

A Simple and Direct Synthesis of 3-Methylene-1, 4-diarylazetidin-2-ones and (E)-3-Arylidene-1-phenylazetidin-2-ones Using Baylis–Hillman Derivatives
Manickam Bakthadoss A B C , Jayakumar Srinivasan B and Raman Selvakumar BA Department of Chemistry, Pondicherry University, Puducherry 605 014, India.
B Department of Organic Chemistry, University of Madras, Guindy Campus, Chennai 600 025, India.
C Corresponding author. Email: bhakthadoss@yahoo.com
Australian Journal of Chemistry 67(2) 295-301 https://doi.org/10.1071/CH13382
Submitted: 30 July 2013 Accepted: 14 October 2013 Published: 2 December 2013
Abstract
Herein we describe a direct method, promoted by potassium tert-butoxide (KOtBu), for the synthesis of highly substituted α-methylene β-lactams and α-arylidene β-lactams from the amino ester intermediates derived from the acetates and bromo derivatives of the Baylis–Hillman adducts. A variety of β-lactams was synthesized in a single step with good yields.
Introduction
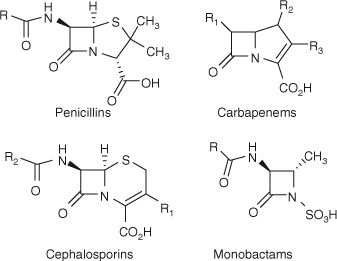
The β-lactam moiety is an important integral part of many natural products and biologically active molecules, the latter being mostly antibiotics and serine protease inhibitors.[1] Penicillins, carbapenems, cephalosporins, and monobactams are some of the β-lactam core-containing antibacterials which have been used therapeutically to date (Fig. 1). Apart from clinical use, β-lactams can also serve as good synthons in the synthesis of many biologically active heterocycles.[2]
|
In recent years, the Baylis–Hillman reaction has emerged as a powerful synthetic tool for the synthesis of diverse classes of multifunctional molecules.[3] Baylis–Hillman adducts can be used as synthons to obtain a wide variety of natural products and biologically active molecules.[4] We have been working on the application of Baylis–Hillman adducts[5] with a view to demonstrate that Baylis–Hillman chemistry is a powerful tool for the synthesis of various important and useful structural frameworks. Due to the multifunctionality present in Baylis–Hillman adducts, the opportunity for converting them into a new class of cyclic compounds is high and is very attractive in the field of organic chemistry. Although Baylis–Hillman adducts have been utilised for numerous applications,[6] the transformation of Baylis–Hillman adducts into β-lactam is very limited.[7] All the literature methods available for the formation of β-lactams involves lactamization, achieved by the coupling of amino and ester groups which is a two step process: the ester group is hydrolyzed to an acid moiety followed by the coupling of acid and amino groups in an intramolecular fashion to obtain the β-lactam core moiety.
Results and Discussion
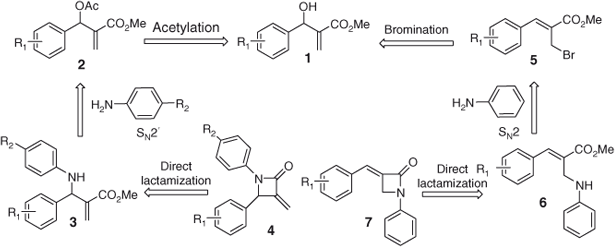
We envisaged that an amino group and ester moiety could be directly cyclized using potassium tert-butoxide (KOtBu) without hydrolyzing the ester group. Therefore, we planned to directly connect the ester moiety and amino group (which is present in the β-position) in an intramolecular fashion using KOtBu in a shorter synthetic sequence to obtain α-methylene β-lactam 4 from the amino ester 3 (Table 1). We also envisage that the α-arylidene β-lactam 7 can be easily achieved from the amino ester 6 (Table 2). According to the retro-synthetic strategy, Baylis–Hillman adducts can be easily converted into the corresponding substituted amino ester 3 by an SN2′ reaction. The amino ester 3 can be easily converted into the corresponding α-methylene β-lactam 4 by direct intramolecular cyclization. Similarly, the amino ester 6 can be easily synthesized from the bromo derivative of Baylis–Hillman adducts by an SN2 reaction, and a direct lactamization of an amino group with an ester group in an intramolecular fashion will lead to the desired α-arylidene β-lactam 7 core as described in Scheme 1.

|

|
|
To execute our idea, we first selected methyl 2-(acetoxy(phenyl)methyl)acrylate (2a), and treated it with 1,4-diazabicyclo[2.2.2]octane (DABCO) in THF at room temperature for 15 min which led to the DABCO salt of Baylis–Hillman acetate. To this DABCO salt, aniline was added and stirred at room temperature for 1 h which successfully furnished the required amino ester precursor, i.e. methyl 2-(phenyl(phenylamino)methyl)acrylate (3a) in 76 % yield. Further treatment of amino ester 3a with KOtBu in dry THF at 0°C for 1 h successfully led to the desired β-lactam, i.e. 3-methylene-1,4-diphenylazetidin-2-one (4a) in 66 % yield (Entry 1, Table 1). Encouraged by this result, we prepared a variety of amino esters using anilines 3b–f and treated them with KOtBu which successfully led to the anticipated substituted α-methylene β-lactams 4b–f in 65–70 % yields (Entries 2–6, Table 1). To further extend the methodology, we also employed p-toluidine for the lactamization reaction. Reaction of methyl 2-(acetoxy(aryl)methyl)acrylates 2a, 2b, 2d, and 2e, with DABCO in THF for 15 min led to the corresponding DABCO salts. To these DABCO salts, p-toluidine was added and the mixture stirred at room temperature for 1 h which led to the anticipated amino esters 3g–j in 72–83 % yields. Further treatment of amino ester precursors 3g–j with KOtBu successfully provided the desired products, i.e. 3-methylene-4-aryl-1-p-tolylazetidin-2-ones 4g–j in 66–71 % yields (Entries 7–10, Table 1).
After successfully synthesizing an array of 3-methylene-1,4-diarylazetidin-2-ones (4a–j), we planned to utilise the same protocol for the synthesis of (E)-3-benzylidene-1-phenylazetidin-2-one (7a) from (E)-methyl 2-((phenylamino)methyl)-3-phenylacrylate (6a).
Treatment of (Z)-methyl 2-(bromomethyl)-3-phenylacrylate (5a) with aniline in the presence of potassium carbonate in CH3CN for 3 h led to the formation of required amino ester precursor 6a in 79 % yield. The amino ester 6a was further treated with KOtBu in dry THF at 0°C for 1 h which successfully led to the desired (E)-3-benzylidene-1-phenylazetidin-2-one (7a) in 65 % yield (Entry 1, Table 2). Encouraged by this result, we prepared a variety of amino ester precursors 6b–f and treated them with KOtBu which smoothly led to the desired substituted α-arylidene β-lactams 7b–f in 63–72 % yields (Entries 2–6, Table 2).
Conclusion
In conclusion, we have successfully developed a first general method for the synthesis of both α-methylene β-lactams and α-arylidene β-lactams in a single step with good yields using Baylis–Hillman adducts. Comparatively, this method is very simple and better than the methods already known in the literature. Since the core unit of β-lactam is an integral part of many biologically active molecules, the derivatives which we have synthesized may also possess similar activity which will be studied in our laboratory in the future.
Experimental
All reagents were purchased from commercial sources and used without further purification. Solvents were distilled before use. Column chromatography was performed on silica gel. IR spectra were recorded on an FTIR-8300 Shimadzu spectrophotometer. 1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded on a Bruker spectrometer using CDCl3 as solvent and tetramethylsilane as an internal standard; chemical shifts are reported in δ (ppm). Mass spectra were recorded on a Jeol-JMS-DX 303 HF mass spectrometer. Elemental analyses were recorded on a Perkin–Elmer 240C-CHN analyzer. Melting points are uncorrected. Thin-layer chromatography (TLC) was performed using glass plates coated with silica gel (ACME, 254F). Spots were visualized using iodine vapour and UV lamp.
Methyl 2-(Phenyl(phenylamino)methyl)acrylate (3a): Typical Procedure
To a stirred solution of methyl 2-(acetoxy(phenyl)methyl)acrylate (2a) (1.17 g, 5 mmol) in THF (8 mL), DABCO (0.56 g, 5 mmol) was added and stirred at room temperature for 15 min. To this solution aniline (0.47 g, 5 mmol) was added and stirred for 1 h at room temperature. After completion of the reaction, as monitored by TLC, the reaction mixture was evaporated under reduced pressure to remove THF. The crude mixture obtained was diluted with water (10 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated under reduced pressure which led to the crude product. The crude product thus obtained was purified by column chromatography (with 4 % ethyl acetate in hexanes) to afford the methyl 2-(phenyl(phenylamino)methyl)acrylate (3a) as a colourless liquid.
Methyl 2-(Phenyl(phenylamino)methyl)acrylate (3a)
Yield: 76 %. Reaction time: 1 h. δH 3.71 (s, 3H), 4.15 (br s, 1H), 5.42 (s, 1H), 5.97 (s, 1H), 6.39 (s, 1H), 6.56–7.39 (m, 10H). δC 51.90, 58.99, 113.48, 117.94, 126.18, 127.52, 127.81, 128.77, 129.18, 140.13140.66, 146.70, 166.68. νmax (KBr)/cm–1 3389.35, 1714.64, 1629.58.
3-Methylene-1,4-diphenylazetidin-2-one (4a): Typical Procedure
To a stirred solution of methyl 2-(phenyl(phenylamino)methyl)acrylate (3a) (0.67 g, 2.5 mmol) in dry THF (10 mL), KOtBu (0.37 g, 2.5 mmol) was added at 0°C. The reaction mixture was stirred for one hour at 0°C and then THF was removed under reduced pressure. The crude product thus obtained was diluted with water (10 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine solution and dried over anhydrous Na2SO4. Solvent was evaporated under reduced pressure and the crude product thus obtained was purified by column chromatography (with 20 % ethyl acetate in hexanes) which afforded the 3-methylene-1,4-diphenylazetidin-2-one (4a) as a colourless solid.
3-Methylene-1,4-diphenylazetidin-2-one (4a)
Yield: 66 %. Reaction time: 1 h. Mp 121–123°C. δH (300 MHz, CDCl3) 5.31 (s, 1H), 5.98 (s, 1H), 6.45–7.29 (m, 11H). δC (75 MHz, CDCl3) 58.65, 113.57, 118.16, 127.59, 127.95, 128.68, 128.84, 129.23, 139.55, 140.35, 146.52, 171.38. νmax (neat)/cm–1 1685, 1602. m/z 235 (M+). Anal. Calc. for C16H13NO: C 81.68, H 5.57, N 5.95. Found: C 81.61, H 5.49, N 6.06 %.
3-Methylene-1-phenyl-4-p-tolylazetidin-2-one (4b)
Yield: 67 %. Reaction time: 1 h. Mp 136–138°C. δH (300 MHz, CDCl3) 2.33 (s, 3H), 5.34 (s, 1H), 6.04 (s,1H), 6.51–7.25 (m, 10H). δC (75 MHz, CDCl3) 21.14, 58.35, 113.53, 118.06, 127.55, 128.43, 129.23, 129.54, 137.39, 137.72, 139.61, 146.58, 171.67. νmax (neat)/cm–1 1686, 1600. m/z 249 (M+). Anal. Calc. for C17H15NO: C 81.90, H 6.06, N 5.62. Found: C 81.95, H 6.12, N 5.59 %.
4-(4-Ethylphenyl)-3-methylene-1-phenylazetidin-2-one (4c)
Yield: 70 %. Reaction time: 1 h. Mp 140–142°C. δH (300 MHz, CDCl3) 1.15 (t, 3H, J 7.5), 2.56 (q, 2H, J 7.5), 5.27 (s, 1H), 6.01 (s, 1H), 6.46 (s, 1H), 6.55–7.21 (m, 9H). δC (75 MHz, CDCl3) 15.38, 28.49, 58.67, 113.81, 118.35, 127.60, 128.31, 128.36, 129.21, 137.35, 139.48, 144.05, 146.26, 171.22. νmax (neat)/cm–1 1686, 1627. m/z 263 (M+). Anal. Calc. for C18H17NO: C 82.10, H 6.51, N 5.32. Found: C 82.06, H 6.55, N 5.28 %.
4-(4-Isopropylphenyl)-3-methylene-1-phenylazetidin-2-one (4d)
Yield: 65 %. Reaction time: 1 h. Mp 148–151°C. δH (300 MHz, CDCl3) 1.16 (d, 6H, J 6.9), 2.82 (sep, 1H, J 6.9), 5.26 (s, 1H), 6.01 (s, 1H), 6.52 (s, 1H), 6.70–7.26 (m, 9H). δC (75 MHz, CDCl3) 23.95, 33.80, 58.32, 113.50, 118.03, 126.91, 127.57, 128.41, 129.22, 137.64, 139.54, 146.57, 148.64, 171.71. νmax (neat)/cm–1 1683, 1625. m/z 277 (M+). Anal. Calc. for C19H19NO: C 82.28, H 6.90, N 5.05. Found: C 82.32, H 6.95, N 5.08 %.
4-(4-Chlorophenyl)-3-methylene-1-phenylazetidin-2-one (4e)
Yield: 68 %. Reaction time: 1 h. Mp 152–154°C. δH (300 MHz, CDCl3) 5.27 (s, 1H), 6.01 (s, 1H), 6.58 (s, 1H), 6.86–7.35 (m, 9H). δC (75 MHz, CDCl3) 58.09, 113.61, 118.43, 128.89, 128.99, 129.10, 129.27, 133.78, 138.87, 139.29, 146.27, 170.91. νmax (neat)/cm–1 1684, 1600. m/z 269 (M+). Anal. Calc. for C16H12ClNO: C 71.25, H 4.48, N 5.19. Found: C 71.21, H 4.43, N 5.23.
4-(4-Fluorophenyl)-3-methylene-1-phenylazetidin-2-one (4f)
Yield: 69 %. Reaction time: 1 h. Mp 158–160°C. δH (300 MHz, CDCl3) 5.29 (s, 1H), 5.98 (s, 1H), 6.46–7.28 (m, 10H). δC (75 MHz, CDCl3) 58.06, 113.72 (d, J 66), 115.54, 115.85, 116.14, 118.46 (d, J 69), 129.28, 131.90 (d, J 36), 143.96, 146.36, 147.37, 172.47. νmax (neat)/cm–1 1232, 1592, 1627, 1697, 3254. m/z 253 (M+). Anal. Cal. for C16H12FNO: C 75.88, H 4.78, N 5.53. Found: C 75.93, H 4.71, N 5.61 %.
3-Methylene-4-phenyl-1-p-tolylazetidin-2-one (4g)
Yield: 67 %. Reaction time: 1 h. Mp 126–128°C. δH (300 MHz, CDCl3) 2.23 (s, 3H), 5.24 (s, 1H), 5.94 (s, 1H), 6.40–7.14 (m, 10H). δC (75 MHz, CDCl3) 20.01, 57.27, 112.45, 116.98, 126.42, 127.31, 128.12, 128.42, 136.30, 136.60, 138.54, 145.48, 170.64. νmax (neat)/cm–1 1237, 1592, 1635, 1717, 3284. m/z 249 (M+). Anal. Calc. for C17H15NO: C 81.90, H 6.06, N 5.62. Found: C 81.86. H 6.09. N 5.68 %.
3-Methylene-1,4-dip-tolylazetidin-2-one (4h)
Yield: 68 %. Reaction time: 1 h. Mp 134–136°C. δH (300 MHz, CDCl3) 2.23 (s, 3H), 2.33 (s, 3H), 5.29 (s, 1H), 6.02 (s, 1H), 6.48–7.25 (m, 9H). δC (75 MHz, CDCl3) 20.38, 21.09, 58.79, 113.78, 127.43, 127.47, 128.33, 129.48, 129.70, 137.49, 137.64, 139.72, 144.24, 171.16. νmax (neat)/cm–1 1234, 1486, 1621, 1700, 3381. m/z 263 (M+). Anal. Calc. for C18H17NO: C 82.10, H 6.51, N 5.32. Found: C 82.14, H 6.48, N 5.27 %.
4-(4-Isopropylphenyl)-3-methylene-1-p-tolylazetidin-2-one (4i)
Yield: 71 %. Reaction time: 1 h. Mp 155–157°C. δH (300 MHz, CDCl3) 1.24 (d, 6H, J 6.9), 2.23 (s, 3H), 2.89 (sep, 1H, J 6.9), 5.31 (s, 1H), 6.03 (s, 1H), 6.49–7.28 (m, 9H). δC (75 MHz, CDCl3) 20.37, 23.90, 33.76, 59.00, 113.90, 126.86, 127.45, 128.30, 129.70, 130.18, 137.98, 139.60, 144.02, 148.43, 170.26. νmax (neat)/cm–1 1236, 1485, 1594, 1706, 3436. m/z 291 (M+). Anal. Calc. for C20H21NO: C 82.44, H 7.26, N 4.81. Found: C 82.38, H 7.31. N 4.87 %.
4-(4-Chlorophenyl)-3-methylene-1-p-tolylazetidin-2-one (4j)
Yield: 66 %. Reaction time: 1 h. Mp 160–162°C. δH (300 MHz, CDCl3) 2.23 (s, 3H), 5.32 (s, 1H), 6.02 (s, 1H), 6.47–7.32 (m, 9H). δC (75 MHz, CDCl3) 20.38, 58.46, 113.83, 127.75, 128.88, 128.95, 129.77, 131.06, 133.68, 139.03, 139.44, 143.98, 170.66. νmax (neat)/cm–1 1242, 1590, 1600, 1716, 3434. m/z 283 (M+). Anal. Calc. for C17H14ClNO: C 71.96, H 4.97, N 4.94. Found: C 71.92, H 4.92, N 4.96 %.
(E)-Methyl 2-((Phenylamino)methyl)-3-phenylacrylate (6a): Typical Procedure
To a stirred solution of aniline (0.47 g, 5 mmol) in dry CH3CN (15 mL), potassium carbonate (1.04 g, 7.5 mmol) was added and stirred well at room temperature. To this solution, (Z)-methyl 2-(bromomethyl)-3-phenylacrylate (5a) (1.27 g, 5 mmol) in dry CH3CN (10 mL) was added drop wise and stirred at room temperature for 3 h. After completion of the reaction, the reaction mixture was evaporated under reduced pressure to remove CH3CN. The crude mixture obtained was diluted with water (10 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and the solvent was evaporated under reduced pressure which led to the crude product. The crude product was purified by column chromatography (4 % ethyl acetate in hexanes) to afford (E)-methyl 2-((phenylamino)methyl)-3-phenylacrylate (6a) as a pale yellow liquid.
Yield: 79 %. Reaction time: 3 h. δH 3.83 (s, 3H), 4.13 (br s, 3H), 6.51–7.46 (m, 10H), 7.89 (s, 1H). δC 41.00, 52.20, 113.47, 117.89, 128.73, 129.18, 129.25, 129.32, 129.56, 134.81, 142.88, 147.78, 168.19. νmax (KBr)/cm–1 3393.60, 1717.79, 1629.58.
(E)-3-Benzylidene-1-phenylazetidin-2-one (7a): Typical Procedure
To a stirred solution of (E)-methyl 2-((phenylamino)methyl)-3-phenylacrylate (6a) (0.67 g, 2.5 mmol) in dry THF (10 mL), KOtBu was added (0.37 g, 2.5 mmol) at 0°C. The reaction mixture was stirred for 1 h at 0°C and then THF was removed under reduced pressure. The crude mass obtained was diluted with water (10 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed with brine and was dried over anhydrous Na2SO4. Solvent was evaporated under reduced pressure and the crude product thus obtained was purified by column chromatography (15 % ethyl acetate in hexanes) which afforded the (E)-3-benzylidene-1-phenylazetidin-2-one (7a) as a yellow solid.
(E)-3-Benzylidene-1-phenylazetidin-2-one (7a)
Yield: 65 %. Reaction time: 1 h. Mp 144–146°C. δH (300 MHz, CDCl3) 4.17 (s, 2H), 6.52–7.48 (m, 10H), 8.03 (s, 1H). δC (75 MHz, CDCl3) 41.05, 113.83, 118.40, 128.33, 128.80, 129.22, 129.63, 129.72, 134.48, 145.00, 147.46, 172.13. νmax (KBr)/cm–1 1234, 1487, 1599, 1722, 3426. m/z 235 (M+). Anal. Calc. for C16H13NO: C 81.68, H 5.57, N 5.95. Found: C 81.72, H 5.61. N 5.91 %.
(E)-3-(2-Methylbenzylidene)-1-phenylazetidin-2-one (7b)
Yield: 63 %. Reaction time: 1 h. Mp 122–124°C. δH (300 MHz, CDCl3) 2.27 (s, 3H), 4.12 (s, 2H), 6.42–7.34 (m, 9H), 8.07 (s, 1H). δC (75 MHz, CDCl3) 19.97, 40.82, 113.95, 118.31, 126.01, 128.98, 129.14, 129.46, 130.34, 133.91, 137.38, 144.09, 147.20, 173.06. νmax (KBr)/cm–1 1234, 1511, 1630, 1711, 3244. m/z 249 (M+). Anal. Calc. for C17H15NO: C 81.90, H 6.06, N 5.62. Found: C 81.92, H 6.09, N 5.58 %.
(E)-3-(4-Methylbenzylidene)-1-phenylazetidin-2-one (7c)
Yield: 68 %. Reaction time: 1 h. Mp 112–114°C. δH (300 MHz, CDCl3) 2.38 (s, 3H), 4.15 (s, 2H), 6.57–7.47 (m, 9H), 8.05 (s, 1H). δC (75 MHz, CDCl3) 21.45, 41.02, 113.71, 118.17, 128.40, 129.55, 129.91, 130.26, 131.69, 140.04, 145.10, 147.67, 172.93. νmax (KBr)/cm–1 1266, 1606, 1631, 1710, 3289. m/z 249 (M+). Anal. Calc. for C17H15NO: C 81.90, H 6.06, N 5.62. Found: C 81.94, H 6.08, N 5.67 %.
(E)-3-(4-Ethylbenzylidene)-1-phenylazetidin-2-one (7d)
Yield: 69 %. Reaction time: 1 h. Mp 136–138°C. δH (300 MHz, CDCl3) 1.25 (t, 3H, J 7.5), 2.68 (q, 2H, J 7.5), 4.17 (s, 2H), 6.58–7.43 (m, 9H), 8.00 (s, 1H). δC (75 MHz, CDCl3) 15.34, 28.81, 41.07, 113.79, 118.20, 127.48, 128.40, 129.26, 130.09, 131.92, 145.33, 146.39, 147.66, 173.40. νmax (KBr)/cm–1 1237, 1600, 1625, 2217, 3415. m/z 263 (M+). Anal. Calc. for C18H17NO: C 82.10, H 6.51, N 5.32. Found: C 82.15, H 6.55, N 5.36 %.
(E)-3-(4-Isopropylbenzylidene)-1-phenylazetidin-2-one (7e)
Yield: 64 %. Reaction time: 1 h. Mp 135–137°C. δH (300 MHz, CDCl3) 1.26 (d, 6H, J 6.9), 2.93 (sep, 1H, J 6.9), 4.16 (s, 2H), 6.58–7.44 (m, 9H), 8.00 (1H). δC (75 MHz, CDCl3) 23.80, 34.08, 41.03, 113.74, 118.23, 126.98, 129.24, 130.12, 132.00, 145.37, 147.66, 151.01, 172.00. νmax (KBr)/cm–1 1246, 1490, 1590, 2210, 3248. m/z 277 (M+). Anal. Calc. for C19H19NO: C 82.28, H 6.90, N 5.05. Found: C 82.32, H 6.87, N 5.09 %.
(E)-3-(2-Methoxybenzylidene)-1-phenylazetidin-2-one (7f)
Yield: 72 %. Reaction time: 1 h. Mp 134–136°C. δH (300 MHz, CDCl3) 3.86 (s, 3H), 4.10 (s, 2H), 6.55–7.41 (m, 9H), 8.16 (s, 1H). δC (75 MHz, CDCl3) 41.39, 55.56, 110.71, 113.84, 118.18, 120.66, 123.66, 128.19, 129.16, 130.34, 131.21, 141.13, 147.66, 157.83, 173.07. νmax (KBr)/cm–1 1239, 1495, 1600, 2210, 3228. m/z 265 (M+). Anal. Calc. C17H15NO2: C 76.96, H 5.70, N 5.28. Found: C 76.92. H 5.73, N 5.31 %.
Supplementary Material
1H and 13C NMR spectra for compounds 3a, 4a–j, 6a and 7a–f are available on the Journal’s website.
Acknowledgements
The authors thank the Department of Science and Technology-Science and Engineering Research Board (DST-SERB) New Delhi, India, for financial support. The authors also thank the Department of Science and Technology-Fund for Improvement of S&T Infrastructure (DST-FIST) for use of the NMR facility. J.S. and R.S. thank the Council of Scientific & Industrial Research (CSIR) for their Senior Research Fellowship.
References
[1] (a) For selected reviews, see: B. K. Hubbard, C. T. Walsh, Angew. Chem. Int. Ed. 2003, 42, 730.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhvFWmt7o%3D&md5=100fc007002802de25d234a0454f7247CAS |
(b) R. Nau, A. Eiffert, Clin. Microbiol. Rev. 2002, 15, 95.
| Crossref | GoogleScholarGoogle Scholar |
(c) G. Veinberg, M. Vorona, I. Shestakova, I. Kanepe, E. Lukevics, Curr. Med. Chem. 2003, 10, 1741.
| Crossref | GoogleScholarGoogle Scholar |
(d) E. L. Setti, R. G. Micetich, Curr. Med. Chem. 1998, 5, 101.
(e) G. I. Georg, Ed., The Organic Chemistry of β-Lactams 1993 (VCH: New York).
(f) G. S. Singh, Mini-Rev. Med. Chem. 2004, 4, 69.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) B. Alcaide, P. Almendros, C. Aragoncillo, Chem. Rev. 2007, 107, 4437.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXotFeitrs%3D&md5=d537edcd33dadb080b6d7c0918d27218CAS | 17649981PubMed |
(b) I. Ojima, Acc. Chem. Res. 1995, 28, 383.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Suffness, Ed., Taxol Science and Applications 1995, pp. 3–25 (CRC Press, Boca Raton, FL).
(d) B. Alcaide, P. Almendros, Curr. Med. Chem. 2004, 11, 1921.
| Crossref | GoogleScholarGoogle Scholar |
(e) I. Ojima, F. Delaloge, Chem. Soc. Rev. 1997, 26, 377.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) D. Basavaiah, B. S. Reddy, S. S. Badsara, Chem. Rev. 2010, 110, 5447.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVKmtbbM&md5=49989754ac3e337fb3a9c67ef45ba5feCAS | 20735052PubMed |
(b) D. Basavaiah, A. J. Rao, T. Satyanarayana, Chem. Rev. 2003, 103, 811.
| Crossref | GoogleScholarGoogle Scholar |
(c) V. Declerck, J. Martinez, F. Lamaty, Chem. Rev. 2009, 109, 1.
| Crossref | GoogleScholarGoogle Scholar |
(d) D. Basavaiah, K. V. Rao, R. J. Reddy, Chem. Soc. Rev. 2007, 36, 1581.
| Crossref | GoogleScholarGoogle Scholar |
(e) V. Singh, S. Batra, Tetrahedron 2008, 64, 4511.
| Crossref | GoogleScholarGoogle Scholar |
(f) D. Basavaiah, P. D. Rao, S. R. Hyma, Tetrahedron 1996, 52, 8001.
| Crossref | GoogleScholarGoogle Scholar |
[4] (a) D. Basavaiah, J. S. Rao, R. J. Reddy, J. Org. Chem. 2004, 69, 7379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXnsVOqur0%3D&md5=d65cbef0a41dca247f38d0bbffff5c00CAS | 15471499PubMed |
(b) B. M. Trost, O. R. Thiel, H.-C. Tsui, J. Am. Chem. Soc. 2002, 124, 11616.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Saquib, M. K. Gupta, R. Sagar, Y. S. Prabhakar, A. K. Shaw, R. Kumar, P. R. Maulik, A. N. Gaikwad, S. Sinha, A. K. Srivastava, V. Chaturvedi, R. Srivastava, B. S. Srivastava, J. Med. Chem. 2007, 50, 2942.
| Crossref | GoogleScholarGoogle Scholar |
(d) V. Singh, R. Saxena, S. Batra, J. Org. Chem. 2005, 70, 353.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Batra, A. K. Roy, A. Patra, A. P. Bhaduri, W. R. Surin, S. A. V. Raghavan, P. Sharma, K. Kapoorb, M. Dikshit, Bioorg. Med. Chem. 2004, 12, 2059.
| Crossref | GoogleScholarGoogle Scholar |
(f) G. W. Amarante, F. Coelho, Tetrahedron 2010, 66, 6749.
| Crossref | GoogleScholarGoogle Scholar |
(g) Y. Zulykama, P. T. Perumal, Tetrahedron Lett. 2009, 50, 3892.
| Crossref | GoogleScholarGoogle Scholar |
(h) D. Basavaiah, M. Krishnamacharyula, R. S. Hyma, P. K. S. Sarma, N. Kumaragurubaran, J. Org. Chem. 1999, 64, 1197.
| Crossref | GoogleScholarGoogle Scholar |
(i) Y.-Q. Jiang, Y.-L. Shi, M. Shi, J. Am. Chem. Soc 2008, 130, 7202.
| Crossref | GoogleScholarGoogle Scholar |
(j) P. Auvray, P. Knochel, J. F. Normant, Tetrahedron Lett. 1986, 27, 5095.
| Crossref | GoogleScholarGoogle Scholar |
(k) A. Weichert, H. M. R. Hoffmann, J. Org. Chem. 1991, 56, 4098.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) M. Bakthadoss, G. Sivakumar, D. Kannan, Org. Lett. 2009, 11, 4466.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOgu7fL&md5=088984701439c2c7abc478384f721861CAS | 19775188PubMed |
(b) M. Bakthadoss, G. Murugan, Eur. J. Org. Chem. 2010, 5825.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Bakthadoss, N. Sivakumar, Synlett 2011, 1296.
| Crossref | GoogleScholarGoogle Scholar |
(d) M. Bakthadoss, N. Sivakumar, A. Devaraj, Synthesis 2011, 611.
| Crossref | GoogleScholarGoogle Scholar |
(e) D. Basavaiah, M. Bakthadoss, S. Pandiaraju, Chem. Commun. 1998, 1639.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. Bakthadoss, N. Sivakumar, Synlett 2009, 1014.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. Bakthadoss, N. Sivakumar, G. Sivakumar, G. Murugan, Tetrahedron Lett. 2008, 49, 820.
| Crossref | GoogleScholarGoogle Scholar |
(h) M. Bakthadoss, N. Sivakumar, A. Devaraj, D. S. Sharada, Synthesis 2011, 2136.
| Crossref | GoogleScholarGoogle Scholar |
(i) D. Basavaiah, M. Bakthadoss, G. Jayapal Reddy, Synthesis 2001, 0919.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) R. Pathak, A. K. Roy, S. Batra, Synlett 2005, 848.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjtVyhu7o%3D&md5=f3f464890d70f369bb9e589aa4777c87CAS |
(b) V. Singh, S. Batra, Synthesis 2006, 63.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. S. Yadav, B. V. S. Reddy, A. P. Sing, A. K. Basak, Synthesis 2008, 469.
| Crossref | GoogleScholarGoogle Scholar |
(d) G.-L. Zhao, J.-N. Huang, M. Shi, Org. Lett. 2003, 5, 4737.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Gowrisankar, H. S. Lee, J. M. Kim, J. N. Kim, Tetrahedron Lett. 2008, 49, 1670.
| Crossref | GoogleScholarGoogle Scholar |
(f) E. S. Kim, K. H. Kim, S. Park, J. N. Kim, Tetrahedron Lett. 2010, 51, 4648.
| Crossref | GoogleScholarGoogle Scholar |
(g) B. Das, J. Banerjee, G. Mahender, A. Majhi, Org. Lett. 2004, 6, 3349.
| Crossref | GoogleScholarGoogle Scholar |
(h) B. Das, K. Damodar, N. Bhunia, B. Shashikanth, Tetrahedron Lett. 2009, 50, 2072.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) R. Buchholz, H. M. R. Hoffmann, Helv. Chim. Acta 1991, 74, 1213.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXmtlWqsLg%3D&md5=091207439d80381e1580bfd785b262feCAS |
(b) S. Kawahara, A. Nakano, T. Esumi, Y. Iwabuchi, S. Hatakeyama, Org. Lett. 2003, 5, 3103.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Lee, S. Y. Moon, G.-S. Hwang, D. H. Ryu, Org. Lett. 2010, 12, 3234.
| Crossref | GoogleScholarGoogle Scholar |