty surface FeOx levels in preference to the surface oxygen vacancy levels. Additional surface oxygen vacancy is not induced by the surface modification in contrast to the Fe-doping, which is probably because the local charge balance is conserved on the surface.
|
Electron Transfer to Molecular Oxygen
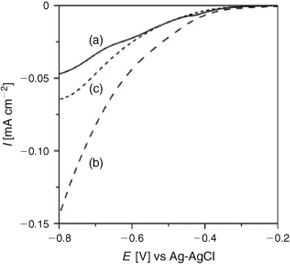
In the oxidative decomposition of organic pollutants, the key to increasing the photocatalytic activity of TiO2 is to enhance the transfer of the excited electrons to O2.[40,41] Current (I)–potential (E) curves were measured for the mesopoporous TiO2 nanocrystalline film-coated fluorine-tin oxide (FTO) (mp-TiO2/FTO) electrodes in an aerated 0.1 m NaClO4 aqueous solution in the dark. Fig. 6 shows the I–E curves of mp-TiO2/FTO, FeOx/mp-TiO2/FTO, and SnO2/mp-TiO2/FTO. In the presence of O2, cathodic currents attributable to the O2 reduction are observed at E < –0.2 V, whereas the current hardly flowed at –0.8 < E < –0.2 V without O2. With the FeOx surface modification, the reduction current at E < –0.55 V greatly increases, i.e., the surface FeOx species mediate the electron transfer from TiO2 to O2. Also, a smaller enhancing effect is observed with the SnO2 surface modification.
|
Simulations of Surface-Modified TiO2
Calculation Methods
For the calculations of surface-modified TiO2 we use the DFT approach with corrections for on-site Coulomb interactions, DFT+U, to describe Fe and Ti oxidation states consistently; no such correction is applied to SnO2, since DFT adequately describes this system.
For modelling TiO2 rutile (110) and anatase (001) surfaces, we use a three dimensional periodic slab model within the VASP code.[42] The valence electrons were described by a plane wave basis set and the cut-off for the kinetic energy is 396 eV. There are four valence electrons for Ti, eight for Fe, four for Sn, and six for O. The exchange-correlation functional was approximated by the Perdew–Wang 91[43] functional. The Monkhorst–Pack scheme was used for K-point sampling with a 2 × 1 × 1 sampling grid.
To describe Ti 3d states the DFT+U approach was used where U = 4.5 eV. The need to introduce the U parameter in order to describe the electronic states of d shells properly is well known.[44,45] Fe 3d states were described with U = 6.5eV and J = 1 eV, which are typical values from the literature.[46] For Sn, the electronic states are consistently described by DFT so no U correction was applied. The DFT+U approach gives a relatively correct d state description but still gives an underestimation of the bandgap and this depends on the precise DFT+U set up. We are aware of this important issue but are primarily concerned with qualitative changes in the bandgap upon surface modification. With this in mind, the simulation results are important for understanding the experimental results.
The rutile (110) surface is terminated by two coordinated bridging oxygens and the surface contains five-fold and six-fold coordinated Ti atoms. The anatase (001) surface is terminated by two coordinated oxygen atoms while the oxygen atoms in the surface are three coordinated. The Ti atoms in the surface are five-fold coordinated. All surfaces have a 12 Å vacuum gap. We used a 4 × 2 surface supercell for both surfaces. For consistency in the calculation we applied the same supercell for the bare TiO2 surface and free clusters. While the anatase (001) surface is not the most stable surface of anatase (which is (101)), the consensus from experimental work is that this surface is the most active for photocatalysis[47,48] and hence we consider this surface in our simulations.
The clusters are positioned on the TiO2 surfaces and the adsorption energy is computed from:
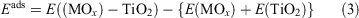
where E((MOx)n – TiO2) is the total energy of the MOx cluster supported on the TiO2 surface and E(MOx) and E(TiO2) are the total energies of the free MOx cluster and the bare surface, respectively.
A negative adsorption energy signifies that the absorption of the metal oxide cluster is favourable. For both oxide–TiO2 systems, several FeOx and SnO2 adsorption configurations were tested and those presented in Fig. 7 are the most stable we have found.
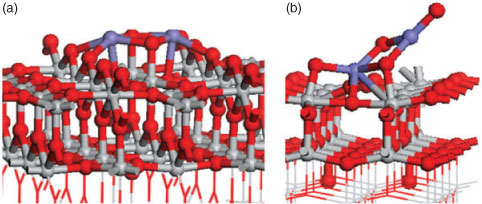
|
Simulation Results
We performed calculations for the following oxide clusters: FeO, (FeO)2, Fe2O3, SnO2, and Sn2O4 and for the purpose of this summary we present only the most stable structures that we have found for (FeO)2 on rutile (110) and Sn2O4 on anatase (001), with further details presented in the literature.[33,49] We adsorbed the oxide nanoclusters on the TiO2 surfaces and ran a full ionic relaxation.
Fig. 7 presents the atomic structure of (FeO)2 on the rutile (110) surface, and Sn2O4 on the anatase (001) surface, with the computed adsorption energies shown. In both composite materials the computed adsorption energies are negative, being –1.84 eV for (FeO)2/TiO2 and –5.29 eV for Sn2O4/TiO2.
These negative adsorption energies indicate the stability of the adsorbed iron and tin oxide clusters when supported at the TiO2 surface. While examining the atomic structures, both materials show an interaction between the surface and the cluster with the formation of new bonds. In the case of (FeO)2 the Fe atoms are bonded to two bridging oxygen atoms with distances of 2.11 and 2.29 Å. Two atoms from the cluster bind to Ti atoms from the surface with a bond length of 1.89 Å. A Sn2O4 cluster creates new bonds with the anatase (001) surface, where two O atoms from the surface have migrated from the surface layer to bind to Sn atoms of the cluster, undergoing a displacement of 1.11 Å. The two O atoms from the cluster bond to Ti from the surface. Thus the growing number of new metal–oxygen bonds results in the stability of the presented structures.
The electronic structure of the composite material can be used to understand its photocatalytic properties. We plot in Fig. 8 the projected electronic density of states (PEDOS) for bare rutile (110) and anatase (001), as well as the Fe 3d states from the (FeO)2 cluster and Ti 3d states from the rutile (110) surface and the Sn 5s states from the Sn2O4 cluster and Ti 3d states from the anatase (001) surface.

|
For (FeO)2-modified TiO2, the DOS shows that the Fe 3d states of adsorbed FeOx lies above the valence band of TiO2 while the conduction band derives from Ti 3d from the TiO2 surface. This result explains the photocatalytic properties found for FeOx–TiO2, because the state from FeOx that lies above the TiO2 valence band gives the bandgap narrowing compared with bare TiO2 surface pushing the light absorption into the visible region. Moreover we can expect improved charge separation because upon visible-light excitation the excited electrons will be found on TiO2 and the holes on the iron oxides.[37]
The behaviour of Sn2O4 on TiO2 anatase (001) is, however, different; the resulting bandgap is unchanged in comparison to the bare TiO2 surface. Examining the PEDOS plot we found that empty tin oxide states lie above the conduction band of the TiO2 surface and there are no tin oxide states found in the VB–CB energy gap of TiO2. Therefore, SnO2 modification of anatase will give no bandgap shift to the visible-light region. The fact that the Sn 5s states are dispersed above the CB of TiO2 has the consequence that these states will be acceptors of electrons during UV-light excitation.
The presented results for both oxides explain the experimental results that show FeOx-modified TiO2 shifts the photocatalytic activity to the visible region, and Sn2O4-supported material enhances only the UV-light activity.
Mechanism for Enhanced Photocatalytic Activity of Surface-Modified TiO2
The energy band diagram scheme of oxide-modified TiO2 derived on the basis of the results above is shown in Scheme 2a for FeOx. As indicated by the VB-XPS measurements and DFT simulation, the loading of the FeOx species mainly modifies the electronic levels of TiO2 around the VB edge. Visible-light absorption triggers electronic excitation from the surface d sub-band, derived from iron oxide, to the CB (derived from TiO2) (p1, Scheme 2). The charge separation is achieved by this visible-light induced surface-to-bulk interfacial electron transfer (IET). On illumination with UV light (p2), the electrons in the VB(TiO2) are excited to the CB(TiO2). In both the cases, without the FeOx surface modification, the excited electrons in the CB(TiO2) are rapidly trapped at the surface oxygen vacancy levels to undergo recombination with the holes. The FeOx surface modification permits the preferential electron transfer from the CB(TiO2) to the surface FeOx levels (p3). The electrons effectively reduce adsorbed O2 by the action of the surface FeOx species as an excellent mediator (p4). As a result of the effective suppression of the recombination by the surface oxygen vacancy levels, the photocatalytic activities drastically increase under illumination of visible and UV light. A feature of the surface modification in the anodic process should also be stressed in that the holes generated in the surface d sub-band take part in the oxidation process without diffusion (p5).[21]

|
On the other hand, the loading of the SnO2 species modifies the unfilled electronic levels, while the Eg is invariant.[37,49] Scheme 2b shows the energy band diagram scheme of SnO2/TiO2 derived from experimental and simulation results. On UV-light irradiation, the electrons in the VB(TiO2) can be promoted to either the CB(TiO2) or the SnO2 states around the CB edge of TiO2. The latter process, i.e., the UV-light induced bulk-to-surface IET, leads to the charge separation. Subsequently, the electrons in the Sn states are transferred to O2 with an aid of the function as a mediator.
Conclusions
This short review summarizes our work on FeOx and SnO2 surface-modified TiO2 photocatalysts. Extremely small metal oxide clusters are formed on the TiO2 surface by the CCC technique with the loading amount strictly controlled. The compatibility of high visible-light and UV-light activities is achieved by the FeOx surface modification, while only UV-light activity somewhat increases with the SnO2 surface modification. Both the effects can be rationalized in terms of the experimental characterization and the DFT calculations. For FeOx supported on TiO2 rutile (110) we: (a) obtained a narrowed bandgap, giving visible-light absorption, when compared with pure TiO2, (b) determined that the top of the VB comes from the Fe 3d states of the cluster and the CB arises from Ti 3d states of the surface, and (c) subsequently observed an expected charge separation and consequent reduction of electron–hole recombination. SnO2 species supported on TiO2 anatase (001) presents the following features: (a) no change in the bandgap, so no visible-light activity as confirmed by the experiment, (b) Sn 5s states dispersed and centred higher than the CB edge, and (c) under UV light excitation these states can be acceptor levels of electrons from the VB (TiO2). Based on these promising results, systematic experimental and simulation studies on the surface modification of TiO2 with various metal oxides are currently in progress.
Acknowledgements
HT is deeply indebted to Dr M. Fujishima, Q. Jin (Kinki University), and S.-i. Okuoka, T. Hattori, Y. Sumida (Nippon Shokubai Co.) who have diligently contributed to this research. MN and AI acknowledge support from the Science Foundation Ireland (SFI) through the Starting Investigator Research Grant Program, project ‘EMOIN’, grant number SFI 09/SIRG/I1620. The authors acknowledge computing resources at Tyndall provided by SFI and by the SFI and Higher Education Authority funded Irish Centre for High End Computing.
References
[1] A. Fujishima, X. Zhang, D. A. Tryk, Surf. Sci. Rep. 2008, 63, 515.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVWhurvP&md5=5d39241c9466700b6bd7b1f38ea3567fCAS |
[2] K. Hashimoto, H. Irie, A. Fujishima, Jpn. J. Appl. Phys. 2005, 44, 8269.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjslGkug%3D%3D&md5=d1d6d003c2a1b1645c58a1dd8e59f538CAS |
[3] A. Y. Ahmed, T. A. Kandiel, T. Oekermann, D. Bahnemann, J. Phys. Chem. Lett 2011, 2, 2461.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFyksrzL&md5=6e6557331311d69368fc5318336ec74cCAS |
[4] O.-O. Prieto-Mahaney, N. Murakami, R. Abe, B. Ohtani, Chem. Lett. 2009, 38, 238.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsVaku7k%3D&md5=2ab0eefb466960ab55d5ec0dc01bef62CAS |
[5] T. Kawahara, Y. Konishi, H. Tada, N. Tohge, J. Nishii, S. Ito, Angew. Chem. Int. Ed. 2002, 41, 2811.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xmt1emsr0%3D&md5=de4f2b679a14dda80ad0388a86f92a61CAS |
[6] R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki, Y. Taga, Science 2001, 293, 269.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlt1yitbc%3D&md5=6b5bfe3d177da3ec1192f5d2c0f60b28CAS |
[7] S. U. M. Khan, M. Al-Shahry, W. B. Ingler, Science 2002, 297, 2243.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XnvFKqsLw%3D&md5=e0c4f2968e874342de2bee90b1363085CAS |
[8] N. Serpone, J. Phys. Chem. B 2006, 110, 24287.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFKrsLzO&md5=2a2f18a7d4e0faa2e1f6e2dac58dc261CAS |
[9] H. Zhang, G. Chen, D. W. Bahnemann, J. Mater. Chem. 2009, 19, 5089.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXosV2js7s%3D&md5=f48094d027ce6fc295b5b836920b5aa3CAS |
[10] M. Anpo, M. Takeuchi, J. Catal. 2003, 216, 505.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjvVCmsrs%3D&md5=6198ff74b411b5d5786ffd111ef2cb77CAS |
[11] M. Kitano, K. Funatsu, M. Matsuoka, M. Ueshima, M. Anpo, J. Phys. Chem. B 2006, 110, 25266.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFKjsrnJ&md5=0cbe5fa814f04e1dd137fcea0a35cff6CAS |
[12] G. Liu, L. Wang, H. G. Yang, H.-M. Cheng, G. Q. Lu, J. Mater. Chem. 2010, 20, 831.
| Crossref | GoogleScholarGoogle Scholar |
[13] H. Tada, A. Hattori, Y. Tokihisa, K. Imai, N. Tohge, S. Ito, J. Phys. Chem. B 2000, 104, 4585.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXis1Wgsrg%3D&md5=66fab68f162b60c87aa4b520004ea225CAS |
[14] A. Hattori, Y. Tokihisa, H. Tada, N. Tohge, S. Ito, K. Hongo, R. Shiratsuchi, G. Nogami, J. Sol-Gel Sci. Technol. 2001, 22, 53.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXns1Gnsbo%3D&md5=b4121b97b1fdbab4472d94f5b73e4dd9CAS |
[15] T. Kawahara, Y. Konishi, H. Tada, N. Tohge, S. Ito, Langmuir 2001, 17, 7442.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXnsFyntbg%3D&md5=5dd2bd4c941136c2df1da671210e42a4CAS |
[16] X. Hu, T. An, M. Zhang, G. Sheng, J. Fu, Res. J. Chem. Environ. 2007, 11, 13.
| 1:CAS:528:DC%2BD1cXhtlaqs7w%3D&md5=7e6870dde94f0adeac8476d78a8d0f7fCAS |
[17] H. Tada, A. Kokubu, M. Iwasaki, S. Ito, Langmuir 2004, 20, 4665.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjt12rsLc%3D&md5=f05219956251b927bd39847a34cc4c28CAS |
[18] H. Kisch, L. Zhang, C. Lange, W. F. Maier, C. Antonius, D. Meissner, Angew. Chem. Int. Ed. 1998, 37, 3034.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXnvFGhsro%3D&md5=1212350e4b8bd7b4b66c7d7b3a7c52bfCAS |
[19] N. Murakami, T. Chiyoya, T. Tsubota, T. Ohno, Appl. Catal. A. 2008, 348, 148.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVagt7nI&md5=c6a90c3da05cf849e521dc36099939eaCAS |
[20] H. Yu, H. Irie, Y. Shimodaira, Y. Hosogi, Y. Kuroda, M. Miyauchi, K. Hashimoto, J. Phys. Chem. C 2010, 114, 16481.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtV2ktb7J&md5=8c40f1d2c69852df1f3ebe91b8913dceCAS |
[21] H. Irie, T. Shibanuma, K. Kamiya, S. Miura, T. Yokoyama, K. Hashimoto, Appl. Catal. B 2010, 96, 142.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkt12rtL0%3D&md5=9934909d108ee7e5714d23cfb8abe082CAS |
[22] H. Irie, S. Miura, K. Kamiya, K. Hashimoto, Chem. Phys. Lett. 2008, 457, 202.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlvFyitbY%3D&md5=b48ebdc31bd6c58cd97370c2ef00ce97CAS |
[23] J. A. Libera, J. W. Elam, N. F. Sather, T. Rajh, N. M. Dimitrijevic, Chem. Mater. 2010, 22, 409.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1alu7jN&md5=97343cd9ca11cd0b13322af44421b1e4CAS |
[24] A. Ismail, Appl. Catal. B: Environ. 2012, 117, 67.
[25] A. Primo, A. Corma, H. Garcia, Phys. Chem. Chem. Phys. 2011, 13, 886.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1WjsLjL&md5=f1211c66c9ad9d25679c8a89efb3d0f9CAS |
[26] J. H. Bang, P. V. Kamat, Adv. Funct. Mater. 2010, 20, 1970.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotVCnt7g%3D&md5=26f291f9032e904a89e21228aa8decbaCAS |
[27] G. Ai, W. Sun, X. Gao, Y. Zhang, L.-M. Peng, J. Mater. Chem. 2011, 21, 8749.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntVOrurg%3D&md5=4a2021c77dd0d123d1943e994bba76f2CAS |
[28] T. Cao, Y. Li, C. Wang, Z. Zhang, M. Zhang, C. Shao, Y. Liu, J. Mater. Chem. 2011, 21, 6922.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlt1SrtLo%3D&md5=bd90499e16c9ac35e3314b43c315ba80CAS |
[29] S. J. Hong, S. Lee, J. S. Jang, J. S. Lee, Energy Environ. Science 2011, 4, 1781.
| 1:CAS:528:DC%2BC3MXmslOjur0%3D&md5=96f3826205c1c885de45642753e1393cCAS |
[30] L. Kong, Z. Jiang, T. Xiao, L. Lu, M. O. Jones, P. P. Edwards, Chem. Commun. 2011, 47, 5512.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlt1Cks7s%3D&md5=0ce0985795b06b127e38c05f61ad38c4CAS |
[31] H. Tada, Q. Jin, H. Nishijima, H. Yamamoto, M. Fujishima, S.-i. Okuoka, T. Hattori, Y. Sumida, H. Kobayashi, Angew. Chem. Int. Ed. 2011, 50, 3501.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktFWmsbk%3D&md5=1ca71cf38740f6ac327350f783d5f399CAS |
[32] Q. Jin, T. Ikeda, M. Fujishima, H. Tada, Chem. Commun. (Camb.) 2011, 47, 8814.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpt1Kqsrc%3D&md5=a4d4038c0c7235af8e91d3ff96494ccaCAS |
[33] M. Fujishima, Q. Jin, H. Yamamoto, H. Tada, M. Nolan, Phys. Chem. Chem. Phys. 2012, 14, 705.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsF2jsrvL&md5=8759307fdf325a555650ea5d83143e2eCAS |
[34] H. Tada, Encyclopedia of Surface and Colloid Science (Ed.: A. T. Hubbard), 2002, (Marcel Dekker, New York).
[35] M. Kitano, K. Funatsu, M. Matsuoka, M. Ueshima, M. Anpo, J. Phys. Chem. B 2006, 110, 25266.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFKjsrnJ&md5=0cbe5fa814f04e1dd137fcea0a35cff6CAS |
[36] V. B. R. Boppana, R. F. Lobo, ACS Catal. 2011, 1, 923.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFylsb8%3D&md5=ff621c5f9d59cc3b302fe9f1de9720aeCAS |
[37] M. Nolan, Phys. Chem. Chem. Phys. 2011, 13, 18194.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Gqsr%2FJ&md5=1c0f15c6cdbf32f3aa74f040a2aee1d3CAS |
[38] M. Grätzel, F. P. Rotzinger, Chem. Phys. Lett. 1985, 118, 474.
| Crossref | GoogleScholarGoogle Scholar |
[39] J. Shi, J. Chen, Z. Feng, T. Chen, Y. Lian, X. Wang, C. Li, J. Phys. Chem. C 2007, 111, 693.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht12itb3P&md5=0d3bc0e5b0f2874a484292ac5c6a8d53CAS |
[40] C. M. Wang, A. Heller, H. Gerischer, J. Am. Chem. Soc. 1992, 114, 5230.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XktVOgsbw%3D&md5=13684a2ded7f096a5acb9ad6d3204fd8CAS |
[41] M. R. Hoffmann, S. T. Martin, W. Choi, D. W. Bahnemann, Chem. Rev. 1995, 95, 69.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjtF2qur4%3D&md5=a2c095c6cd961275f5015f655349caeaCAS |
[42] G. Kresse, J. Hafner, Phys. Rev. B Condens. Matter 1994, 49, 14251.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXkvFKrtL4%3D&md5=4b4dba0f72a5b37d1b29e60348ee1ae6CAS |
[43] J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh, C. Fiolhais, Phys. Rev. B 1993, 46, 6671.
| Crossref | GoogleScholarGoogle Scholar |
[44] B. J. Morgan, G. W. Watson, J. Phys. Chem. C 2010, 114, 2321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnt1SqsA%3D%3D&md5=d660214f58a8d2fc87dbb4ec17695046CAS |
[45] M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker, G. W. Watson, Surf. Sci. 2005, 576, 217.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXoslCjsg%3D%3D&md5=323287ac1cc8420e6bcca6ac29b2928aCAS |
[46] P. Liao, E. A. Carter, J. Mater. Chem. 2010, 20, 6703.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXps1CgsLY%3D&md5=d4a696abbf141d35f7b6f7548f73efc7CAS |
[47] G. Liu, J. C. Yu, G. Q. Lu, H.-M. Cheng, Chem. Commun. 2011, 47, 6763.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntlagsrc%3D&md5=fb1d3f8aae124e5775d42440dbbba5d1CAS |
[48] D. Q. Zhang, G. S. Li, X. F. Yang, J. C. Yu, Chem. Commun. 2009, 45, 4381.
| Crossref | GoogleScholarGoogle Scholar |
[49] M. Nolan, Chem. Commun. 2011, 47, 8617.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVyjsr8%3D&md5=8c368b8a144cfe4a8aaa08d11144d64dCAS |