

Electrochemical Restructuring of Copper Surfaces Using Organic Additives and Its Effect on the Electrocatalytic Reduction of Nitrate Ions†
Ali Balkis A and Anthony P. O’Mullane B CA School of Applied Sciences, RMIT University, GPO Box 2476V, Melbourne, Vic 3001, Australia.
B School of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, GPO Box 2434, Brisbane, Qld 4001, Australia.
C Corresponding author. Email: anthony.omullane@qut.edu.au
Australian Journal of Chemistry 68(8) 1213-1220 https://doi.org/10.1071/CH15191
Submitted: 16 April 2015 Accepted: 15 May 2015 Published: 16 June 2015
Abstract
This work describes the fabrication of nanostructured copper electrodes using a simple potential cycling protocol that involves oxidation and reduction of the surface in an alkaline solution. It was found that the inclusion of additives, such as benzyl alcohol and phenylacetic acid, has a profound effect on the surface oxidation process and the subsequent reduction of these oxides. This results in not only a morphology change, but also affects the electrocatalytic performance of the electrode for the reduction of nitrate ions. In all cases, the electrocatalytic performance of the restructured electrodes was significantly enhanced compared with the unmodified electrode. The most promising material was formed when phenylacetic acid was used as the additive. In addition, the reduction of residual oxides on the surface after the modification procedure to expose freshly active reaction sites on the surface before nitrate reduction was found to be a significant factor in dictating the overall electrocatalytic activity. It is envisaged that this approach offers an interesting way to fabricate other nanostructured electrode surfaces.
Introduction
The electrochemical formation, characterization, and utilization of nanostructured materials are areas of burgeoning interest.[1] In particular, metals such as Au, Pt, Pd have been extensively studied due to their excellent (electro)-catalytic properties as well as their interesting fundamental electrochemical behaviour in the absence of an analyte over wide pH ranges. The latter property offers significant insights into surface cleanliness, structure, and activity in addition to determining the electrochemically active surface area of an electrode. In comparison to the metals mentioned previously, copper has received less attention, but is still a remarkably active catalyst and electrocatalyst as well as being the choice of interconnect material in the microelectronics industry.[2] The electrodeposition of copper has been widely studied for the latter application where smooth, defect-free coatings are required. To achieve this, the electroplating bath consists of a variety of components in addition to the source of copper ions and background electrolyte such as accelerators, levellers, and brighteners.[2] However, for electrocatalytic applications, creating highly active copper with a high surface area in a nanostructured form is highly desirable. Several approaches have been used to create such materials using electrochemical approaches including dynamic hydrogen bubble templating,[3] electrodeposition of Cu nanoparticles onto high surface area support materials,[4] or planar electrodes.[5] For the hydrogen bubble templating method, additives are often used to control the pore size distribution via perturbing the hydrogen evolution and/or metal deposition rate.[3b,6]
A different approach that has been used to create highly active materials is a surface rebuilding method. This has been reported for gold where the electrode is subjected to vigorous oxygen evolution to form an oxide, which is then reduced under vigorous hydrogen evolution conditions to fabricate a porous structure. This is done quite quickly, at a frequency of ~50 Hz for 6000 s. The resultant porous gold electrode showed good activity for ethanol, glucose, and ascorbic acid oxidation.[7] This type of approach was later used to restructure the surface of a copper electrode.[8] Pt has been roughened using a repetitive square wave potential cycle at 1 kHz involving the oxidation of the surface and its subsequent reduction for use as electrodes for neural stimulation.[9] Highly active copper surfaces have also been produced via the reduction of electrochemically grown Cu(OH)2 films.[10] In addition, copper nanoparticles have been produced via reduction of CuCl powder in an ionic liquid to produce particles of ~10 nm in diameter.[11] Another way to create active sites on metal surfaces, which has been reported, is to restructure nanoparticles such as Pt via the application of a square wave potential cycle in the presence of ascorbic acid to generate high-index facets at the surface,[12] or Pt nanorods if exposed to air before the electrochemical modification.[13] Therefore, in principle, electrochemically restructuring an electrode via an oxide formation and reduction process in the presence of additives may offer an interesting route for creating a nanostructured surface. In this work, copper was chosen given the recent attention it has received as an electrocatalyst for a wide variety of reactions including CO2 reduction,[14] non-enzymatic glucose detection,[15] and nitrate detection and/or removal,[10,16] to name a few. The latter in particular is an important topic as the imbalance in the nitrogen cycle caused by human activities has led to high levels of nitrate in groundwater[16b] that can have serious environmental effects such as eutrophication of lakes, seas, and rivers[16d] and health effects for humans such as liver damage or possibly cancer.[16c,16d] In addition, several processes that involve the removal of nitrate are based on biological and physicochemical methods (e.g. catalytic reduction by hydrogen, reverse osmosis, ion exchange resins, electrodialysis).[16c] However, these approaches can be costly, time consuming, and generate harmful by-products. On the contrary, electrochemical methods have shown promise for the efficient conversion of nitrate into harmless nitrogen (N2) gas, as well as offering a method to monitor nitrate levels.
Here, we explore the effect of electrochemically restructuring a copper electrode in alkaline electrolyte via application of a simple potential cycling protocol involving oxide formation and reduction in the absence and presence of additives such as benzyl alcohol and phenylacetic acid, which are known to influence the electrodeposition[17] and corrosion[18] of metals, respectively. The activated electrode is then investigated for its electrocatalytic properties toward nitrate reduction.
Results and Discussion
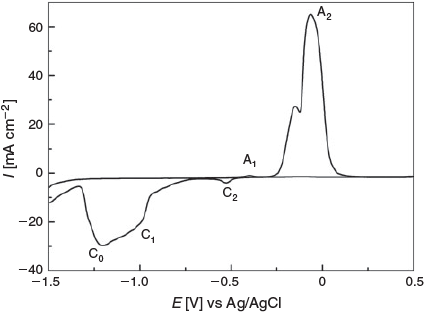
The electrochemical behaviour of copper electrodes in alkaline solution has been well documented.[10,19] Fig. 1 illustrates a typical cyclic voltammogram recorded for a Cu electrode in 1 M NaOH. On the anodic sweep, two distinct peaks can be distinguished. Peak A1 is attributed to the oxidation of Cu to Cu2O and peak A2 reflects the onset of a mixture of two processes, as evidenced by the appearance of a shoulder on the main peak, namely, the oxidation of both Cu and Cu2O to a mixture of Cu(OH)2 and CuO. On the cathodic sweep, peak C2 is the counterpart of process A2. The magnitude of this process is significantly lower than that of A2 because a passivating layer of Cu2O is formed when CuO or Cu(OH)2 is being reduced that is quite insulating and hence inhibits the reduction of CuO and Cu(OH)2 at this potential. At ~–0.98 V, the Cu2O is reduced (peak C1), and thus allows the bulk of CuO to be reduced at ~–1.21 V, which is represented as a broad peak C0. However, the reduction of hydrated oxides formed on the positive sweep has also been considered to be included in this C0 response. Previous work by Burke et al.[19b,19c] demonstrated that Cu can be activated electrochemically and thermally to produce significantly different voltammetric responses, in particular for the reduction of oxides formed on the surface of Cu. Generally, this was observed after thermally treating a Cu electrode via resistive heating in an inert atmosphere or polarization in the hydrogen evolution region for several hours.
|
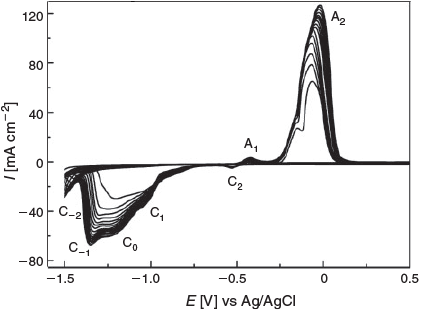
Fig. 2 illustrates the cyclic voltammetric response for the same Cu electrode cycled in the oxide formation and reduction region (–1.5 to 0.5 V at 10 mV s–1) at room temperature for 20 cycles. It can be clearly seen that peaks A1 and A2 grow in magnitude upon repetitive cycling similarly to processes C1 and C0. Interestingly, peaks A1 and C2 grow at a much slower rate, which is not too surprising given that the processes relating to these peaks involve growing a passivating Cu2O layer. However, the major difference upon repetitive cycling of the potential is the appearance and growth of new processes at ~–1.35 V (C–1) and –1.47 V (C–2). Burke et al. have attributed these processes to the reduction of hydrous oxides formed on active Cu sites created during thermal or electrochemical activation procedure.[19b,19c,20] This type of behaviour has also been observed by our group on highly porous Cu electrodes where it was confirmed that these processes are not due to the re-deposition of ionic Cu2+ ions that may have been liberated during the process of Cu oxidation at more positive potentials.[21] Bond et al. have provided further evidence that these processes involve an active Cu/hydrous oxide transition by large-amplitude Fourier transform (FT) ac voltammetry experiments.[19a] Therefore, it appears that upon the repetitive growth and reduction of oxides on Cu that a significantly different surface can be produced via a very simple protocol. When the oxide formed on the positive sweep is reduced, it creates highly active surface atoms, which in principle will have a significant influence on electrocatalytic activity. The active sites created via electrochemical reduction of copper oxide formed on foils by oxidation in air at 500°C for 12 h was reported to be highly effective for the reduction of CO2.[14b] It is also a dynamic process, whereby significant changes in the electrochemical behaviour of Cu are seen upon repetitive cycling, which then reaches a near steady-state after 20 cycles. A similar type of behaviour was also observed when Cu electrodes were treated by a two-step protocol, whereby the electrodes were extensively cycled (3000 times) at a scan rate of 10 V s–1 into the oxide formation region and subsequently reduced via the application of 20 potential cycles in a region where oxide formation did not occur.[10]
|
The surface of the Cu electrode was then investigated by scanning electron microscopy (SEM) after the application of 1, 5, 10, and 20 potential cycles between –1.50 and 0.50 V at 10 mV s–1. It can be seen that even after 1 cycle of potential that the relatively smooth Cu surface (Fig. S1, Supplementary Material) has been converted into a surface containing nanosized spherical-like particles of ~5–500 nm in diameter (Fig. 3a). Upon additional cycling, these features grow in size and coalesce into a film after 5–10 cycles (Fig. 3b, c), and after 20 cycles, a multilayer film can be observed where large protrusions of crystallites from the surface are evident (Fig. 3d). It should be noted that lower magnification images of all surfaces, shown in Fig. S2 (Supplementary Material), indicate the homogeneity of the surface modification procedure. This change in morphology from relatively smooth to a rough surface is consistent with an electrode whose surface area increases as demonstrated by the cyclic voltammetry data (Fig. 2), which showed an increase in magnitude of the current response upon potential cycling.

|
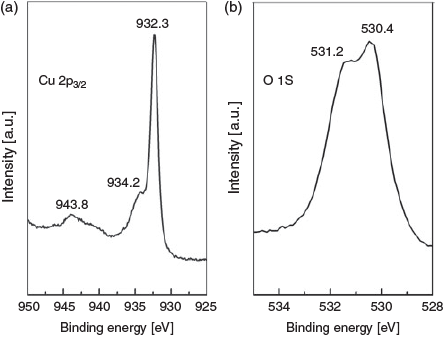
X-Ray photoelectron spectroscopy (XPS) analysis of the surface after 20 cycles shows that both Cu2O and CuO are formed on the surface. The Cu 2p3/2 core level spectrum shows a peak centred at 932.3 eV, which is attributed to Cu and Cu2O, which are indistinguishable[10] (Fig. 4a). There is a distinct peak at 934.2 eV that is due to the formation of CuO on the surface.[10] There is no real change in the XPS data upon repetitive cycling (Fig. S3, Supplementary Material), and all peaks remain at the same binding energy. The O 1S core level spectrum for the Cu electrode cycled 20 times is shown in Fig. 4b. Peaks at 530.4 and 531.2 eV were observed that are attributed to Cu2O and Cu(OH)2 formation.[10] Therefore, the surface of the electrode consists of Cu and residual mixed oxides consisting of Cu2O, CuO, and Cu(OH)2 after the application of this repetitive cycling protocol. From analysis of the electrode after 20 cycles using energy-dispersive X-ray spectroscopy (EDX), it was found that the sample consisted of 4.5 wt-% oxygen, indicating that the electrode was mostly Cu.
|
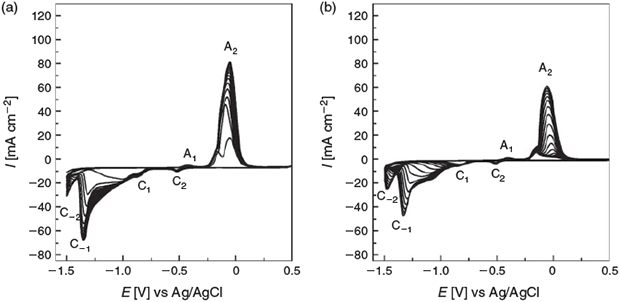
The use of additives in the chemical synthesis of colloidal materials is extensive because the additives are used as reductants, growth-directing agents, and capping agents to prevent aggregation. However, in comparison, this approach is under-utilized in the area of electrodeposition of nanostructured materials. As mentioned in the Introduction, there was an interesting report where ascorbic acid was used in the synthesis of high-energy facets on Pt to create highly active electrocatalysts.[12] In that work, electrodeposited Pt nanospheres were converted into tetrahexahedral nanocrystals via a square wave potential protocol; however, no mention was given to the role or influence of ascorbic acid, which is a reducing agent that is used quite commonly in the synthesis of colloidal materials. In this work, we chose to use benzyl alcohol, which has been used as a mild reducing agent for the synthesis of Pd nanoparticles[22] as well as an additive for controlling Zn and Cd deposition,[17,23] however, with the aim that it is not electrochemically oxidized in the potential range where the Cu electrodes were cycled, as opposed to ascorbic acid which is oxidized quite readily at metallic electrodes. It has also been used as an additive in the electroplating of CuSn alloys to ensure a lustrous finish.[24] Fig. 5 shows the cyclic voltammetric behaviour of a Cu electrode cycled in 1 M NaOH containing 0.1 M benzyl alcohol (Fig. 5a) and 0.1 M phenylacetic acid (Fig. 5b). The latter was chosen to examine the effect of the functional group on the restructuring process as well as its ability to suppress the corrosion of metals.[18] It is initially evident that the current being passed in the presence of these additives is lower than that in pure NaOH electrolyte (Fig. 2) at all cycle numbers studied. However, the reduction of the oxides on the negative sweep is distinctly different in all cases and in particular the cathodic peak at ~–1.50 V emerges, indicating the formation of an additional active Cu/hydrous oxide transition state on the electrode surface. This is particularly apparent for the case of phenylacetic acid where process C–2 is very prominent. Even though the magnitude of this process is quite similar in all cases (Fig. 2 and Fig. 5a, b are shown on the same scale), the relative intensity of peak C–2 in comparison with that of all the other peaks, in particular A2, is higher when phenylacetic acid is used. It can also be seen that the peak is more defined when phenylacetic is used.
|
Therefore, restructuring Cu in the presence of these additives has a significant effect on their voltammetric behaviour, which is reflected in the surface morphology shown in Fig. 6. When benzyl alcohol is used as the additive, there is some evidence of surface roughening after 1 cycle (Fig. 6a) that becomes more apparent after 5 cycles, as shown in Fig. 6b, which illustrates a film of crystallites of ~300 nm in diameter. After 10 cycles, these have grown in size to ~1 µm in diameter (Fig. 6c) and after 20 cycles to particles of 1–5 µm in diameter (Fig. 6d). It should be noted that these structures consist of much smaller particles that are aggregated together. When phenylacetic acid was used, a similar trend was observed as seen for benzyl alcohol in terms of growth in size of the structures; however, their morphology was distinctly different. After 1 cycle (Fig. 6e), surface roughening can be observed, and after 5 cycles both spherical, but more elongated structures, were formed (Fig. 6f). Upon additional cycling (Fig. 6 g, h), these structures grew larger, but did not appear to constitute aggregates of smaller particles as seen for the other additives.

|
The change in the surface of the film can be clearly seen by the digital images taken of the surface that was restructured in phenylacetic acid after various cycles. It can be seen that the shiny reddish copper surface is transformed initially to a yellow colour after 5 cycles which after 10 cycles then transforms to a darker colour indicative of the presence of some oxides on the surface[25] as well as a nanostructured surface (Fig. 7). In all cases, there was no evidence of a blue film that would indicate Cu(OH)2 formation.

|
XPS analysis of these surfaces shown in Fig. S3 (Supplementary Material) revealed that the samples comprised Cu, Cu2O, and CuO as evidenced by the Cu 2p3/2 core level spectra showing peaks at 932.4 and 934.3 eV, respectively, as well as the satellite shake-up peaks at 944 eV. Analysis of the O 1S spectra confirms the presence of Cu2O and some Cu(OH)2. Therefore, in all cases, the presence of the additive did not significantly influence the surface composition, but more so the morphology of the deposit and the surface area given the changes in the magnitude of the response recorded during the repetitive cycling process. This suppression of the overall magnitude of the response indicates that the extent of oxide formation in the presence of the additives is decreased or inhibited. In contrast, the responses observed for the reduction of these oxides on the cathodic sweep are quite different. As discussed previously by Burke et al., the composition of these oxides is difficult to ascertain and the oxides are generally considered as hydrous oxide-type species.[19b–d] To form oxides on Cu, the electrode must have access to OH– ions and water for oxide growth to continue. Generally, quite thick films can be produced on Cu via repetitive cycling at much higher potential limits (~2.0 V versus RHE (reversible hydrogen electrode)) or polarization in the oxygen evolution region.[19d] Given that the response here increases in magnitude upon cycling (Fig. 2) indicates that densely packed oxide films are not produced and are readily reduced in the reverse sweep. In the presence of benzyl alcohol and phenylacetic acetic acid, the oxide formation is inhibited. indicating that these additives compete with OH– ions adsorbing on the surface which is the first step in copper oxide formation. This perturbs the oxide growth process, and ultimately affects the coverage of oxide and its subsequent reduction. It is known in the electroplating field that benzyl alcohol influences the deposition of metals and alloys such as Cd,[17,23] Zn,[23] Cu, and CuSn.[24] The main use of benzyl alcohol is to suppress hydrogen evolution via surface adsorption and control the grain size of the deposit. Therefore, adsorption of benzyl alcohol onto the surface of Cu during repetitive cycling is consistent with the decrease in the overall magnitude of the current passed during repetitive cycling when compared with when benzyl alcohol is absent. A similar phenomenon is observed for phenylacetic acid that inhibits the oxide formation process to an even greater extent. This is consistent with phenylacetic acid being used as a corrosion inhibitor for Al in both alkaline and acidic media where adsorption on the surface occurs through the carbonyl group.[18] Due to its structure, the phenyl group can lie flat on the surface and maximize its efficiency for corrosion inhibition. Interestingly, pharmaceutical drugs based on phenylacetic acid, such as 2-(2,6-dichloranilino) phenylacetic acid, have been shown to inhibit the corrosion of mild steel via surface adsorption, but without a change in the corrosion mechanism.[26] In essence, oxide formation on Cu is a corrosion process and therefore phenylacetic acid simply acts as a corrosion inhibitor which again perturbs the oxide formed on the electrode surface and its subsequent reduction.
The restructured Cu electrodes were then tested for their electrocatalytic properties for the reduction of nitrate ions in alkaline electrolyte. Fig. 8a illustrates the linear sweep voltammograms recorded for these electrodes in 1M NaOH solution containing 0.1 M KNO3. It should be noted that the current responses are normalized via the charge associated with peak A2. The charge passed for peak A2 on the 1st cycle of the Cu electrode in 1 M NaOH was equated to the geometric area of 0.126 cm2. Using this correlation the surface area was then calculated for each of the restructured samples by calculating the charge passed for peak A2 for cycle number 20 in each instance. The data in the absence of nitrate ions are also presented (Fig. 8b) and were also normalized to the surface area as opposed to the geometric area in Figs. 2 and 5. It can again be seen here that the magnitude and peak position of the hydrous oxide reduction peaks are significantly different for all the surfaces investigated and the response for the sample restructured in phenylacetic acid shows the highest amount of residual oxides normalized to the electrochemically active surface area. Therefore, even though the surface area of the material formed with this additive is the lowest, the relative contribution from residual surface oxides is highest in this case. The scale chosen in Fig. 8b illustrates that the response recorded in the presence of nitrate ions is indeed an electrocatalytic process and not just associated with residual oxide reduction. For an unmodified copper foil, the voltammogram in the absence of nitrate displays one prominent reduction peak associated with process C1. There is no clear evidence of any other oxide or hydrous oxide reduction response until the end of the sweep. In the presence of nitrate ions, the peak associated with process C1 is shifted to a more negative potential and increases in magnitude due to the concomitant reduction of nitrate and oxide on the surface. Once the oxide is removed, the current increases gradually which corresponds to the reduction of nitrate to nitrite.[10] At ~–1.3 V, there is a significant increase in current due to the reduction of nitrite ions from the first step. It is interesting to note that this increase corresponds to the point where process C–1 was observed in 1 M NaOH only (Fig. 2). These observations are consistent with the incipient hydrous oxide adatom mediator (IHOAM) model of electrocatalysis[27] which describes that dissolved oxidants such as NO3– ions are reduced at active metal sites (M*), in this case Cu*, which supply the electrons for the reduction process to occur. Cu* is then immediately converted into an incipient oxide species which is subsequently reduced back to the active state Cu* by the current flowing in the external circuit. Therefore, the reduction of nitrate ions is akin to a surface-confined electrocatalytic process involving a Cu*/Cu incipient oxide couple that is kinetically fast enough to maintain a catalytic process. The involvement of such a redox couple was confirmed to be quasi-reversible by Bond et al. using FT ac voltammetry, whereby the electrocatalytic behaviours of smooth Cu electrodes and cathodically polarized electrodes were investigated.[19a] Interestingly, in that study, even when a dc cyclic voltammetric response indicated only double layer charging, the Fourier transform ac technique revealed Faradaic components in the higher harmonics related to the Cu*/incipient oxide transition. Therefore, though a clear oxide reduction response cannot be seen in the cyclic voltammogram shown in the absence of nitrate ions at ~–1.3 V (Fig. 8b), this does not mean that such a process is not present. A similar study was carried out with gold electrodes whereby the FT ac technique revealed Au*/hydrous oxide transitions not visible by dc voltammetry methods.[28] The reaction products for the second step involving nitrite reduction are not the focus of the current study and will be analyzed in future by batch electrolysis. However, previous reports have indicated a complex process where species such as NH3, N2O, NO and N2 are formed which is highly dependent on the surface conditions.[16c]
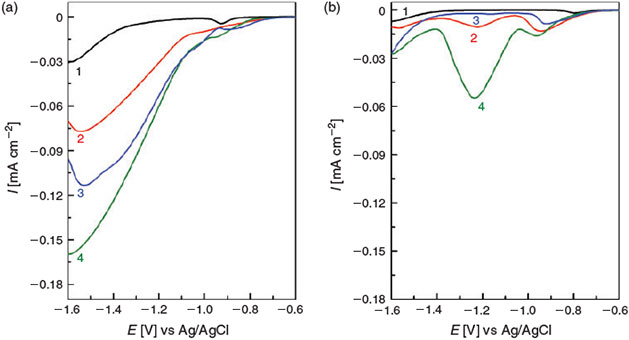
|
Significantly, for all the restructured electrodes, the current density for nitrate reduction is substantially greater than that obtained with the unmodified electrode. The nitrate reduction current measured at –1.6 V increased in the following order in the presence of Cu restructured in NaOH < benzyl alcohol < phenylacetic acid. Therefore once active Cu* mediators are exposed, they are highly active for nitrate reduction, in particular in the potential region after nitrate has been reduced to nitrite, i.e. below ~–1.0 V. The higher concentration of residual oxides on the surface in the C–2 region also appears to be linked to the sustained reduction of the nitrite species. It should be noted that in all cases, the nitrate to nitrite conversion process is also enhanced significantly at the restructured electrodes, indicating that the concentration of Cu* active atoms at electrochemically oxidized and then reduced Cu electrodes is promoted when compared with that when a pristine Cu electrode is employed. However, the concomitant reduction of residual oxides is also a contributing factor to the current, in particular in the case of phenylacetic acid. Therefore, this observation suggests that the presence of additives during the restructuring process affects the nature of the oxide formed on the electrode surface whose subsequent reduction to expose Cu* dictates the overall electrocatalytic performance, in particular at potentials from –1.2 to –1.6 V. These data also indicate that having residual oxide on the surface is not detrimental to electrocatalytic reduction reactions. This is reasonable because a freshly reduced oxide will expose highly reactive Cu* centres that then participate in the reaction. The nature of the active site will then depend on the type of oxide material as suggested by the data in Fig. 8b, which shows the difference in the oxide reduction profile at each of the Cu electrodes. Burke et al. have demonstrated that the formation of incipient oxides on the surface of Cu is a complex process, and is dependent on the treatment of the electrode surface, as confirmed in this study. The exact composition of these incipient oxides is still open to debate and the surface is unlikely to consist purely of Cu2O, CuO, or Cu(OH)2, but also CuO·OH, Cu(OH)·H2O, and CuO(OH–)0.5[19d] whose reduction to Cu* is expected to produce a range of metal clusters and adatoms on the surface with significantly different energies which control the electrocatalytic performance of the electrode. This was indicated in this study by the differences in the current recorded for the reduction of nitrate ions. This is also consistent with previous work on CO2 reduction on copper electrodes prepared from copper oxides, whereby the concomitant reduction of the Cu2O film during CO2 reduction was the key aspect to selectively produce methanol while also maintaining high electrocatalytic activity.[14c] This work also demonstrates that care should be taken when investigating the electrocatalytic behaviour of Cu electrodes and in particular nanostructured Cu, in that performing a cyclic voltammetry analysis such as that shown in Figs. 1 and 2 can significantly influence the performance of the material given the extensive changes to the surface even after 1 cycle. This is expected to be particularly influential in the case of Cu nanoparticles created either chemically or electrochemically which is the focus of future research.
Conclusion
The results presented here show that copper electrodes can be restructured in alkaline solution in a simple manner by applying a repetitive potential cycling protocol involving oxide formation and reduction to generate a nanostructured Cu surface with residual surface oxides. The presence of additives, such as benzyl alcohol and phenylacetic acid, perturbs the oxide growth process and hence the morphology of the deposit once these oxides are reduced. In all cases, the cyclic voltammetric behaviour was distinctly different compared with the 1st cycle of a Cu electrode in alkaline solution, and significant hydrous oxide reduction responses were observed at lower potentials indicating the formation of an active surface. These restructured electrodes showed enhanced performance for the electrocatalytic reduction of nitrate compared with an unmodified Cu electrode, and the best performing material was found when phenylacetic acid was used as an additive. In all cases, nitrate reduction was initiated once residual oxides were removed from the surface, thus exposing fresh active sites that participated in the electrocatalytic reaction. This approach offers an interesting way to nanostructure an electrode surface and could be applied to many other electrode surfaces and electrocatalytic applications.
Experimental
Chemicals
All the chemicals were analytical grade reagents and used as received without further purification. Aqueous solutions of phenylacetic acid (Ajax Finechem), benzyl alcohol (Merck), and NaOH (Aldrich) were made up with deionized water (resistivity 18.2 MΩ cm) purified by use of a Milli-Q reagent deionizer (Millipore). Cu foil (thickness = 0.025 mm, 99.98 % trace metals basis; Aldrich) was used as the substrate.
Sample Preparation
The restructured Cu electrode was achieved using a cyclic voltammetry protocol (1 to 20 cycles recorded at 10 mV s–1 from –1.5 to 0.5 V versus Ag/AgCl) in a solution containing either: (a) 1 M NaOH, (b) 0.1 M benzyl alcohol in 1 M NaOH (the benzyl alcohol solution was prepared by sonicating for 15 min at 40°C and left to cool to room temperature), or (c) 0.1 M phenylacetic acid in 1 M NaOH (the phenylacetic acid solution was prepared by sonicating for 15 min at 40°C and left to cool to room temperature). The restructured surface was washed thoroughly with Milli-Q water and dried with nitrogen gas before surface analysis or any further electrochemical experiments.
Electrochemical Measurements
Cyclic voltammetry experiments were conducted at (22 ± 2)°C with a CH Instruments (CHI 760C) electrochemical analyzer in a electrochemical cell (BAS) that allowed reproducible positioning of the working, reference, and counter electrodes, and a nitrogen inlet tube. The copper electrodes (0.126 cm2 – copper sheets (2 × 0.65 × 0.0025 cm)) were masked off using Kapton tape and were rinsed with Milli-Q water, acetone, and then in ethanol, and dried with nitrogen gas. The reference electrode was Ag/AgCl (aqueous 3 M KCl). For all of the electrochemical experiments carried out, an inert graphite rod (diameter = 5 mm, Johnson Matthey Ultra ‘F’ purity grade) was used as the counter electrode to prevent any possible contamination from dissolution of the counter electrode.[29] Electrocatalysis experiments were performed on the restructured samples that were subjected to 20 cycles in either 1 M NaOH, or 1 M NaOH containing benzyl alcohol or phenylacetic acid. These samples were removed from the electrolyte and washed with Milli-Q water and dried before the nitrate reduction experiments. The latter were carried out using linear sweep voltammetry from –0.8 to –1.6 V at 100 mV s–1 to avoid any further oxide formation before the experiment.
Surface Characterization
SEM measurements were performed on a FEI Nova scanning electron microscope equipped with an AMETEK EDX system (Nova 200), operating at an accelerating voltage of 30 kV. Prior to SEM imaging, the samples were thoroughly rinsed with Milli-Q water and dried under a flow of nitrogen. XPS analysis was performed using a Thermo K-Alpha instrument at pressures better than 10–9 Torr, with the data being referenced to the adventitious C 1s binding energy of 285 eV.
Supplementary Material
SEM image of unmodified Cu electrode, lower magnification images of restructured electrodes, and XPS spectra of restructured electrodes using benzyl alcohol and phenylacetic acid additives are available on the Journal’s website.
Acknowledgements
AOM gratefully acknowledges funding from the Australian Research Council (Future Fellowship FT110100760). Also, the authors acknowledge the instrument and technical support of the RMIT Microscopy and Microanalysis Facility.
References
[1] (a) S. E. F. Kleijn, S. C. S. Lai, M. T. M. Koper, P. R. Unwin, Angew. Chem., Int. Ed. 2014, 53, 3558.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXjt12lsro%3D&md5=d4d521a72ae24885c6689df5c0a4f9e2CAS |
(b) A. P. O’Mullane, Nanoscale 2014, 6, 4012.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Zhou, J. H. Park, F.-R. F. Fan, A. J. Bard, J. Am. Chem. Soc. 2012, 134, 13212.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. S. Bandarenka, M. T. M. Koper, J. Catal. 2013, 308, 11.
| Crossref | GoogleScholarGoogle Scholar |
(e) R. Gilliam, D. Kirk, S. Thorpe, Electrocatalysis 2011, 2, 1.
| Crossref | GoogleScholarGoogle Scholar |
(f) B. J. Plowman, S. K. Bhargava, A. P. O’Mullane, Analyst (Cambridge, U.K.) 2011, 136, 5107.
| Crossref | GoogleScholarGoogle Scholar |
(g) Y.-G. Zhou, E. J. E. Stuart, J. Pillay, S. Vilakazi, R. Tshikhudo, N. V. Rees, R. G. Compton, Chem. Phys. Lett. 2012, 551, 68.
| Crossref | GoogleScholarGoogle Scholar |
[2] Copper Electrodeposition for Nanofabrication of Electronics Devices (Eds K. Kondo, R. N. Akolkar, D. P. Barkey, M. Yokoi) 2014 (Springer: New York).
[3] (a) I. Najdovski, A. P. O’Mullane, J. Electroanal. Chem. 2014, 722–723, 95.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. Nam, R. Kim, D. Han, J. Kim, H. Kwon, Electrochim. Acta 2011, 56, 9397.
| Crossref | GoogleScholarGoogle Scholar |
(c) I. Najdovski, P. R. Selvakannan, S. K. Bhargava, A. P. O’Mullane, Nanoscale 2012, 4, 6298.
| Crossref | GoogleScholarGoogle Scholar |
(d) N. D. Nikolić, G. Brankovic, M. G. Pavlovic, K. I. Popov, J. Electroanal. Chem. 2008, 621, 13.
| Crossref | GoogleScholarGoogle Scholar |
(e) J.-H. Kim, R.-H. Kim, H.-S. Kwon, Electrochem. Commun. 2008, 10, 1148.
| Crossref | GoogleScholarGoogle Scholar |
(f) N. D. Nikolić, K. I. Popov, L. J. Pavlovic, M. G. Pavlovic, Surf. Coat. Technol. 2006, 201, 560.
| Crossref | GoogleScholarGoogle Scholar |
[4] (a) X. Kang, Z. Mai, X. Zou, P. Cai, J. Mo, Anal. Biochem. 2007, 363, 143.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXislWks7o%3D&md5=ab4923829d6e4b426a94e6db5e784ff9CAS | 17288983PubMed |
(b) J. Yang, W.-D. Zhang, S. Gunasekaran, Biosens. Bioelectron. 2010, 26, 279.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) L. Huang, E.-S. Lee, K.-B. Kim, Colloids Surf., A 2005, 262, 125.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmtFCrtr0%3D&md5=4a744588faa6be525a3acb27bdc44475CAS |
(b) J. Luo, H. Zhang, S. Jiang, J. Jiang, X. Liu, Microchim. Acta 2012, 177, 485.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. C. Tang, X. K. Meng, S. Vongehr, Electrochem. Commun. 2009, 11, 867.
| Crossref | GoogleScholarGoogle Scholar |
[6] K. Tan, M.-B. Tian, Q. Cai, Thin Solid Films 2010, 518, 5159.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnt12qsrk%3D&md5=b3773498778062e52fedd0185b1f01eeCAS |
[7] W. Huang, M. Wang, J. Zheng, Z. Li, J. Phys. Chem. C 2009, 113, 1800.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXls1akug%3D%3D&md5=f07162965d3607a84bd4b56c5558e420CAS |
[8] H. Qiu, L. Lu, L. Xue, X. Huang, Electrochim. Acta 2010, 55, 6081.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotlWktrc%3D&md5=ea8f18d1ba537bc73c8dc3d27db9f77dCAS |
[9] A. Weremfo, P. Carter, D. B. Hibbert, C. Zhao, Langmuir 2015, 31, 2593.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXisF2isrk%3D&md5=66d1ebd5eae517b93088fad3b090a5f2CAS | 25669232PubMed |
[10] D. Reyter, M. Odziemkowski, D. Belanger, L. Roue, J. Electrochem. Soc. 2007, 154, K36.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnsVOksLc%3D&md5=17648def032cef9eedd20eff3d328fd7CAS |
[11] L. Yu, H. Sun, J. He, D. Wang, X. Jin, X. Hu, G. Z. Chen, Electrochem. Commun. 2007, 9, 1374.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlslGitbg%3D&md5=e28365ac897f632a630f131039550e9dCAS |
[12] N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding, Z. L. Wang, Science 2007, 316, 732.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXkvVWrsLY%3D&md5=a86dd14385e1b7da5e9919f2a94d41eeCAS | 17478717PubMed |
[13] Z.-Y. Zhou, N. Tian, Z.-Z. Huang, D.-J. Chen, S.-G. Sun, Faraday Discuss. 2009, 140, 81.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) C. Costentin, M. Robert, J.-M. Saveant, Chem. Soc. Rev. 2013, 42, 2423.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXivFKrurc%3D&md5=32f2f675eb5d764ee03e7392fda383ecCAS | 23232552PubMed |
(b) C. W. Li, M. W. Kanan, J. Am. Chem. Soc. 2012, 134, 7231.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz, J. C. Flake, J. Electrochem. Soc. 2011, 158, E45.
| Crossref | GoogleScholarGoogle Scholar |
(d) R. Reske, M. Duca, M. Oezaslan, K. J. P. Schouten, M. T. M. Koper, P. Strasser, J. Phys. Chem. Lett. 2013, 4, 2410.
| Crossref | GoogleScholarGoogle Scholar |
[15] (a) S. Cherevko, C.-H. Chung, Talanta 2010, 80, 1371.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGhu77I&md5=afab41a697b0c1dd47dbe01944473c8eCAS | 20006101PubMed |
(b) S. Sattayasamitsathit, P. Thavarungkul, C. Thammakhet, W. Limbut, A. Numnuam, C. Buranachai, P. Kanatharana, Electroanalysis 2009, 21, 2371.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Zhao, F. Wang, J. Yu, S. Hu, Talanta 2006, 70, 449.
| Crossref | GoogleScholarGoogle Scholar |
[16] (a) J. Davis, M. J. Moorcroft, S. J. Wilkins, R. G. Compton, M. F. Cardosi, Analyst (Cambridge, U.K.) 2000, 125, 737.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXitV2hsLs%3D&md5=9221e76931ab31b6f15620f6ffa20e94CAS |
(b) M. Duca, M. T. M. Koper, Energy Environ. Sci. 2012, 5, 9726.
| Crossref | GoogleScholarGoogle Scholar |
(c) G. E. Badea, Electrochim. Acta 2009, 54, 996.
| Crossref | GoogleScholarGoogle Scholar |
(d) R. Abdallah, F. Geneste, T. Labasque, H. Djelal, F. Fourcade, A. Amrane, S. Taha, D. Floner, J. Electroanal. Chem. 2014, 727, 148.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. S. Lima, M. O. Salles, T. L. Ferreira, T. R. L. C. Paixão, M. Bertotti, Electrochim. Acta 2012, 78, 446.
| Crossref | GoogleScholarGoogle Scholar |
[17] J. Vijayakumar, S. Mohan, Trans. Inst. Met. Finish. 2013, 91, 165.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXnvVOhurc%3D&md5=8f6558856f8f11052e2ac990383c8426CAS |
[18] S. M. Hassan, M. N. Moussa, M. M. El‐Tagoury, A. A. Radi, Anti-Corros. Methods Mater. 1990, 37, 8.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXkt1Oksbc%3D&md5=3f4011024b04fd3eca2ede5917e55c3bCAS |
[19] (a) M. J. A. Shiddiky, A. P. O’Mullane, J. Zhang, L. D. Burke, A. M. Bond, Langmuir 2011, 27, 10302.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVKrtL8%3D&md5=3795d54adadd4d9329d07c38376fe23bCAS |
(b) L. D. Burke, L. M. Kinsella, A. M. O’Connell, Russ. J. Electrochem. 2004, 40, 1105.
| Crossref | GoogleScholarGoogle Scholar |
(c) L. D. Burke, J. A. Collins, J. Appl. Electrochem. 1999, 29, 1427.
| Crossref | GoogleScholarGoogle Scholar |
(d) L. D. Burke, M. J. G. Ahern, T. G. Ryan, J. Electrochem. Soc. 1990, 137, 553.
| Crossref | GoogleScholarGoogle Scholar |
[20] (a) L. D. Burke, J. A. Collins, M. A. Horgan, L. M. Hurley, A. P. O’Mullane, Electrochim. Acta 2000, 45, 4127.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXnsF2lu7c%3D&md5=e17d2bc099536fc65818f36e340f55ffCAS |
(b) L. D. Burke, T. G. Ryan, J. Electrochem. Soc. 1990, 137, 1358.
| Crossref | GoogleScholarGoogle Scholar |
[21] M. Mahajan, S. K. Bhargava, A. P. O’Mullane, Electrochim. Acta 2013, 101, 186.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslOrsrnF&md5=a6c28b40de0d6c566e7db927be641d01CAS |
[22] K. Esumi, T. Itakura, K. Torigoe, Colloids Surf., A. 1994, 82, 111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhslCgur4%3D&md5=18204a9e437cdece91243dff9b714db6CAS |
[23] T. C. Franklin, T. Williams, T. S. N. Sankara Narayanan, R. Guhl, G. Hair, J. Electrochem. Soc. 1997, 144, 3064.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXmslakt7o%3D&md5=a223795ab7cb7e4e9d9353113dcd5bdaCAS |
[24] G. I. Medvedev, N. A. Makrushin, O. V. Ivanova, Russ. J. Appl. Chem. 2004, 77, 1104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXot1Ohtro%3D&md5=6a6f442046402ba7679d8aed62ad1126CAS |
[25] N. Fredj, T. D. Burleigh, J. Electrochem. Soc. 2011, 158, C104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXislamt7k%3D&md5=fc2b04c6b6bb9922e08c72dc49d6fcc2CAS |
[26] R. S. Abdel Hameed, H. I. AlShafey, A. H. Abu-Nawwas, Int. J. Electrochem. Sci. 2014, 9, 6006.
| 1:CAS:528:DC%2BC2cXhs1yrtbvK&md5=9a00152eb4a10f7fa987fda1911d3f01CAS |
[27] L. D. Burke, J. K. Casey, J. A. Morrissey, M. M. Murphy, Bull. Electrochem. 1991, 7, 506.
| 1:CAS:528:DyaK38Xlt1Sgtr8%3D&md5=ec602b246dce408879698d8e42d3ca9aCAS |
[28] B. Lertanantawong, A. P. O’Mullane, W. Surareungchai, M. Somasundrum, L. D. Burke, A. M. Bond, Langmuir 2008, 24, 2856.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhslKntr0%3D&md5=a06e2f9afd74c2d15506f09e7925d3e8CAS | 18266392PubMed |
[29] D. Burke, A. O’Mullane, V. Lodge, M. Mooney, J. Solid State Electrochem. 2001, 5, 319.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlvVGmsbk%3D&md5=ceec50382ff5d6d2878bc5d59ac22117CAS |
† The author, Anthony O’Mullane, is the winner of the 2014 RACI Citation Award.