

Chemical Activation in Azide and Nitrene Chemistry:
Methyl Azide, Phenyl Azide, Naphthyl Azides, Pyridyl Azides, Benzotriazoles, and Triazolopyridines
Curt Wentrup A
A School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia.
Email: wentrup@uq.edu.au
Australian Journal of Chemistry 66(8) 852-863 https://doi.org/10.1071/CH13283
Submitted: 2 June 2013 Accepted: 24 July 2013 Published: 8 August 2013
Abstract
Chemical activation (the formation of ‘hot’ molecules due to chemical reactions) is ubiquitous in flash vacuum thermolysis (FVT) reactions, and awareness of this phenomenon is indispensable when designing synthetically useful gas-phase reactions. Chemical activation is particularly prevalent in azide chemistry because the interesting singlet nitrenes are high-energy intermediates, and their reactions are highly exothermic. Consequently, chemical activation is observed in the isomerization of methylnitrene CH3N to methylenimine (methanimine) CH2=NH, facilitating the elimination of hydrogen to form HCN or HNC. Rearrangements of phenylnitrene, 1- and 2-naphthylnitrenes, and 2-, 3- and 4-pyridylnitrenes afford cyanocyclopentadiene, 3- and 2-cyanoindenes, and 2- and 3-cyanopyrroles, all showing the effects of chemical activation by undergoing facile interconversion of isomers. Chemical activation can often be reduced or removed entirely by increasing the pressure, thereby promoting collisional deactivation. Larger molecules having more degrees of freedom are better able to dissipate excess energy; therefore the effects of chemical activation are less pronounced or completely absent in the formation of 3-cyanoindole and 1-cyanobenzimidazoles from 3- and 4-quinolylnitrenes and 4-quinazolinylnitrenes, respectively. In compounds possessing nitro groups, chemical activation can cause the loss of the nitro group at nominal temperatures far below those normally needed to cleave the C-NO2 bond.
Introduction
Chemical activation (CA) is the phenomenon of acceleration of chemical reactions due to excess thermal energy carried over from preceding reaction steps.[1] In other words the ‘hot’ molecules formed in exothermic reactions may contain sufficient vibrational energy to overcome the activation barrier for the next step(s) at temperatures where the reactions would not usually occur. Not only the exothermicity, but the energy difference between the preceding transition state and the ground state of the newly formed hot molecule can be used in this way. The phenomenon is most important for small molecules, because larger molecules have many more vibrational modes over which the excess energy can be redistributed.
Chemical activation is usually only observed for reactions in the gas-phase at low pressure, because collisions with other molecules will help deactivate the hot molecules. Thus, it can be expected in many flash vacuum thermolysis (FVT) reactions. It is important to consider this possibility, because so-called ‘control experiments’ prove nothing if CA is involved in the reaction under investigation but not in the control. In practise, the unactivated process may take place at temperatures several hundred degrees above that of the chemically activated one.
It is also noted that reactions in crossed molecular beams can lead to vibrationally excited molecules and radicals, which can therefore undergo secondary isomerizations. The exact collision energies and available excess energies in the observed reaction channels can often be determined in such experiments.[2]
In a thermal reaction such as FVT at low pressure, molecules are activated by collisions with the wall rather than with other molecules. But collisions can also serve to deactivate hot molecules. Thus, performing an FVT reaction at a higher pressure, for example in a stream of Ar or N2, may deactivate chemically activated molecules sufficiently, so that the ‘unactivated’ chemistry is observed. A change from e.g. 10–3 hPa to 1 hPa can be sufficient. Examples will be presented below.
Although CA is typically a low-pressure gas-phase phenomenon, there are occasional suggestions of chemical activation in solution. The case of an unusually reactive benzyne derivative was reported recently.[3] The aryne was formed in an intramolecular [2+4] cycloaddition reaction of a triyne and estimated to be exothermic by ~50 kcal mol–1 (1 kcal mol–1 = 4.186 kJ mol–1), and it is suggested that this excess energy is the reason for the increased reactivity.[4]
Methyl Azide – Methylnitrene – Methanimine – HCN + H2
Methylnitrene has a triplet ground state and a singlet–triplet energy splitting of ~31 kcal mol–1, and the singlet is an extremely fleeting intermediate.[5–7] The thermal and photochemical decomposition of methyl azide CH3N3 1 has been investigated since 1933, but the early studies, particularly as regards the possible formation of methylnitrene CH3N and methylenimine CH2=NH (methanimine; formaldimine) gave contradictory and inconclusive results. Leermakers and others postulated the formation of methylenimine, but it was not directly identified.[8] Darwent and others denied the formation of CH2=NH altogether.[9] However, 1,2-shifts of H and substituents are ubiquitous in carbene and nitrene chemistry,[10,11] so it would be expected that methylenimine CH2=NH was a primary product of methyl azide decomposition.
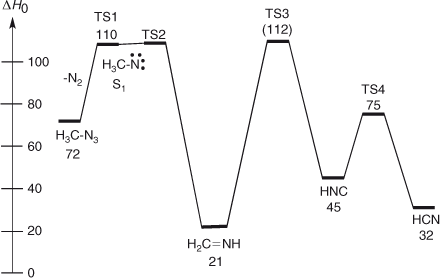
In 1979, Fischer, Wentrup, and Winnewisser (FWW) investigated the FVT of methyl azide 1 by real-time, online millimeterwave spectroscopy.[12–14] The azide was pyrolyzed in the temperature range 500–900°C at 10–2 hPa, and the products were led directly into the evacuated and continuously pumped 250 × 10 cm glass cell of the millimeterwave spectrometer.[15] Methylenimine was unambiguously identified as a product by monitoring characteristic frequencies,[16] J = 61,5 ← 61,6; F = 5 ← 5, F = 7 ← 7 (110896.94 (25) MHz) and J = 61,5 ← 61,6; F = 6 ← 6 (110899.28(25) MHz). The intensities of the methylenimine signals increased over the temperature range 500–900°C. Moreover, signals due to HCN were also present,[17] weakly at 500°C, but strongly at 900°C. FWW interpreted this in terms of chemical activation of the methylenimine by 89 ± 5 kcal mol–1 arising from the activation energy for denitrogenation of azidomethane[18] ~38 ± 2 kcal mol–1 plus the exothermicity of the reaction ~51 ± 3 kcal mol–1[19,20] (Eqns 1 and 2 and Fig. 1). This amount of CA is hardly enough by itself for the elimination of H2, but the additional thermal energy available on FVT at 500–900°C allows the decomposition to take place.[12–14] Temperatures above 1000°C are required to decompose the unactivated methylenimine to HCN under these conditions.[21]
|
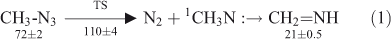

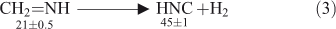

This thermochemical estimate is in excellent agreement with a CASPT2 calculation, giving an energy difference of 82.9 kcal mol–1 between 1CH3N and CH2NH and a low barrier of 2.5 ± 1 kcal mol–1 for the 1,2-H shift in the singlet nitrene.[6]
The microwave spectrum of methylenimine generated by pyrolysis of methyl azide at 490°C (and of aminoacetonitrile at 740°C) was also reported by Brown, Godfrey, and Winkler.[22] Formation of methylenimine by pyrolysis of methyl azide and its chemically activated decomposition to HCN and H2 was confirmed by photoelectron spectroscopy in detailed studies by Bock and Dammel and others,[23] and more recently by cavity ring down spectroscopy by Dagdigian et al.[24] However, on the basis of semiempirical calculations, Bock and Dammel concluded that methylnitrene (singlet or triplet) was not an intermediate, i.e. that the formation of methylenimine was concerted with N2 loss from methyl azide, and this was supported by early ab initio calculations.[25] More recently, multiconfigurational ab initio calculations (CASSCF and CAS/MP2) have revealed that methylnitrene (singlet and/or triplet) is indeed an intermediate, which undergoes isomerization to methylenimine via a very low barrier (1.4 kcal mol–1), and a second reaction channel leads directly from singlet methylnitrene to HCN + H2 (barrier 19.4 kcal mol–1).[26] The methylenimine can undergo chemically activated decomposition to HCN and H2 as well. The calculated barrier is 107 kcal mol–1, so in the absence of CA this reaction will be extremely slow at temperatures below 1000°C. The decomposition of methylenimine to HNC and H2 (Eqn 3) has a much smaller calculated activation barrier of 91 kcal mol–1 — close to the amount of chemical activation available (89 ± 5 kcal mol–1; see Fig. 1) — which raises the question why HNC has not been observed.[26,27] In fact, it has long been known that matrix photolysis of methylenimine at 4 K affords mainly HNC with little HCN.[28] A likely explanation is that HNC is in fact formed in the FVT reactions, but it too is chemically activated and rearranges rapidly to HCN.[27,29] The barrier for this process (Eqn 4) is ~30 kcal mol–1,[30] and the enthalpy of formation difference between HNC and HCN is close to 14 kcal mol–1.[30,31] This means that the thermal decomposition of methyl azide under FVT conditions can yield chemically activated CH2=NH and chemically activated HNC, which finally isomerizes to HCN (Eqns 1–4). The tautomerization of HNC to HCN will also be facilitated by collisions with the walls of the pyrolysis tube in FVT experiments. For the same reason all our attempts to observe the millimeterwave spectrum of some other unstable and easily tautomerizable molecules such as HOCN and H2N-C≡C-H failed because of wall-catalyzed isomerization to HNCO and HN=C=CH2/CH3CN,[32] respectively.
The formation of singlet methylnitrene and its isomerization to methylenimine were revealed in an investigation of the collision-free photolysis of azidomethane.[33] The hot methylenimine underwent two distinct decay reactions, one to HNC via 1,1-H2 elimination, and one to HCN via sequential elimination of H atoms, first from the NH function. Much more energy is available in these photochemical reactions, and elimination of H atoms is not likely to be competitive under FVT conditions. Also, the observation of distinct pathways for HNC and HCN formation in the photolysis does not preclude the very favourable HNC → HCN isomerization under FVT conditions (Eqn 4).
Ab initio trajectory calculations also predicted H2 elimination to produce HNC and both molecular (H2 elimination) and radical (H atom elimination) routes to HCN.[34]
Phenyl Azide – Phenylnitrene – Cyanocyclopentadiene
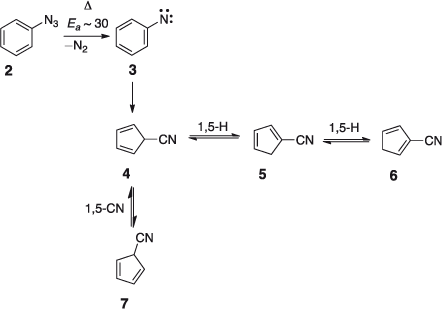
The open-shell singlet phenylnitrene S1 3 has been characterized spectroscopically.[35] It lies ~15 kcal mol–1 above the ground state triplet To 3.[36] FVT of phenyl azide 2 at high temperatures affords cyanocyclopentadiene 5; high temperature is necessary because under milder conditions the nitrene is deactivated to the triplet ground state, which yields azobenzene and aniline as the major products.[37] The activation energy for denitrogenation of phenyl azide in solution has been reported as 32.6[38] or 30.0[39] kcal mol–1. The activation barrier for ring contraction of the open shell singlet S1 phenylnitrene in the gas-phase is calculated as 32 kcal mol–1 at the CASPT2 level (see Scheme 1 and Fig. 2).[40]
|
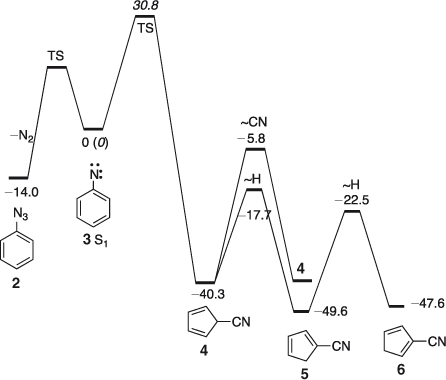
|
The 5-cyano-5H-cyclopentadiene 4 initially formed lies ~40 kcal mol–1 below the S1 nitrene and ~71 kcal mol–1 below the transition state for ring contraction. Therefore 4 can be activated by as much as 71 kcal mol–1. This is far more than the energy required for cycloperambulation in the cyanocyclopentadiene (23–27 kcal mol–1 for H-shifts and ~34 kcal mol–1 for CN-shifts calculated at the B3LYP/6–31G* level) (see Scheme 1 and Fig. 2). Labelling experiments have demonstrated that, in fact, all ring carbon atoms in cyanocyclopentadiene become completely scrambled due to these rapid sigmatropic shifts of H and CN.[41]
Napthyl Azides – Naphthylnitrenes
A mixture of 2- and 3-cyanoindenes 17 and 16 is obtained on FVT of 1- and 2-naphthyl azides 8 and 22 as well as the triazoles and tetrazoles 10, 11, 21, and 24 at 340–400°C (Scheme 2). The triazoles and tetrazoles have been shown to be precursors of quinolyl- and isoquinolylcarbenes, which rearrange to the 1- and 2-naphthylnitrenes via ring expansion to cyclic carbodiimides 12 and 19.[42] The nitrenes finally undergo ring contraction to the cyanoindenes (Scheme 2).
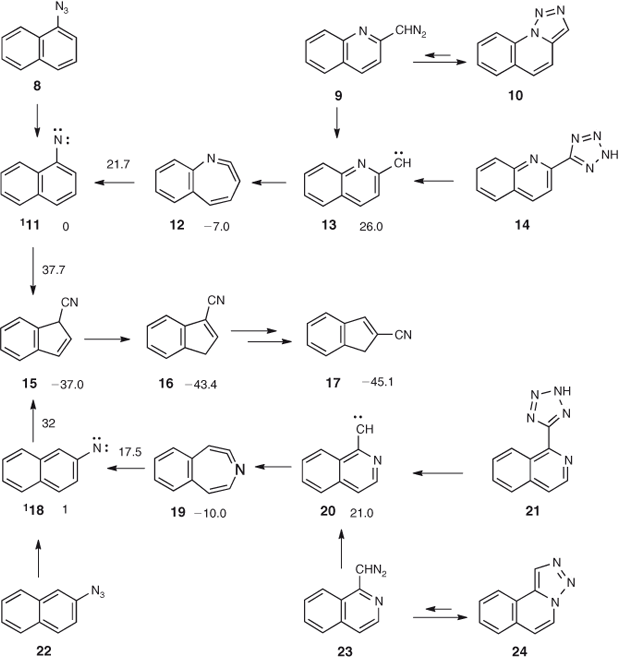
|
Experiments at different temperatures and pressures reveal that the product ratios are strongly affected by chemical activation. The calculated energies of 3- and 2-cyanoindene 16 and 17 are 36 and 38 kcal mol–1 below the cyclic ketenimine 12, respectively (B3LYP/6–31+G* energies).[43] With calculated activation energies for ring contraction of 32–38 kcal mol–1 for the two nitrenes, the initially formed 3-cyano-3H-indene 15 may be activated by as much as 75 kcal mol–1 and the 3-cyanoindene itself by 81 kcal mol–1 (see Scheme 3), regardless whether they are generated from the azides, the triazoles, or the tetrazoles. This CA will cause interconversion of the cyanoindenes by means of sigmatropic shifts of H and CN (Scheme 3). The highest activation barrier in this scheme, ~53 kcal mol–1 above the energy of 3-cyanoindene 16, corresponds to the 1,5-shift of CN, from C3 to C2, so even at this transition state there are still at least 25 kcal mol–1 of excess energy available. 2-Cyanoindene 17 is a little lower in energy than 3-cyanoindene, so under conditions of reversibility, 2-cyanoindene will be the major product, and this is exactly what is found experimentally, from all precursors, on FVT at 10–3 hPa. However, when N2 is used as a carrier gas at 1 Torr, 3-cyanoindene becomes the principal ring contraction product in all cases due to collisional deactivation, so it can be concluded that 3-cyanoindene is the primary product. Since the cyanoindenes have more degrees of freedom than cyanocyclopentadiene 5 (3N – 6 = 48 and 30 vibrational modes, respectively), they are able to distribute some excess energy more readily; consequently it becomes easier to deactivate the cyanoindenes than cyanocyclopentadiene collisionally. Furthermore, the isomerization of the cyanoindenes has to go through non-aromatic ortho-quinonoid intermediates (Scheme 3, Fig. 3), which raise the activation barriers compared with cyanocyclopentadiene. When pure, isolated samples of 2- or 3-cyanoindenes are subjected to FVT (i.e. in the absence of chemical activation), their interconversion requires much higher temperatures (800–1000°C).[44,45]
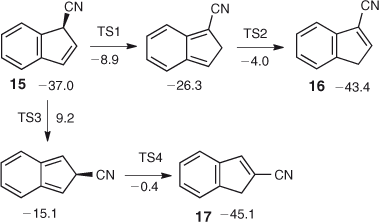
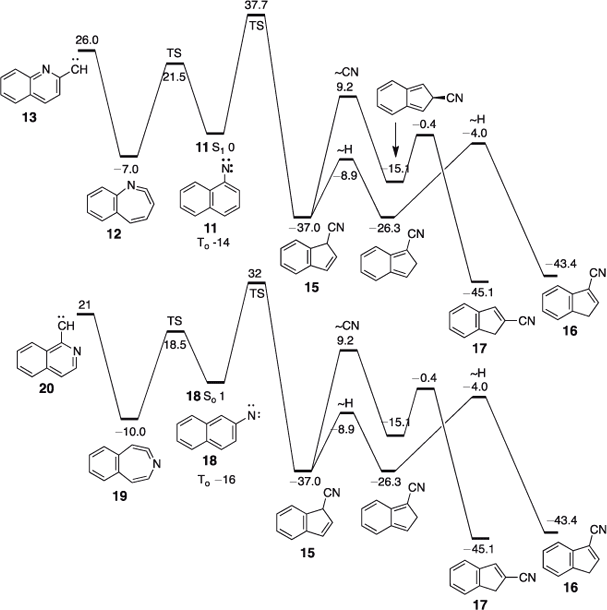
|
Note also that, as usual,[37,46] the singlet carbenes are of higher energy than the isomeric, open-shell singlet nitrenes (by 20–26 kcal mol–1 in Scheme 3).
3-Pyridylnitrene – 2- and 4-Pyrimidinylcarbenes – Cyanopyrroles
FVT of 3-azidopyridine 25 affords a mixture of 3- and 2-cyanopyrroles 30 and 31, with the 3-cyanopyrrole isomer dominating.[45,47] For the S1 3-pyridylnitrene 26 the lowest energy route to ring contraction is via ring opening to the nitrile ylide 28,[48] which has been observed directly (Scheme 4, Fig. 4).[49] Ylide 28 may be formed either directly from the nitrene or via the cyclic ketenimine 27 (Scheme 4). There is only a small, calculated barrier of 14–16 kcal mol–1 for the ring opening of either the nitrene or the ylide, and another small barrier of 10.2 kcal mol–1 for the ring closure of the ylide to form 3-cyano-3H-pyrrole 29 (Fig. 4). Nevertheles, this 3-cyano-3H-pyrrole will carry CA to the extent of ~51 kcal mol–1 – sufficient for the subsequent interconversion of the two cyanopyrroles by 1,5-shifts of H and CN, which have calculated barriers of 23–33 kcal mol–1 relative to 29 (Fig. 4 and Scheme 5).[50]
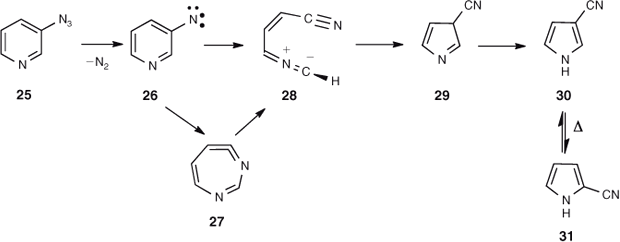
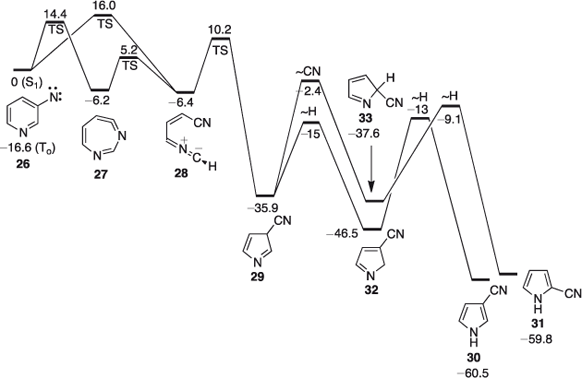
|
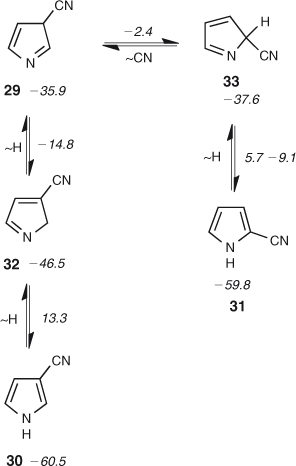
|
FVT of 2-(5-tetrazolyl)pyrimidine 34 also affords a mixture of 2- and 3-cyanopyrroles (Scheme 6). Similarly, the tetrazoles 35 and 36 afford mixtures of three principal dimethylcyanopyrroles 37–39 (Scheme 6). Here too, the formation of mixtures of cyanopyrroles is ascribed to chemical activation, which causes sigmatropic shifts of H, CN, and CH3 in the product pyrroles. Cyanopyrroles often act as ‘thermodynamic sinks’, so they can be expected as final products even though they may be formed by different mechanisms from different types of precursor (see below).
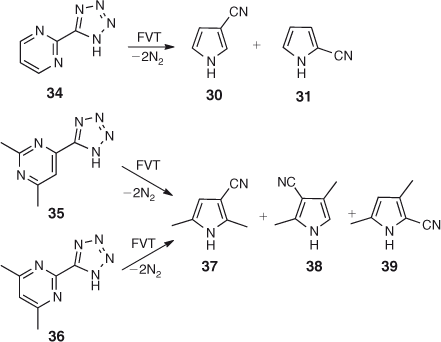
First, the pyrimidinylcarbenes generated by double N2 loss from the tetrazoles rearrange to 3-pyridylnitrenes 42 and 46 via ring expansion to cyclic carbodiimides 41 and 45 (Scheme 7). Second, the pyridylnitrenes and/or the seven-membered ring carbodiimides undergo ring contraction. As before, the lowest energy pathways proceed via the nitrile ylides 43 and 48 (Scheme 7). The initially formed 3-cyano-3H-pyrroles 44 and 49 can be activated by ~46 kcal mol–1, but, in addition, because the carbenes are higher in energy than the nitrene,[37,46] the reactions are easier when starting from the tetrazoles and afford good yields of the pyrroles at 400°C/10–3 hPa. Mixtures of 37, 38, and 39 are invariably obtained from both tetrazole precursors under these conditions due to the sigmatropic shifts of H, CN, and CH3. However, already a pressure increase to 1 hPa N2 leads to more distinct product mixtures, with dimethylcyanopyrrole 37 as the primary reaction product of 35, and the isomeric dimethylcyanopyrroles 38 and 39 as the primary products from 36. FVT of the individual dimethylcyanopyrroles 37 and 38 requires a temperature of 800°C for isomerization to occur (see section on benzotriazoles below).[51,52]

Tetrazolo[1,5-a]pyridine – 2-Azidopyridine – 2-Pyridylnitrene
Tetrazolo[1,5-a]pyridine 50 is calculated to be 4 kcal mol–1 lower in energy than the anti-2-azidopyridine 51 and 0.4 kcal mol–1 below the syn-2-azidopyridine 52. The azide forms cannot be detected in solution at room temperature but are readily formed on mild FVT at 100–200°C and isolated at low temperatures.[53] The azide cyclizes back to the tetrazole at ~-10°C. FVT of the tetrazole/azide mixture at 370–500°C causes decomposition to the open-shell singlet S1 2-pyridylnitrene 53 (Scheme 8). By analogy with phenyl azide and 3-pyridyl azide (see above), the activation energy for decomposition of the azide is expected to be 30–32 kcal mol–1.
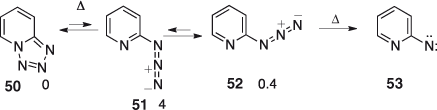
|
Three different routes to cyanopyrroles from 2-pyridylnitrene 53 have been identified,[50] whereby the overall lowest energy path proceeds via ring opening to the biradical/vinylnitrene 56 (Scheme 9). Depending on the route and the method of calculation (B3LYP and CASPT2), CA will be present to the extent of at least 52 kcal mol–1 in the initially formed 2-cyano-2H-pyrrole 33, or ~74 kcal mol–1 in the 2-cyanopyrrole 31 itself (Scheme 9).[50] Not surprisingly therefore, the principal reaction product is again a mixture of 2- and 3-cyanopyrroles, even under the mildest possible conditions (400–500°C/10–3 hPa) (Scheme 9).[50,54]
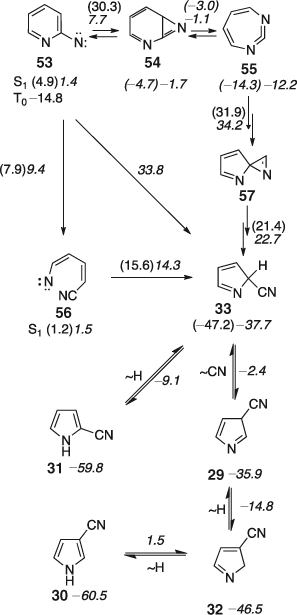
|
6-Nitrotetrazolo[1,5-a]pyridine – 2-Azido-5-nitropyridine – 5-Nitro-2-pyridylnitrene
The C–NO2 bond dissociation energy in nitrobenzene has been determined as 70 ± 3 kcal mol–1 (log A = 17.0 ± 0.5),[55] and this has been supported by a laser-powered homogeneous pyrolysis experiment[56] and by calculations using several different methods.[57] Fields and Meyerson generated phenyl radicals by pyrolysis of nitrobenzene at 600°C,[58] but this was in a flow reactor with contact times of the order of 2–30 s (typically 9 s), which are ~1000 times longer than those typically used in FVT experiments. Therefore, one does not expect the C–NO2 bond to break under FVT conditions below 700–1000°C.
In agreement with this, FVT of 3-(p-nitrobenzyl)benzo[c]cinnoline at 900°C/0.02 hPa (contact time ~10 ms) only gave products resulting from loss of the NO2 group (phenanthrene, anthracene, and phenylacetylene).[59] In contrast, FVT of 4-nitro-2,5-dimethylisoxazole in N2 flow at 400°C (0.1 hPa) afforded 1-cyano-1-nitroacetone in quantitative yield, i.e. without any loss of the NO2 group.[60]
In contrast to tetrazolo[1,5-a]pyridine 50 (Scheme 8), 6-nitrotetrazolo[1,5-a]pyridine 58T exists in equilibrium with a small amount of the azide valence isomer 58A at room temperature.[61] Chollet observed that the FVT of this material at 400°C/10–3 hPa afforded a 15–20 % yield of 2-cyanopyrrole 31 as well as NO2 as the only low molecular weight products (Scheme 10).[62] NO2 was identified by its IR spectrum[63] in the gas-phase. Loss of NO2 is not expected at such a low temperature under FVT conditions. Therefore, the loss of NO2 and the fact that only 2-cyanopyrrole – not 3-cyanopyrrole – was identified, suggest that CA caused the breaking of the C–NO2 bond to form a 2-cyanopyrrolyl radical, which by hydrogen abstraction afforded the observed product. The 2-cyanopyrrole so formed will no longer be chemically activated and therefore does not undergo isomerization to 3-cyanopyrrole.
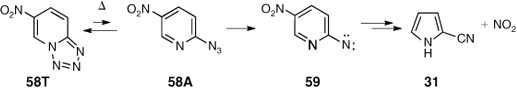
In contrast, the nitro group remains intact on FVT of 4-azido-7-nitro-2-phenylquinazoline/ 8-nitro-5-phenyltetrazolo[1,5-c]quinazoline 60A/60T at 400–500°C to yield 5-nitro-2-phenylbenzimidazole-1-carbonitrile 64 quantitatively (Scheme 11).[64] The large molecule with 78 fundamental vibrational modes allows for more efficient redistribution of the excess energy and hence no apparent effect of CA, even though the reaction product will carry some 70 kcal mol–1 of excess energy originating from the ring expansion of the S1 nitrene 61. The reaction also takes place photochemically, where it has been shown to proceed via the directly observed and spectroscopically characterized intermediates 62 and 63 (Scheme 11).
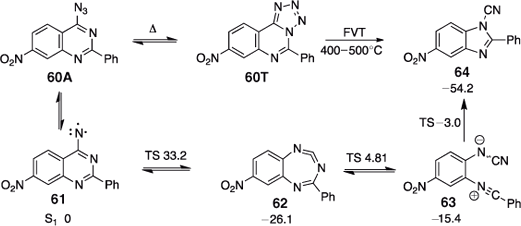
4-Azidopyridines and 4-Pyridylnitrenes
FVT of 4-azidopyridine 65 also affords a mixture of 3- and 2-cyanopyrroles 30 and 31 (Fig. 5). Here, the 3-isomer is expected to be the first-formed, but CA of ~68 kcal mol–1 ensures that a mixture is obtained.[65] The concerted mechanism for ring contraction (Fig. 5) is analogous to that proposed for phenylnitrene above, and the calculated amount of CA is also very similar (compare Figs. 2 and 5). Note that much less CA is generated in the reaction of 3-pyridylnitrene 26 (~51 kcal mol–1, Fig. 4) because this nitrene has an especially favourable pathway open, namely the ring opening to the nitrile ylide, which can only take place when there is a meta-relationship between a nitrene or carbene centre and a ring nitrogen atom.[48]
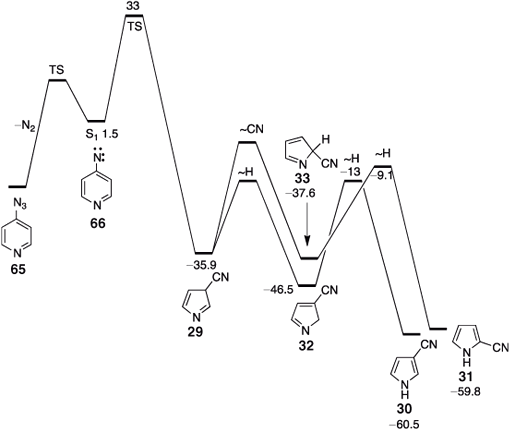
|
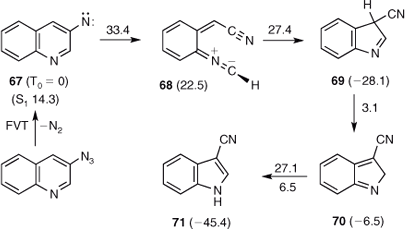
It is noteworthy that 3-azidoquinoline affords 3-cyanoindole 71 cleanly and in high yield without any appreciable isomerization to 2-cyanoindole.[66] The reaction mechanism (Scheme 12) has been elucidated under matrix-isolation photolysis conditions, and the same mechanism is expected to apply under FVT conditions. The calculated energies indicate that there would be enough energy available for isomerization, namely ~60 kcal mol–1 for the 3-cyano-3H-indole 69 arising from ring opening/ring contraction in 3-quinolylnitrene 67 (Scheme 12). The indoles have many more degrees of freedom than the pyrroles over which to dissipate the excess vibrational energy, with the result that the effects of CA are not apparent.
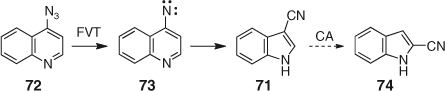
Similarly, 4-quinolylnitrene 73 yields nearly pure 3-cyanoindole 71 on FVT with matrix isolation of the product, but partial rearrangement to 2-cyanoindole 74 is observed under preparative FVT conditions (Scheme 13). The mechanism is similar to that described for 1-naphthylnitrene 11 and 4-pyridylnitrene 66 above (Figs. 3 and 5). The CA causing partial isomerization to 2-cyanoindole can again be removed by increasing the pressure.[67]
|
Triazolopyridines and Benzotriazoles
Interestingly, and synthetically usefully, the pure, unisomerized 2- and 3-cyanopyrroles 31 and 30 can be obtained in nearly quantitative yields by FVT of triazolopyridines 75 and 77, respectively, at 600°C (Scheme 14).[52] Benzotriazole (79a) similarly gives cyanocyclopentadiene 81a in virtually quantitative yield (Scheme 14).
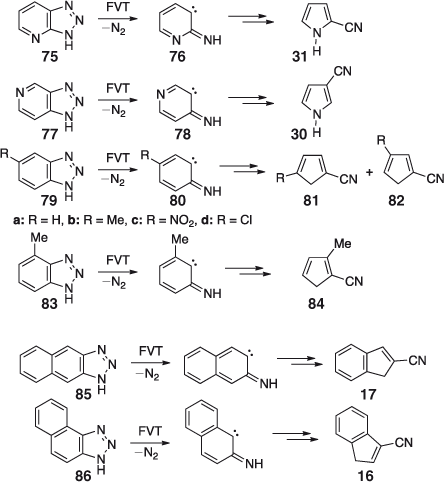
Unisomerized methylcyanocyclopentadienes 81b and 84 are obtained from the corresponding 4- and 5-methylbenzotriazoles 79b and 83 at 500°C (a small amount of the tautomer 82b accompanies 81b) under mild conditions (500°C) and with a low degree of conversion, but interconversion of the nitriles takes place at 600–700°C.[52] Naphtho[4,5-a]- and naphtho[4,5-b]triazoles 85 and 86 yield the respective cyanoindenes 17 and 16 on FVT at 500–600°C (Scheme 14), but interconversion of these nitriles does take place on FVT at 700–1000°C due to thermally activated substituent migration.[52] All these reactions are highly exothermic; the initially formed ketenimine can carry ~50 kcal mol–1 excess energy in the benzotriazole case, and 60 kcal mol–1 in the triazolopyridine case as illustrated in Scheme 15. The final products, cyanocyclopentadiene 5 and 3-cyanopyrrole 30 are even more highly activated, by 70 and 90 kcal mol–1, respectively. As clearly revealed in the examples described above, this would be more than sufficient to isomerize a cyanopyrrole or cyanocyclopentadiene. The apparent absence of CA under mild FVT conditions is ascribed to the fact that the initial products are not nitriles but ketenimines 87 and 88, formed in a Wolff-type rearrangement.[68] The ketenimines cannot easily undergo any sigmatropic rearrangements. The tautomerization to cyanopyrroles is likely to take place during workup and/or during collisions with the walls of the pyrolysis tube, which would help dissipate the excess energy.
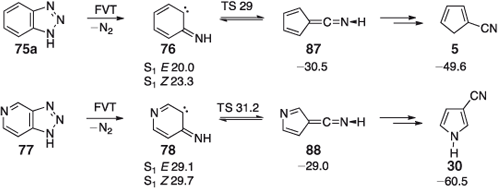
|
That chemical activation is nevertheless present is revealed by the nitro-group effect: FVT of the NO2-substituted benzotriazole (79c) leads to loss of the NO2 group, and even in the case of the chloro-substituted benzotriazole (79d) there is a 20 % loss of Cl under the mildest possible conditions for reaction to occur (550°C/0.1 hPa). In contrast, the stronger F–C bond stays intact.[52]
Conclusion
Chemical activation (CA) is ubiquitous in azide gas-phase chemistry because the nitrenes are high-energy intermediates, and their reactions are highly exothermic. Whereas the nitrenes usually have triplet ground states (To), preparatively interesting rearrangements take place in the singlet nitrenes (S1). The effects of CA is observed in the isomerization of methylnitrene CH3N to methylenimine (methanimine) CH2=NH, where it causes elimination of hydrogen to form HCN or HNC already at FVT temperatures above 500°C, whereas a temperature above 1000°C is required in the absence of CA. FVT of the relevant aryl azides at 400–500°C generates phenylnitrene, 1- and 2-naphthylnitrenes, and 2-, 3-, and 4-pyridylnitrenes, which undergo ring contraction to cyanocyclopentadiene, 3- and 2-cyanoindenes, and 2-and 3-cyanopyrroles, respectively; CA causes facile interconversion of isomers by means of sigmatropic shifts of both H and CN (and CH3 when present), so that mixtures of nitriles are invariably obtained in each case. However, CA can be reduced or removed entirely by increasing the pressure, thereby promoting collisional deactivation. Using N2 or Ar as carrier gas at a pressure of 1 hPa is usually sufficient to remove most of the CA. Because larger molecules have more degrees of freedom, they are better able to dissipate the excess energy, with the result that chemical activation is less apparent or completely absent in the formation of 3-cyanoindole and 1-cyanobenzimidazoles from 3- and 4-quinolylnitrenes and 4-quinazolinylnitrenes, respectively. In compounds possessing nitro groups, chemical activation can cause the loss of the nitro group at nominal temperatures far below those normally needed to cleave the C–NO2 bond, as seen in the example of 5-nitro-2-pyridylnitrene.
Supplementary Material
Details of the computational methods used and general experimental procedures are available on the Journal’s website.
Acknowledgements
This work was generously supported by The University of Queensland and by the National Computing Infrastructure facility (NCMAS g01) supported by the Australian Government. I am indebted to my students and co-workers, whose names are given in the references, particularly to Dr David Kvaskoff for computational work.
References
[1] (a) P. J. Robinson, K. A. Holbrook, Unimolecular Reactions 1972 (Wiley: New York, NY).(b) C. Wentrup, Reactive Molecules 1984, pp. 225–230 (Wiley-Interscience: New York, NY).
(c) B. S. Rabinovitch, M. C. Flowers, Q. Rev. Chem. Soc. 1964, 18, 122.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) N. Balucani, L. Cartechini, M. Alagia, P. Casavecchia, G. G. Volpi, J. Phys. Chem. A 2000, 104, 5655.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXktVaqt7s%3D&md5=fe2639c706a8348fd5ca201420159f85CAS |
(b) H. Umemoto, N. Terada, K. Tanaka, J. Chem. Phys. 2000, 112, 5762.
| Crossref | GoogleScholarGoogle Scholar |
[3] R. Hoye, B. Baire, D. Niu, P. H. Willoughby, B. P. Woods, Nature 2012, 490, 208.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVyrsrzN&md5=de90c3896fd219becf9a5363e7b33ea7CAS |
[4] R. W. Hoffmann, K. Suzuki, Angew. Chem. Int. Ed. 2013, 52, 2655.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXntFeqsQ%3D%3D&md5=448912fbf0676ce7c82df60dfe940746CAS |
[5] M. J. Travers, D. C. Cowles, E. P. Clifford, G. B. Ellison, P. C. Engelking, J. Phys. Chem. 1999, 111, 5349.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXls1Cntbo%3D&md5=a75bb80866448f5fe7331a62389ae2a5CAS |
[6] C. R. Kemnitz, C. B. Ellison, W. L. Karney, W. T. Borden, J. Am. Chem. Soc. 2000, 122, 1098.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXms1Wnsw%3D%3D&md5=887dfffaeb3081a98a2d2b557e130121CAS |
[7] C. Richards, C. Meredith, S.-J. Kim, G. E. Quelch, H. F. Schaefer, J. Chem. Phys. 1994, 100, 481.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhtVKiu7k%3D&md5=26437af170cd519440f7eb591bba1598CAS |
[8] (a) J. A. Leermakers, J. Am. Chem. Soc. 1933, 55, 3098.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA3sXlsFOisA%3D%3D&md5=83279f483547d8d910c45a0a24a68901CAS |
(b) F. O. Rice, C. J. Grelecki, J. Phys. Chem. 1957, 61, 830.
| Crossref | GoogleScholarGoogle Scholar |
(c) W. Pritzkow, D. Timm, J. Prakt. Chem. 1966, 32, 178.
| Crossref | GoogleScholarGoogle Scholar |
[9] (a) C. L. Currie, B. DeB. Darwent, Can. J. Chem. 1963, 41, 1552.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3sXktVWis78%3D&md5=2f3fef28eb82813dad79bbd28a95d738CAS |
(b) M. S. O’Dell, B. DeB. Darwent, Can. J. Chem. 1970, 48, 1140.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. Koch, Tetrahedron 1967, 23, 1747.
| Crossref | GoogleScholarGoogle Scholar |
[10] C. Wentrup, C. O. Kappe, M. W. Wong, Pure Appl. Chem. 1995, 67, 749.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXlvVOqs7c%3D&md5=bd21ac686f2847f3a0485f159207a3fcCAS |
[11] (a) Azides and Nitrenes (Ed. E. F. V. Scriven) 1984 (Academic Press: Orlando, FL).
(b) S. Bräse, K. Banert, Organic Azides – Synthesis and Applications 2008 (Wiley: Chichester, UK).
(c) Nitrenes and Nitrenium Ions (Eds D. E. Falvey, A. D. Gudmundsdottir) 2013 (Wiley: Hoboken, NJ).
[12] S. Fischer, Diploma Thesis: Darstellung von Nitriliminen durch thermische Zersetzung von N-Heterocyclen und Untersuchung ihrer Stabilisierungsreaktionen in Lösung und in der Gasphase 1980, Philipps-Universität Marburg, Germany.
[13] S. Fischer, C. Wentrup, M. Winnewisser, DFG Schwerpunktprogramm Erzeugung und Stabilisierung reaktiver anorganischer Moleküle, Bad Honnef, Germany, 18–19 Oct 1982. Available at https://www.researchgate.net/publication/234051408_Identification_of_Methylenimine_in_the_Thermolysis_of_Methyl_Azide._Chemical_Activation_Causing_Fragmentation_to_H2_and_HCN (accessed 1 June 2013).
[14] C. Wentrup, Gas-Phase and Matrix Studies, in Azides and Nitrenes (Ed. E. F. V. Scriven) 1984, pp. 396–399 (Academic Press: Orlando, FL).
[15] M. Winnewisser, B. P. Winnewisser, Z. Naturforsch. C 1974, 29a, 633.
[16] (a) The microwave spectrum of CH2=NH was first obtained by H-abstraction from methylamine and by pyrolysis of methylamine and ethane-1,2-diamine at 900–1000°C/~10–2 hPa: D. R. Johnson, F. J. Lovas, Chem. Phys. Lett. 1972, 15, 65.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE38XltVOgsL0%3D&md5=b7b099ce3b7b69a66128b2b7fae73b74CAS |
(b) R. Pearson, F. J. Lovas, J. Chem. Phys. 1977, 66, 4149.
| Crossref | GoogleScholarGoogle Scholar |
(c) This method permitted the identification of methylenimine as an interstellar molecule in Sagittarius B2: P. D. Godfrey, R. D. Brown, B. J. Robinson, M. W. Sinclair, Astrophys. Lett. 1973, 13, 119.
(d) See also: L. Dore, L. Bizzocchi, C. Degli Esposti, Astron. Astrophys. 2012, 544, A19.
| Crossref | GoogleScholarGoogle Scholar |
[17] Millimeterwave spectrum of HCN: F. DeLucia, W. Gordy, Phys. Rev. 1969, 187, 58.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3cXhslWktw%3D%3D&md5=c940e7a97b2082087103a940a7b52cf9CAS |
[18] (a) Activation energy for MeN3 decomposition: 37 kcal mol–1: J. L. Franklin, V. H. Dibeler, R. M. Reese, M. Krauss, J. Am. Chem. Soc. 1958, 80, 298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG1cXjvV2nsQ%3D%3D&md5=99d66c35ee4d3013ced7d6334621488dCAS |
(b) 40.5 kcal mol–1: Ref [8b].
(c) 38.4 kcal mol–1: C. C. Chen, M. J. McQuaid, J. Phys. Chem. A 2012, 116, 3561.
| Crossref | GoogleScholarGoogle Scholar |
[19] (a) Enthalpy of formation of CH3N3: 67 kcal mol–1: W. Benson, F. R. Cruickshank, D. M. Golden, G. R. Haugen, H. E. O’Neal, A. S. Rogers, R. Shaw, R. Walsh, Chem. Rev. 1969, 69, 279.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF1MXktleqtb0%3D&md5=8dc9f9c7dd20967d7649a610115cb6eaCAS |
(b) 71.0 kcal mol–1: D. W. Rogers, F. J. McLafferty, J. Chem. Phys. 1995, 103, 8302.
| Crossref | GoogleScholarGoogle Scholar |
(c) 72.2–73.8 kcal mol–1: M. J. McQuaid, B. M. Rice, Computational Chemistry-Based Enthalpy-of-Formation, Enthalpy-of-Vaporization, and Enthalpy-of-Sublimation Predictions for Azide-Functionalized Compounds 2006, Army Research Laboratory ARL-TR-3770. Available at http://www.arl.army.mil/arlreports/2006/ARL-TR-3770.pdf (accessed 1 June 2013).
[20] (a) Enthalpy of formation of CH2NH:26 kcal mol–1: D. J. DeFrees, W. J. Hehre, J. Phys. Chem. 1978, 82, 391.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1cXht1Snsrg%3D&md5=1243e89559db78b2e62f119ab054f858CAS |
(b) 21± 4 kcal mol–1: J. L. Holmes, F. P. Lossing, P. M. Mayer, Chem. Phys. Lett. 1992, 198, 211.
| Crossref | GoogleScholarGoogle Scholar |
(c) 21.1 ± 0.5 kcal mol–1: G. de Oliveira, J. L. Martin, I. K. C. Silwal, J. F. Liebman, J. Comput. Chem. 2001, 22, 1297.
| Crossref | GoogleScholarGoogle Scholar |
[21] (a) For alternative preparations, low temperature isolation and NMR characterization of CH2NH see: J.-C. Guillemin, J.-M. Denis, Angew. Chem. Int. Ed. 1982, 21, 690.
(b) J.-C. Guillemin, J.-M. Denis, Angew. Chem. Suppl. 1982, 1515.
| Crossref | GoogleScholarGoogle Scholar |
(c) J.-C. Guillemin, J.-M. Denis, J. Chem. Soc., Chem. Commun. 1985, 951.
| Crossref | GoogleScholarGoogle Scholar |
(d) J.-C. Guillemin, J.-M. Denis, M. Bogey, J. L. Destombes, Tetrahedron Lett. 1986, 27, 1147.
| Crossref | GoogleScholarGoogle Scholar |
[22] R. D. Brown, P. D. Godfrey, D. A. Winkler, Aust. J. Chem. 1982, 35, 667.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XktlSmu70%3D&md5=6560a9299db6c0c9781e2a84e6785287CAS |
[23] (a) H. Bock, R. Dammel, Angew. Chem. Int. Ed. 1987, 26, 504.
| Crossref | GoogleScholarGoogle Scholar |
(b) H. Bock, R. Dammel, J. Am. Chem. Soc. 1988, 110, 5261.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Bock, R. Dammel, L. Horner, Chem. Ber. 1981, 114, 220.
| Crossref | GoogleScholarGoogle Scholar |
(d) For the identification of the photoelectron spectrum of CH3N in this reaction see: J. Wang, Z. Sun, X. Zhu, X. Yang, M. Ge, D. Wang, Angew. Chem. Int. Ed. 2001, 40, 3055.
| Crossref | GoogleScholarGoogle Scholar |
[24] (a) A. Teslja, B. Nizamov, P. J. Dagdigian, J. Phys. Chem. A 2004, 108, 4433.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjsVSksL8%3D&md5=7fe91acec07e1f64526c178364f95155CAS |
(b) D. M. Wong, P. J. Dagdigian, Spectrochim. Acta A 2007, 67, 1019.
| Crossref | GoogleScholarGoogle Scholar |
[25] (a) J. Demuynck, D. J. Fox, Y. Yamaguchi, H. F. Schaefer, J. Am. Chem. Soc. 1980, 102, 6204.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXls1OnsrY%3D&md5=750db326cb53a1dbe5bcb9108161a0e9CAS |
(b) J. A. Pople, K. Raghavachari, M. J. Frisch, J. S. Binkley, P. v. R. Schleyer, J. Am. Chem. Soc. 1983, 105, 6389.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. T. Luke, J. A. Pople, M.-B. Krogh-Jespersen, Y. Apeloig, M. Kami, J. Chandrasekhar, P. v. R. Schleyer, J. Am. Chem. Soc. 1986, 108, 270.
| Crossref | GoogleScholarGoogle Scholar |
[26] (a) J. F. Arenas, J. I. Marcos, J. C. Otero, A. Sanchez-Galvez, J. Soto, J. Chem. Phys. 1999, 111, 551.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXktVShtL0%3D&md5=6a1b0a4274547d59ce58e4e3fc15e0b7CAS |
(b) J. F. Arenas, J. I. Marcos, J. C. Otero, I. L. Tocon, J. Soto, Int. J. Quantum Chem. 2001, 84, 241.
| Crossref | GoogleScholarGoogle Scholar |
(c) See also: M. Besora, J. N. Harvey, J. Chem. Phys. 2008, 129, 044303.
| Crossref | GoogleScholarGoogle Scholar |
[27] D. W. McPherson, M. L. McKee, P. B. Shevlin, J. Am. Chem. Soc. 1983, 105, 6493.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXlsVKrtb4%3D&md5=0059838871b41c72c0d99281534a61a0CAS |
[28] (a) D. E. Milligan, M. E. Jacox, J. Chem. Phys. 1963, 39, 712.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3sXkt1Gqsbc%3D&md5=51a0eb1fa4813df91371cec3893b2449CAS |
(b) D. E. Milligan, M. E. Jacox, J. Chem. Phys. 1967, 47, 278.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. E. Jacox, D. E. Milligan, J. Mol. Spectrosc. 1975, 56, 333.
| Crossref | GoogleScholarGoogle Scholar |
[29] M. T. Nguyen, D. Sengupta, T.-K. Ha, J. Phys. Chem. 1996, 100, 6499.
| Crossref | GoogleScholarGoogle Scholar |
[30] (a) M. T. Nguyen, P. J. Groarke, S. Malone, F. Hegarty, J. Chem. Soc., Perkin Trans 2 1994, 807.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXivFOku7k%3D&md5=8ec0e107b3fa4af8e67143fa75364ffbCAS |
(b) T. Barger, A. M. Wodtke, J. M. Bowman, Astrophys. J. 2003, 587, 841.
| Crossref | GoogleScholarGoogle Scholar |
[31] Enthalphy of formation of HNC = 40 ± 1 kcal mol–1: A. Hansel, C. Schering, M. Glantschnig, W. Lindinger, E. E. Ferguson, J. Chem. Phys. 1998, 109, 1748.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkslegsLo%3D&md5=94ce32247ec8b8c61b5387cfd178b525CAS |
[32] C. Wentrup, H. Briehl, P. Lorencak, U. J. Vogelbacher, H. W. Winter, A. Maquestiau, R. Flammang, J. Am. Chem. Soc. 1988, 110, 1337.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXhtFWkt7Y%3D&md5=12b2720113c4d3993810dce8118d6d0fCAS |
[33] (a) C. Larson, Y. Ji, P. Samartzis, A. M. Wodtke, S.-H. Lee, J. J.-M. Lin, C. Chaudhuri, T.-T. Ching, J. Chem. Phys. 2006, 125, 133302.
| Crossref | GoogleScholarGoogle Scholar | 17029455PubMed |
(b) A. Quinto-Hernandez, J. Doehla, W.-T. Huang, C.-Y. Lien, W.-Y. Lin, J. J.-M. Lin, A. M. Wodtke, J. Phys. Chem. A 2012, 116, 4695.
| Crossref | GoogleScholarGoogle Scholar |
[34] J. Zhou, H. B. Schlegel, J. Phys. Chem. A 2009, 113, 9958.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVSgtb3M&md5=9e5886e1796da7574e52d0cd9770622dCAS | 19739680PubMed |
[35] N. P. Gritsan, M. S. Platz, Chem. Rev. 2006, 106, 3844.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XosVChsbg%3D&md5=829955a7a2e71dbaf892920fe79e3782CAS | 16967923PubMed |
[36] (a) M. J. Travers, D. C. Cowels, E. P. Clifford, G. B. Ellison, J. Am. Chem. Soc. 1992, 114, 8699.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XlvFWhs70%3D&md5=075a825008cf0b65a8fc94880b748fceCAS |
(b) N. R. Wijeratne, M. DaFonte, A. Ronemus, P. J. Wyss, D. Tahmassebi, P. G. Wenthold, J. Phys. Chem. A 2009, 113, 9467.
| Crossref | GoogleScholarGoogle Scholar |
[37] C. Wentrup, Top. Curr. Chem. 1976, 62, 173.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1cXls1Wg&md5=e2cf1d56d9298e8b1d7c5eb16d165b63CAS | 941142PubMed |
[38] L. K. Dyall, Aust. J. Chem. 1975, 28, 2147.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXlvFagsr4%3D&md5=62dc524231f0471c3c285fa64755d48bCAS |
[39] L. K. Dyall, P. A. S. Smith, Aust. J. Chem. 1990, 43, 997.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXkvFKru74%3D&md5=199674a377c1a3c81eb6b4fcc7630548CAS |
[40] D. Kvaskoff, P. Bednarek, L. George, S. Pankarakshan, C. Wentrup, J. Org. Chem. 2005, 70, 7947.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXpt1WhsLc%3D&md5=a3d270e4ae61d06c5f3b1f336e6b7e90CAS | 16277314PubMed |
[41] C. Thétaz, C. Wentrup, J. Am. Chem. Soc. 1976, 98, 1258.
| Crossref | GoogleScholarGoogle Scholar |
[42] A. Maltsev, T. Bally, M.-L. Tsao, M. S. Platz, A. Kuhn, M. Vosswinkel, C. Wentrup, J. Am. Chem. Soc. 2004, 126, 237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXps1yms7s%3D&md5=85c3e8b6329e2d05e414e20ba34b0b72CAS | 14709089PubMed |
[43] N. M. Lan, R. Burgard, C. Wentrup, J. Org. Chem. 2004, 69, 2033.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXht12gt70%3D&md5=c669af3cc9912e9118ee517c4d23eb49CAS | 15058950PubMed |
[44] C. Wentrup, W. D. Crow, Tetrahedron 1971, 27, 880.
[45] C. Wentrup, N. M. Lan, A. Lukosch, P. Bednarek, D. Kvaskoff, Beilstein J. Org. Chem. 2013, 9, 743.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmvFaqtbc%3D&md5=a40577eef216fc27d612e817b3590fccCAS | 23766786PubMed |
[46] C. R. Kemnitz, W. L. Karney, W. T. Borden, J. Am. Chem. Soc. 1998, 120, 3499.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXit12lurs%3D&md5=e4d613faf7910a8e5723276bf65d88a5CAS |
[47] The activation energy for thermolysis of 3-pyridyl azide in solution is ~30 kcal mol–1: L. K. Dyall, M. W. Wong, Aust. J. Chem. 1985, 38, 1045.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28Xkt1ers7k%3D&md5=f14f6f2e652acadcd29656db9e49a1c1CAS |
[48] C. Wentrup, Acc. Chem. Res. 2011, 44, 393.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktFegu7s%3D&md5=f78694801b8c0c3c9299b61421afd3d4CAS | 21452850PubMed |
[49] P. Bednarek, C. Wentrup, J. Am. Chem. Soc. 2003, 125, 9083.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXltFKqsLs%3D&md5=889269d61a7333254de41001258187b8CAS | 15369365PubMed |
[50] D. Kvaskoff, P. Bednarek, C. Wentrup, J. Org. Chem. 2010, 75, 1600.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsF2rtbY%3D&md5=db800b984879029296222f3b95df774cCAS | 20131858PubMed |
[51] C. Wentrup, Tetrahedron 1974, 30, 1301.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2cXltVCnsLc%3D&md5=ef8b7c6b4a4f34ee6664af335d172846CAS |
[52] C. Wentrup, W. D. Crow, Tetrahedron 1970, 26, 3965.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3cXltV2ktr0%3D&md5=9ea56ca730b8e903ba2f03381f78c131CAS |
[53] C. Wentrup, H.-W. Winter, J. Am. Chem. Soc. 1980, 102, 6159.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXlvVKgs74%3D&md5=fe6b25809f7e9ab9f9f749c1d3419c55CAS |
[54] A. McCluskey, C. Wentrup, J. Org. Chem. 2008, 73, 6265.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovVWjt7w%3D&md5=369ee4e2e5eacf8e7db025477c9a00b7CAS | 18646825PubMed |
[55] (a) V. G. Matveev, V. V. Dubikhin, G. M. Nazin, Bull. Acad. Sci. USSR, Div. Chem. Sci. 1978, 27, 474.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. B. Pedley, R. D. Naylor, S. P. Kirby, Thermochemical Data of Organic Compounds 1986, 2nd edn (Chapman and Hall: New York, NY).
[56] K. E. Lewis, D. F. McMillen, D. M. Golden, J. Phys. Chem. 1980, 84, 227.
| Crossref | GoogleScholarGoogle Scholar |
[57] (a) S. Xu, M. C. Lin, J. Phys. Chem. B 2005, 109, 8368.
(b) M. F. Lin, Y. T. Lee, C.-K. Ni, S. Xu, M. C. Lin, J. Chem. Phys. 2007, 126, 64310.
| Crossref | GoogleScholarGoogle Scholar |
(c) G. Fayet, L. Jounert, P. Rotureau, C. Adamo, J. Phys. Chem. A 2008, 112, 4054.
| Crossref | GoogleScholarGoogle Scholar |
(d) C. Qi, Q.-H. Lin, Y.-Y. Li, S.-P. Pang, R.-B. Zhang, J. Mol. Struct. Theochem. 2010, 961, 97.
| Crossref | GoogleScholarGoogle Scholar |
(e) M. L. Hause, N. Herath, R. Zhu, M. C. Lin, A. G. Suits, Nat. Chem. 2011, 3, 932.
| Crossref | GoogleScholarGoogle Scholar |
[58] (a) E. K. Fields, S. Meyerson, J. Am. Chem. Soc. 1967, 89, 724.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2sXnsFOmsQ%3D%3D&md5=d454c7d66594553c6d367e623b9f9389CAS |
(b) E. K. Fields, S. Meyerson, J. Am. Chem. Soc. 1967, 89, 3224.
| Crossref | GoogleScholarGoogle Scholar |
[59] Y. A. Ibrahim, N. A. Al-Awadi, K. Kaul, Tetrahedron 2001, 57, 7377.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXmtFGhu7o%3D&md5=18c68cc441bf1075741d95bdbacd0327CAS |
[60] J. D. Perez, D. A. Wunderlin, J. Org. Chem. 1987, 52, 3637.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXkslOltbk%3D&md5=9976646531f37ff3e4a5d60367544aefCAS |
[61] C. K. Lowe-Ma, R. A. Nissan, W. S. Wilson, J. Org. Chem. 1990, 55, 3755.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXktVKhtr8%3D&md5=f07175d13c4a830a7c0c58ac7da71d0bCAS |
[62] A. Chollet, Diploma Thesis: Contribution à l’Etude des Réarrangements en Phase Gazeuse de Carbènes et de Nitrènes Aromatiques et Hétéroaromatiques 1974, University of Lausanne, Switzerland.
[63] R. H. Pierson, A. N. Fletcher, E. St Clair Gantz, Anal. Chem. 1956, 28, 1218.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG28XntlSqtw%3D%3D&md5=22484dc8990fa4c265afeb36ebf42423CAS |
[64] D. Kvaskoff, M. Vosswinkel, C. Wentrup, J. Am. Chem. Soc. 2011, 133, 5413.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsVGjtLw%3D&md5=a7de98bb4ada1fc71a87e58aa4becb6bCAS | 21417327PubMed |
[65] C. Wentrup, A. Reisinger, D. Kvaskoff, Beilstein J. Org. Chem. 2013, 9, 754.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmvFaqtbY%3D&md5=0434ec9df9b94d5f9890ef83ded09374CAS | 23766787PubMed |
[66] D. Kvaskoff, U. Mitschke, C. Addicott, P. Bednarek, J. Finnerty, C. Wentrup, Aust. J. Chem. 2009, 62, 275.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsVKqtr4%3D&md5=1212b94586c4920bd128c256095efd13CAS |
[67] C. Addicott, H. Lüerssen, M. Kuzaj, D. Kvaskoff, C. Wentrup, J. Phys. Org. Chem. 2011, 24, 999.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpsFajt74%3D&md5=20bfcfd4d17690dbb04a4909fb4f82efCAS |
[68] (a) Direct observation of C-cyclopentadienylidenemethanimines from benzotriazoles: In FVT: A. Maquestiau, D. Beugnies, R. Flammang, B. Freiermuth, C. Wentrup, Org. Mass Spectrom. 1990, 25, 197.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXltlWnsr4%3D&md5=a7705f3382d88f38baef6f8cdfa71f6bCAS |
(b) In matrix photolysis: M. Kiszka, I. R. Dunkin, J. Gebicki, H. Wang, J. Wirz, J. Chem. Soc. Perkin 2 2000, 2420.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Tomioka, N. Ichikawa, K. Komatsu, J. Am. Chem. Soc. 1992, 114, 8045.
| Crossref | GoogleScholarGoogle Scholar |