

Masked Ketenes as Dienophiles in the Diels–Alder Reaction*
Emily G. Mackay A C and Christopher G. Newton B CA Organisch-Chemisches Institut, Westfälische Wilhelms Universität, Corrensstrasse 40, 48149 Münster, Germany.
B Laboratory of Asymmetric Catalysis and Synthesis, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland.
C Corresponding authors. Email: mackay@wwu.de; christopher.newton@epfl.ch

Emily Mackay is a post-doctoral fellow in the research group of Professor Armido Studer at the Westfälische Wilhelms-Universität in Münster. She was awarded the Ludwig Leichhardt Memorial Fellowship from the Alexander von Humboldt Foundation in 2016. She received her Ph.D. from the Australian National University in 2014, where she researched domino Diels–Alder approaches to steroid synthesis, under the supervision of Professor Michael Sherburn. Her research interests include the application of the Diels–Alder reaction in complex settings and functionalization reactions using electron catalysis. |

Christopher Newton obtained his undergraduate degree from Victoria University, New Zealand, and his Ph.D. from the Australian National University under the supervision of Professor Michael Sherburn. His thesis focussed on the synthesis of novel reactive hydrocarbons and their application in the Diels–Alder reaction. In mid-2015, he began a post-doctoral stay as an EPFL Fellow in the group of Professor Nicolai Cramer, Switzerland, working in the fields of ligand design and catalytic enantioselective C–H functionalization. |
Australian Journal of Chemistry 69(12) 1365-1374 https://doi.org/10.1071/CH16428
Submitted: 22 July 2016 Accepted: 12 August 2016 Published: 5 September 2016
Abstract
The Diels–Alder reaction is one of the most powerful, well-established, and versatile reactions in organic chemistry; however, its application in certain settings remains a challenge as a result of functional group incompatibility. In this review, we examine the methods in which masked ketenes can be employed as dienophiles, taking particular note of applications in complex settings.
Introduction
The Diels–Alder (DA) reaction is one of the most important transformations in synthetic organic chemistry. It has played a central role in the development of reactivity theory,[1–3] and has enjoyed a great number of elegant and creative applications in both the academic and industrial setting.[4–8] Despite almost a century of research,[9] the DA reaction remains a highly active area of study, and current fields of interest include the development of new catalytic asymmetric reactions,[10] incorporation of DA reactions into domino sequences,[11,12] the elucidation of natural product biosyntheses,[13–17] and the development of diene-transmissive DA reactions.[18–22]
Although the DA reaction has been used to access a vast array of structural motifs, the reaction is not without limitation. For example, the need to electronically match the diene and dienophile often necessitates the incorporation of superfluous functionality that must be removed or modified during subsequent steps.[23,24] In a similar vein, some functionality is not tolerated and must be masked during the cycloaddition. One such example, and the focus of the present review, is the incompatibility of ketenes as dienophiles. Ketenes react via a hetero-DA–Claisen rearrangement sequence,[25,26] as exemplified by the reaction between butadiene (1) and ketene (2),[27] yielding formal [2+2] cycloadduct 3-vinylcyclobutan-1-one (3) (via intermediate 4, Scheme 1).
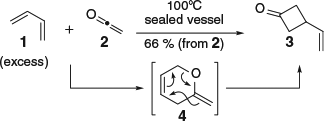
|
Many ingenious examples of masked ketenes as dienophiles have been developed, and this topic has been the focus of two reviews: the first by Ranganathan and coworkers in 1977,[28] and the second by Aggarwal and coworkers in 1999,[29] and as such, the history and development of this field have already been covered. Our intention here is to revisit masked ketene equivalents within the confines of complex molecule synthesis, to best highlight their utility and relevance. Although we have made all efforts to conduct a comprehensive evaluation of the entire field, we focus herein on what we consider to be the most important developments and applications. We have elected to limit our study to the most common mode of reactivity, namely normal electron-demand DA reactions incorporating carbodienes, thus best enabling a comparative study. In addition, we restrict discussion to the most step-economic methodologies,[30] specifically those examples that require two or fewer steps post DA to unmask the ketone functionality.
Masked Ketenes as Dienophiles in the Diels–Alder Reaction
Vinyl Acetates
Vinyl acetate derivatives constitute a large family of masked ketene dienophiles, the first of which was developed by Bartlett and Tate in 1956 (Scheme 2).[31] In their report, cyclopentadiene (5) was reacted with 1-cyanovinyl acetate (6) at 100°C, providing DA adduct 7 in 62 % yield. Ketone 8 was subsequently accessed in excellent yield, albeit under quite forcing conditions, by exposure to a refluxing solution of aqueous NaOH.
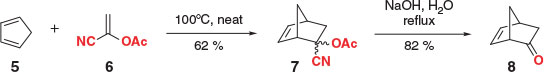
|
Several structural variations to 1-cyanovinyl acetate (6) have since been reported (Fig. 1). For example, replacement of the nitrile group with an ester (dienophile 9) or ketone (dienophile 10) are both tolerated; however, in both cases the unmasking sequence requires more steps than the parent cyano-dienophile.[32] Modifications of the acetate functionality are also known: in 1997, MaGee reported the incorporation of a more electron-withdrawing 2,4-dinitrobenzoate moiety (dienophile 11), providing an increase in DA reactivity of ~5–6 times (comparable reactivity with acrylonitrile (35), discussed in the section Acrylonitriles).[33] In addition, Vogel and coworkers demonstrated that by the incorporation of chiral auxiliaries,[34–39] such as in tartaric acid-derived dienophile 12,[34] enantiomerically pure ketones were accessible via this methodology.
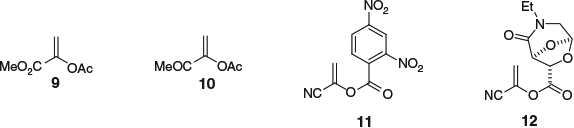
|
The majority of applications involving vinyl acetates as masked ketenes utilize the parent 1-cyanovinyl acetate (6) (Scheme 3). In 1994, Bull et al. applied this methodology to the synthesis of several steroid derivatives containing bridged bicyclic D-rings.[40] In the example depicted, oestrone-derived furan 13 was reacted with 1-cyanovinyl acetate (6) to provide the DA adduct 14 in 32 % yield, along with other isomers (17 %). The ketone was then unmasked under basic conditions, with concurrent cleavage of the second acetate moiety, yielding α-hydroxyester 15 in 82 % yield.

|
In 2007, Hansen et al.[41] demonstrated that the core framework of (–)-colchicine[42] could be accessed via a DA reaction between fused furan 16 and 1-cyanovinyl acetate (6), which, following hydrolysis of the crude mixture of cycloadducts, gave ketones 18 as a 1 : 1 mixture of atropisomers in 77 % yield over the two steps.
Notably, vinyl acetates as masked ketene equivalents are best suited to reaction with cyclic dienes. Only a few examples that employ acyclic dienes have been reported, and significant isomerization of the alkene into conjugation is observed in all cases as a result of the basic conditions employed during the unmasking procedure.[32,33]
Acrylonitriles
2-Chloroacrylonitrile was first employed as a masked ketene in the DA reaction by Paasivirta and Kreiger in 1965.[43,44] Arguably the most famous application came 4 years later from the Corey laboratory within the total synthesis of several prostaglandin natural products (Scheme 4).[45,46] In their report, unstable 5-substituted cyclopentadiene 19 was reacted with 2-chloroacrylonitrile (20), in the presence of a copper Lewis acid catalyst, providing bicyclic adduct 21 as a mixture of diastereoisomers in over 90 % yield. Basic conditions were used to generate ketone 22, presumably via an α-chloro amide intermediate,[47] which was ultimately converted into (±)-prostaglandin-F2α. Notably, Corey’s synthesis of ketone 22 (or related derivatives) has become a benchmark by which the efficiency of many new ketene equivalents is measured.

|
In 1972, Evans demonstrated that under non-catalyzed conditions, 2-chloroacrylonitrile (20) is notably more reactive in the DA reaction than 1-cyanovinyl acetate (6), and reacts with a higher degree of orientational selectivity with unsymmetrical dienes.[48] In the same study, Evans also furthered their utility by developing alternative unmasking reaction conditions, namely Na2S in refluxing ethanol, conditions that are frequently reported to be higher-yielding.[48,49]
Alternative unmasking conditions were also established by Banwell et al., who in studies directed towards the synthesis of paclitaxel, observed that both KOH and Na2S were unsuitable, necessitating development of an alternative protocol (Scheme 5).[50] In this example, a DA reaction between diene 23 and 2-chloroacrylonitrile (20) was performed at high temperature, providing cycloadduct 24 in excellent yield. Using conditions adapted from an earlier paper by Selikson and Watt,[51] the organolithium derived from 24 was treated with O2 to yield an α-cyanohydroperoxide intermediate, followed by cyanohydrin formation with SnCl2, and finally treatment with NaOH gave the final product, ketone 25, in 58 % yield.

|
While conducting structure–activity relationship studies on morphine analogues, Lewis et al. reported a thermal DA reaction between thebaine (26) and 2-chloroacrylonitrile (20), providing a 4 : 1 crude mixture of endo (with respect to the more electron-withdrawing nitrile substituent) and exo isomers 27 and 28, which were subsequently isolated in 43 and 7 % yields respectively after crystallization.[52] Formation of ketone 29 was achieved under standard basic conditions, and the analgetic activity of this substrate, along with a handful of other derivatives, was explored, leading to the discovery of two substrates more potent than morphine.[53]
2-Chloroacrylonitrile (20) has also found application as a ketene equivalent in the industrial sector as part of a multigram route to a preclinical candidate.[54] In this example from Abele and coworkers, the masked ketene underwent a DA reaction with cyclic diene 30 on 800-g scale, utilizing (2,2,6,6-tetramethyl-piperidin-1-yl)oxyl (TEMPO) as a polymerization inhibitor, to provide intermediate cycloadduct 31 as a mixture of isomers. The silyl enol ether was cleaved on treatment with a catalytic quantity of acid in the presence of ethylene glycol, followed by in situ protection of the intermediate ketone to yield ketal 32 in 88 % overall yield from diene 30. Finally, hydrolysis under basic conditions, conducted on a 300-g scale, provided the target 33 in 51 % yield.
In 1968, Brown demonstrated that selective chlorination of acrylonitrile DA adducts was possible, thus establishing a new masked ketene dienophile.[55] In a representative example from Maiti,[56] cyclohexadiene (34) was reacted neat with acrylonitrile (35) at elevated temperatures, followed by exposure of cycloadduct 36 to PCl5 in the presence of excess pyridine, to deliver the ketone precursor 37 in good overall yield (Scheme 6). Despite Brown reporting the overall sequence to be higher-yielding than analogous examples with 2-chloroacrylonitrile,[55] and the fact that acrylonitrile is a more reactive dienophile,[54] the additional chlorination step appears to be a large enough deterrent to prevent this methodology from becoming widely adopted.
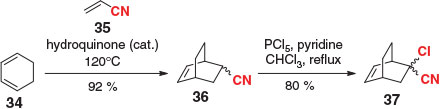
|
As with vinyl acetate derivatives, the use of 2-chloroacrylonitrile (20) as a ketene equivalent is restricted to reaction with cyclic (or 1,1-disusbtituted[57–59]) dienes, as a result of the basic conditions required for their unmasking. When acyclic dienes are employed, elimination of HCN is a competitive process,[60,61] representing a significant limitation to this methodology.
Nitroalkenes
To the best of our knowledge, the first examples of masked ketenes as dienophiles in the DA reaction were disclosed by Wildman and Wildman in 1951.[62] In their report, symmetrical butadienes 38 were reacted with β-nitrostyrenes 39 in hot toluene (Scheme 7). The DA cycloadducts 40 were then subjected to a Nef reaction, yielding a small library of non-conjugated (and non-bridged) ketones 41, a motif that is not accessible via the methodologies discussed earlier.

|
In 1974, Ranganathan et al. reported the first example that utilized nitroethylene as a masked ketene, while simultaneously demonstrating that this dienophile exhibits significantly higher DA reactivity than known alternatives, namely acrylonitrile (35), 2-chloroacrylonitrile (20), and 2-chloroacryloyl chloride (61) (see below), making it more suitable for sensitive or unreactive dienes.[63] However, despite the advantage of their higher DA reactivity, nitroalkenes as ketene equivalents frequently suffer from difficulties associated with the Nef reaction.[64–67] Nevertheless, successful reports in the context of target-oriented synthesis are known (Scheme 8). In 1985, Myers and Corey[68] reacted 2-hydroxy-5-iodobenzoic acid (43)-derived cyclohexadiene 44 in a DA reaction with nitroethylene (45), employing N,N-dibornylamine as a buffer owing to the acid sensitivity of 44. The reaction proceeds with complete orientational and stereoselectivity, yielding nitroalkane 46 in 76 % yield. Following lactone hydrolysis and olefin isomerization to carboxylic acid 47, potassium ruthenate oxidation served to both oxidize the primary alcohol and induce a novel oxidative Nef reaction. Finally, esterification with diazomethane delivered ester 48, which was subsequently converted into the plant hormone (±)-antheridiogen-An.

|
More recently, Thomson et al. disclosed an elegant example of nitroethylene as a masked ketene within an enantioselective total synthesis of (–)-maoecrystal V from 4,4-dimethylcyclohexenone (49).[69] Treatment of α,β-unsaturated ketone 50 with dithium diisopropylamide (LDA) and trimethylsilyl chloride (TMSCl) provided activated diene 51, which underwent a DA reaction with nitroethylene (45), followed by silyl enol ether hydrolysis, to give ketone 52 in 55 % yield. A two-step procedure was used to unmask the ketone, beginning with dithiane protection, followed by in situ reduction of the nitro group to yield amine 53. Finally, oxidation with 2-iodoxybenzoic acid (IBX)[70,71] revealed the ketone 54 in 86 % yield. From this intermediate, only four steps were required to complete the synthesis of the natural product.
The final example of nitroethylene as a masked ketene comes from a 2015 total synthesis of the pseudopterosin (–)-G–J aglycone by Sherburn et al.[72] In this report, highly unreactive Z-substituted diene 56 (accessed in only six steps from crotonaldehyde (55)) was subjected to a high-pressure DA reaction, providing tri-cycle 57 in 82 % yield as a single regio- and diastereoisomer. The ketone was unmasked via treatment with potassium tert-butoxide (tBuOK) and dimethyldioxirane (DMDO),[73] providing the target material 58 in 57 % yield. Oxidation to the catechol was achieved in two steps, thus completing the shortest synthesis of a pseudopterosin to date.[74] As identified by Sherburn and coworkers, this retrosynthetic disconnection of the aglycone via its 1,2-diketone tautomer 59 formally represents a DA reaction between diene 56 and ethylene dione (60), a reagent for which a synthetic equivalent has yet to be developed.
Although not yet realized, there is opportunity for an enantioselective sequence incorporating nitroalkenes as masked ketenes, as enantioselective DA reactions of nitroethylene[75] and substituted derivatives[76,77] are known.
2-Chloroacryloyl Chloride
In further studies directed towards the synthesis of prostaglandin natural products (see Scheme 4 for earlier work), Corey et al. reported the development of an improved, highly reactive ketene equivalent (comparable in reactivity with maleic anhydride) that avoids the need for a strong base during the unmasking procedure (Scheme 9).[78] Thus 2-chloroacrolyl chloride (61) was reacted (under non-catalyzed conditions) with substituted cyclopentadiene 19 at 0°C, which, following concentration of the reaction under reduced pressure, yielded DA adduct 62 in quantitative yield. The crude material was subsequently heated with sodium azide in anhydrous dimethoxyethane (DME), followed by the addition of ethanoic acid, providing the target ketone 22 in excellent yield. Mechanistically, the reaction proceeds via an initial acyl substitution, giving intermediate azide 63. A Curtius rearrangement then provides isocyanate 64, which under the aqueous reaction conditions breaks down to provide amine 65. The ketone is formed via subsequent elimination and hydrolysis. This interesting approach has not found much favour in the literature, perhaps in part owing to the intermediacy of a potentially dangerous azide, or as a result of difficulties associated with the synthesis[79] and handling of the dienophile. Notably, Corey also demonstrated 2-chloroacrolyl chloride (61) could be successfully employed with acyclic dienes, with little to no isomerization of the alkene into conjugation during ketone unmasking.
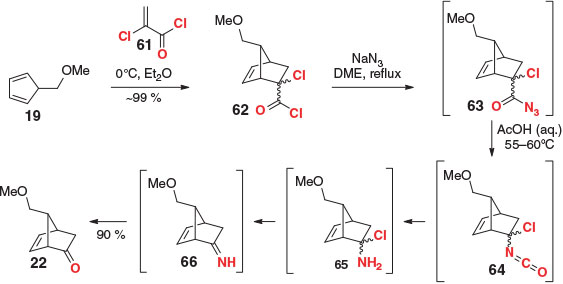
|
Acrylates
Although examples of acrylates as masked ketene equivalents have been known for many years, their application typically requires several synthetic steps to reveal the ketone.[80–83] However, in 2007, Yamamoto and coworkers published an efficient unmasking procedure as part of a formal synthesis of platensimycin (Scheme 10).[84] Utilizing their earlier described DA reaction between 2-methylcyclopenta-1,3-diene (67) and methyl acrylate (68), employing 69 as a Brønsted acid-assisted chiral Lewis catalyst, cycloadduct 70 was accessed in 92 % yield, and with excellent enantio-, diastereo-, and regioselectivity.[85] The ketone was subsequently unmasked in a one-pot operation by treatment with LDA and PhNO, followed by warming in the presence of LiOH. Mechanistic studies[86] indicate the reaction proceeds via initial formation of N-hydroxylamine 71, followed by intramolecular transesterification to spiro-oxazetidinone intermediate 72. Extrusion of CO2 to yield imine 73, followed by hydrolysis to ketone 74, completes the unmasking. This intermediate was converted in eight steps to complex polycycle 75, thus intercepting an intermediate from Nicolaou’s 2006 total synthesis of platensimycin.[87] Yamamoto’s unmasking procedure has since been applied in simple systems by a handful of other researchers;[88,89] however, attempts in more complex settings have not been met with success.[82]

|
Vinylsulfonyl Chloride
An elegant strategy to control the orientational selectivity of DA reactions with unsymmetrical dienes was disclosed by Metz, Fleischer, and Fröhlich.[90] In their study, depicted in Scheme 11, a sulfonyl tether was introduced via a simple substitution reaction of alcohol 76 with vinylsulfonyl chloride (77), providing intermediate 78, which underwent an intramolecular DA reaction in situ to yield tri-cycle 79 in 64 % yield. The ketone was then revealed in a two-step sequence, by lithiation and subsequent trapping with 2-methoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (MeO-Bpin), providing tri-cycle 80. This intermediate was not isolated, but rather treated with meta-chloroperoxybenzoic acid (m-CPBA), resulting in a chemoselective oxidation to the target ketone 81. Notably, the authors demonstrated that reaction of diene 76 with 1-cyanovinyl acetate (6) provided a complex mixture of orientational regioisomers, highlighting the advantage of their new strategy. However, this approach necessitates appropriate proximate functionality, limiting its application. Despite this restriction, the strategy has since been implemented by Winterfeldt et al. in an enantioselective total syntheses of (–)-myltaylenol.[91]

|
Reversing Orientational Selectivity
All of the ketene equivalents presented thus far convert the most electron-withdrawing group of the dienophile into the carbonyl. Challenges arise when the inverse orientational selectivity of the DA reaction is desired, as demonstrated by Overman and coworkers in their synthesis of (±)-didehydrostemofoline (asparagamine A) and (±)-isodidehydrostemofoline (Scheme 12).[92] In their approach, an ideal route from pyrrole 82 to key intermediate 84 would employ a DA reaction with nitroethylene (45) (or equivalent); however, owing to the mismatched electronic properties of the two components, 83 is not accessible. Instead, a less direct route was taken, using dienophile 85. In this case, the nitro group behaved as a removable activating group, while the α,β-unsaturated ester acted as the masked ketene. Following the DA reaction, the double bond was hydrogenated, giving intermediate 86 in 73 % yield. Four steps were necessary to remove the nitro group, protect the alcohol, and reduce the ester, providing alcohol 87 in 51 % yield. A final oxidation–silyl enol ether formation–ozonolysis sequence delivered the target 84 in 72 % yield. A further 19 steps were required to furnish (±)-didehydrostemofoline and (±)-isodidehydrostemofoline, thus completing the first total syntheses of these molecules.
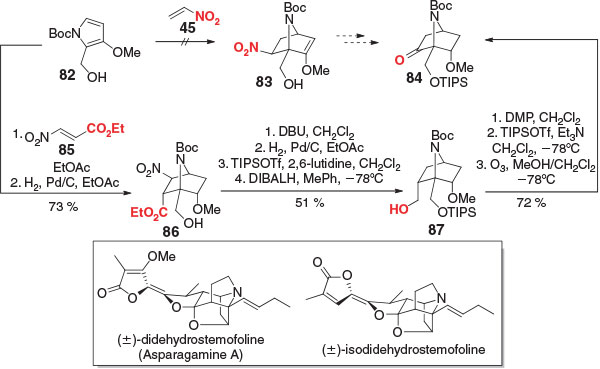
|
Allenes have also been applied as masked ketenes via ozonolysis of the exocyclic alkene post DA reaction; thus, appropriately functionalized allenic dienophiles could enable (formally) inverse orientational selectivity to that typically seen with standard masked ketenes. Although not yet applied with an unsymmetrical diene, a potentially suitable dienophile was introduced by Pavri and Trudell in their formal synthesis of (±)-epibatidine (Scheme 13).[93] In their study, a DA reaction between tert-butyloxycarbonyl (BOC)-protected pyrrole 88 and sulfonyl allene 89 gave the endo cycloadduct 90 as the sole product in 45 % yield. A regioselective hydrogenation of the 1,2-disubstituted alkene yielded the exocyclic alkene 91, allowing for ozonolysis, followed by removal of the sulfonyl activating group via treatment with aluminium amalgam, providing bi-cycle 92 in 47 % yield, and thus intersecting an intermediate from the 1993 total synthesis by Fletcher et al. of the natural product.[94] Although this example demonstrates that allenes can be effective masked ketenes in DA reactions, the requisite hydrogenation before unmasking limits their utility.

|
Conclusions and Future Prospects
Several known ketene equivalents have not been discussed throughout the present review, such as acrylic acid (93),[95,96] silyl enol ether 94,[97] α-thioacrylate 95,[98] phenylseleno acetylene 96,[99] and acetoxymaleic anhydride (97),[100] to name only a few (Scheme 14). As far as we are aware, none of these examples (or any other masked ketene equivalents not covered) have been applied outside simple settings. For some dienophiles, it is easy to understand why: 96 and 97 require three and four steps respectively for ketone unmasking, whereas silyl enol ether 94 exhibits low DA reactivity, necessitating the use of strong Lewis acids for reaction. For others, however, it is less clear why they have not been widely adopted, and perhaps they still have much to offer. In particular, we believe vinyl boranes 98 and their more highly oxidized derivatives hold great promise as masked ketenes in the DA reaction. Although much attention has been paid by Singleton and coworkers to their use as both versatile and reactive alkenyl alcohol equivalents in the DA reaction, via conversion of cycloadducts adducts 99 to 100,[101–104] a simple oxidation to ketone 101 would represent their application as a masked ketene.

|
In conclusion, we hope the examples highlighted within this review demonstrate the power of masked ketenes in the DA reaction. Since the first report by Wildman and Wildman, several creative and well-executed examples have been reported, enabling the synthesis of several highly complex and biologically important natural products. However, we also hope that this survey serves to highlight the limitations of current methods. Specifically, the low DA reactivity of many ketene equivalents often necessitates reasonably forcing conditions, creating potential incompatibility issues. Although this problem has been partially addressed with the advent of more reactive ketene equivalents, most notably nitroethylene, challenges associated with the Nef reaction limit the utility of this dienophile. As yet, a true one-pot procedure where the DA reaction and unmasking can be performed together has not been disclosed; however, Corey’s 2-chloroacrolyl chloride (61) is perhaps compatible. Furthermore, development of an ethylene dione (60) equivalent would be a valuable extension to this field, thus enabling the rapid synthesis of catechols via the DA reaction. Other areas that would benefit greatly from further research include masked ketenes with an inverse polarity to those currently in existence, as well as advancements to intramolecular and asymmetric variants, enabling their application in more complex settings.
* Christopher G. Newton is the recipient of the 2015 RACI Mander Best Ph.D. Thesis in Organic Chemistry Award.
Acknowledgements
E.G.M. acknowledges support from the Alexander von Humboldt Foundation (Ludwig Leichhardt Memorial Fellowship). C.G.N. acknowledges support from the ‘EPFL Fellows’ fellowship program co-funded by Marie Skłodowska-Curie, Horizon 2020 grant agreement no. 665667.
References
[1] R. B. Woodward, R. Hoffmann, Angew. Chem. Int. Ed. Engl. 1969, 8, 781.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3cXjsFymtA%3D%3D&md5=4492a6f02238b26d16014b1b25541464CAS |
[2] A. Streitwieser, Science 1981, 214, 627.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXmtF2gtLo%3D&md5=8678cb88668bb82ae9a3aa93d5d551bbCAS | 17839634PubMed |
[3] I. Fleming, Molecular Orbitals and Organic Chemical Reactions (Reference edn) 2011 (John Wiley & Sons, Ltd: Chichester).
[4] K. C. Nicolaou, S. A. Snyder, T. Montagnon, G. Vassilikogiannakis, Angew. Chem. Int. Ed. 2002, 41, 1668.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XktFymtbw%3D&md5=3723c63257537dba2e0822c0bd8f6dd5CAS |
[5] E. G. Mackay, M. S. Sherburn, Synthesis 2014, 1.
| Crossref | GoogleScholarGoogle Scholar |
[6] J.-A. Funel, S. Abele, Angew. Chem. Int. Ed. 2013, 52, 3822.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjtlaks78%3D&md5=301fe599918769153525502a1d3fd097CAS |
[7] M. M. Heravi, V. F. Vavsari, RSC Adv. 2015, 5, 50890.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXos1ektLY%3D&md5=198318e9496bc26c3663fad888e844baCAS |
[8] E. J. Corey, Angew. Chem. Int. Ed. 2002, 41, 1650.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XktFymtb8%3D&md5=fec49009681f0c614edcb889178b6feaCAS |
[9] O. Diels, K. Alder, Liebigs Ann. 1928, 460, 98.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaB1cXpsFyn&md5=d9485e3f29cd933e1d90c3a2b35cec5bCAS |
[10] T. Gatzenmeier, M. van Gemmeren, Y. Xie, D. Höfler, M. Leutzsch, B. List, Science 2016, 351, 949.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XivFOnt7o%3D&md5=7a285577892c258007127c0e101582d6CAS | 26917765PubMed |
[11] E. G. Mackay, M. Nörret, L. S. M. Wong, I. Louis, A. L. Lawrence, A. C. Willis, M. S. Sherburn, Org. Lett. 2015, 17, 5517.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhvVejtbfK&md5=d378d50dcac3c8d7a37ef3481d3926d9CAS | 26523759PubMed |
[12] J. E. Sears, D. L. Boger, Acc. Chem. Res. 2016, 49, 241.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XitVygsL8%3D&md5=faf189a6cf5a96d0dd530bdf0b1f8275CAS | 26813287PubMed |
[13] S. L. Drew, A. L. Lawrence, M. S. Sherburn, Chem. Sci. 2015, 6, 3886.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXnvFWnsLw%3D&md5=97af34977ddfdacefcd5eb43f2a59bc3CAS |
[14] R. T. Larson, R. P. Pemberton, J. M. Franke, D. J. Tantillo, R. J. Thomson, J. Am. Chem. Soc. 2015, 137, 11197.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtlygt73N&md5=270d1e01fd5f60b4abbbfca28834a2b8CAS | 26305231PubMed |
[15] C. Wan, J. Deng, H. Liu, M. Bian, A. Li, Sci. China Chem. 2014, 57, 926.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXptlKqt7g%3D&md5=b88321b054df1af8c30f641bcb503b6aCAS |
[16] R. S. Harvey, E. G. Mackay, L. Roger, M. N. Paddon-Row, M. S. Sherburn, A. L. Lawrence, Angew. Chem. Int. Ed. 2015, 54, 1795.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXitVygt7jO&md5=1ec9cb5f80359e8d9ab19cfe26ee8904CAS |
[17] S. L. Drew, A. L. Lawrence, M. S. Sherburn, Angew. Chem. Int. Ed. 2013, 52, 4221.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjsVKruro%3D&md5=b90af3f34600f92aaa0164c0c05b1419CAS |
[18] E. G. Mackay, M. S. Sherburn, Pure Appl. Chem. 2013, 85, 1227.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXpslKnsL4%3D&md5=dd3edec26dc6ad2055447b72297721ceCAS |
[19] H. Hopf, M. S. Sherburn, Angew. Chem. Int. Ed. 2012, 51, 2298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xitlygsr0%3D&md5=1abb9ff20dde85e646053abf19baeebcCAS |
[20] M. S. Sherburn, Acc. Chem. Res. 2015, 48, 1961.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtFWqt77P&md5=f696f3cb89dc744e4ba2733337614472CAS | 26151489PubMed |
[21] C. G. Newton, M. S. Sherburn, in Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry (Eds H. Hopf, M. S. Sherburn) 2016, pp. 413–443 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).
[22] K. M. Cergol, C. G. Newton, A. L. Lawrence, A. C. Willis, M. N. Paddon-Row, M. S. Sherburn, Angew. Chem. Int. Ed. 2011, 50, 10425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFOls7rI&md5=e0887c575f2511c38e68020bf4d3ff5dCAS |
[23] M. E. Jung, F. Perez, C. F. Regan, S. W. Yi, Q. Perron, Angew. Chem. Int. Ed. 2013, 52, 2060.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXntFyqtA%3D%3D&md5=0f6e7523ce9b7c7950957ac9cdd84cb0CAS |
[24] E. Taarning, R. Madsen, Chem. – Eur. J. 2008, 14, 5638.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXoslamsLw%3D&md5=9ec15ab836badc9f329c7751136f5ed5CAS | 18470851PubMed |
[25] S. Yamabe, T. Dai, T. Minato, T. Machiguchi, T. Hasegawa, J. Am. Chem. Soc. 1996, 118, 6518.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xjs1Grsbo%3D&md5=ee01e2ba9ef91a45c436e477b9f9101fCAS |
[26] U. Salzner, S. M. Bachrach, J. Org. Chem. 1996, 61, 237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XhtlGqtg%3D%3D&md5=a5e1f06d6030ddbd74a17fd94b5ad838CAS |
[27] E. Vogel, K. Müller, Liebigs Ann. 1958, 615, 29.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG1MXit12ntw%3D%3D&md5=7ee8b774da957fdc045f970a357a15dfCAS |
[28] S. Ranganathan, D. Ranganathan, A. K. Mehrotra, Synthesis 1977, 289.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2sXksFarsb0%3D&md5=24b2bb8e0e795764b2e9eab9d472bc47CAS |
[29] V. K. Aggarwal, A. Ali, M. P. Coogan, Tetrahedron 1999, 55, 293.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXltVaitA%3D%3D&md5=14253506c0694fc2c429acb7d1972bd6CAS |
[30] P. A. Wender, V. A. Verma, T. J. Paxton, T. H. Pillow, Acc. Chem. Res. 2008, 41, 40.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVOntL3E&md5=cb67027ea5105f047a9a1045007f6504CAS | 18159936PubMed |
[31] P. D. Bartlett, B. E. Tate, J. Am. Chem. Soc. 1956, 78, 2473.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG2sXhs1al&md5=9a0f408e4f6daf4902776ab0c5d3cb4fCAS |
[32] P. S. Wharton, B. T. Aw, J. Org. Chem. 1966, 31, 3787.
| Crossref | GoogleScholarGoogle Scholar |
[33] D. MaGee, M. Lee, Synlett 1997, 786.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlt1ahsLk%3D&md5=88b6950005dd56175c115c7f2f2f3cdcCAS |
[34] J. -L. Reymond, P. Vogel, J. Chem. Soc. Chem. Commun. 1990, 1070.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXot1ahsQ%3D%3D&md5=487c139038f99720a7ff9437ed386cf7CAS |
[35] P. Vogel, D. Fattori, F. Gasparini, C. Le Drian, Synlett 1990, 173.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXht1aqs7Y%3D&md5=daa09780054ee70720be2f158de67baeCAS |
[36] J. L. Reymond, P. Vogel, Tetrahedron Asymmetry 1990, 1, 729.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXktVyntr8%3D&md5=2b3fba98949c279e9c3e79b12b52507fCAS |
[37] F. Claret, P. -A. Carrupt, P. Vogel, Helv. Chim. Acta 1987, 70, 1886.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXktlOlu78%3D&md5=93f8f63c5eedab986705f99d722dd79aCAS |
[38] E. Vieira, P. Vogel, Helv. Chim. Acta 1983, 66, 1865.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXosFGhsw%3D%3D&md5=8e25d6a32845a40b7381ead034438351CAS |
[39] P. Kernen, P. Vogel, Tetrahedron Lett. 1993, 34, 2473.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXltFOlu7o%3D&md5=03e76b5c9000edfe630b0e50e951aca4CAS |
[40] J. R. Bull, C. Grundler, H. Laurent, R. Bohlmann, A. Müller-Fahrnow, Tetrahedron 1994, 50, 6347.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXmsFKisrc%3D&md5=66f8e200f5ec583397707fb2bb617accCAS |
[41] P. Uebelhart, C. Weymuth, A. Linden, H.-J. Hansen, Helv. Chim. Acta 2007, 90, 659.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXkvFequ7s%3D&md5=be3c797ff0715f3c0c1ed51710292e95CAS |
[42] T. Graening, H. -G. Schmalz, Angew. Chem. Int. Ed. 2004, 43, 3230.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXlsF2gs7c%3D&md5=ad796615c85681a369f1e7136bddad0bCAS |
[43] J. Paasi-virta, H. Krieger, Suom. Kemistil. 1965, B38, 182.
| 1:CAS:528:DyaF28XislWlug%3D%3D&md5=359df0962e42b5154dbd2bbb022ef375CAS |
[44] H. Krieger, K. Manninen, J. Paasi-virta, Suom. Kemistil. 1966, B39, 8.
[45] E. J. Corey, N. M. Weinshenker, T. K. Schaaf, W. Huber, J. Am. Chem. Soc. 1969, 91, 5675.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF1MXltVWgtLg%3D&md5=30d97e527f05b44fb951a0a46b98257cCAS | 5808505PubMed |
[46] K. C. Nicolaou, E. J. Sorensen, Classics in Total Synthesis: Targets, Strategies, Methods 1996 (Wiley-VCH: Weinheim).
[47] C. S. Shiner, A. M. Fisher, F. Yacoby, Tetrahedron Lett. 1983, 24, 5687.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhsFKhtLk%3D&md5=456a5ab335c60af6a0c9eb9fe5094cadCAS |
[48] D. A. Evans, W. L. Scott, L. K. Truesdale, Tetrahedron Lett. 1972, 13, 121.
| Crossref | GoogleScholarGoogle Scholar |
[49] A. Coop, D. Y. Maeda, Heterocycles 2001, 55, 1147.
| Crossref | GoogleScholarGoogle Scholar |
[50] M. G. Banwell, G. R. Clark, D. C. R. Hockless, S. Pallich, Aust. J. Chem. 2001, 54, 691.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xjt1Cjsr0%3D&md5=6df8e2fe11736c5c58d691fc5fa32f74CAS |
[51] S. J. Selikson, D. S. Watt, J. Org. Chem. 1975, 40, 267.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXos1Wltw%3D%3D&md5=d7a279a735c0bdf2a383e78fadbfa9f4CAS |
[52] J. W. Lewis, M. J. Readhead, I. A. Selby, A. C. B. Smith, C. A. Young, J. Chem. Soc. C 1971, 1158.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXktVKitrY%3D&md5=b313ca5e4697a2c2420702cb28209af7CAS |
[53] J. W. Lewis, M. J. Readhead, A. C. B. Smith, J. Med. Chem. 1973, 16, 84.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3sXpt1Cjtw%3D%3D&md5=85ae39ea5bdf35ab634658a42cd7e4e5CAS | 4682204PubMed |
[54] J. -A. Funel, G. Schmidt, S. Abele, Org. Process Res. Dev. 2011, 15, 1420.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovFWhtbo%3D&md5=f2cc24544599a7abff88c8e41cb758d7CAS |
[55] P. K. Freeman, D. M. Balls, D. J. Brown, J. Org. Chem. 1968, 33, 2211.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF1cXkt1Krtrs%3D&md5=80f81211fd265ff8d09397f3ef7756ebCAS |
[56] S. N. Maiti, R. Ling, J. Yip, C. Gao, D. Nguyen, U.S. Patent 20110288063 A1 2011.
[57] K. C. Nicolaou, C. K. Hwang, E. J. Sorensen, C. F. Clairborne, J. Chem. Soc. Chem. Commun. 1992, 1117.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XmtFaktrw%3D&md5=c87dbd49861d7f3ac832ae0717925b80CAS |
[58] K. C. Nicolaou, J. J. Liu, Z. Yang, H. Ueno, E. J. Sorensen, C. F. Claiborne, R. K. Guy, C. K. Hwang, M. Nakada, P. G. Nantermet, J. Am. Chem. Soc. 1995, 117, 634.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXivFyjsLo%3D&md5=41b29ba069d1705cf48f21d10e601005CAS |
[59] T. Masamune, A. Fukuzawa, A. Furusaki, M. Ikura, H. Matsue, T. Kaneko, A. Abiko, N. Sakamoto, N. Tanimoto, M. Akio, Bull. Chem. Soc. Jpn. 1987, 60, 1001.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXnsFKlsQ%3D%3D&md5=d9b037c1d349ce44d10563dd7b677924CAS |
[60] J. E. Baldwin, M. Otsuka, P. M. Wallace, J. Chem. Soc. Chem. Commun. 1985, 1549.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XitVKqu7g%3D&md5=538d3bdbf18e43b2bf73604f93ff2368CAS |
[61] E. J. Alvarez-Manzaneda, R. Chahboun, E. Cabrera, E. Alvarez, A. Haidour, J. M. Ramos, R. Alvarez-Manzaneda, M. Hmamouchi, H. Bouanou, J. Org. Chem. 2007, 72, 3332.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjsVOrsb8%3D&md5=d40561caa1000225d9726a70c03ab38bCAS | 17388632PubMed |
[62] W. C. Wildman, R. B. Wildman, J. Org. Chem. 1952, 17, 581.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG3sXptFer&md5=1cb958b93a56cf041217b1f849a756a2CAS |
[63] S. Ranganathan, D. Ranganathan, A. K. Mehrotra, J. Am. Chem. Soc. 1974, 96, 5261.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2cXltVCrs7s%3D&md5=72a550aa023c132588cea67392e8a723CAS |
[64] G. Mehta, D. Subrahmanyam, J. Chem. Soc., Perkin Trans. 1 1991, 395.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXitVOmsb4%3D&md5=7c49e01e7c724eba9034e5e006f843d7CAS |
[65] C. M. Moorhoff, L. A. Paquette, J. Org. Chem. 1991, 56, 703.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXotFSksw%3D%3D&md5=cb3f5c8558342778998306a84e0aaf34CAS |
[66] H. E. Zimmerman, P. Wang, J. Org. Chem. 2002, 67, 9216.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XovFykurs%3D&md5=71e6dc8331006df66bcc766aabe75d61CAS | 12492323PubMed |
[67] K. E. Lazarski, D. X. Hu, C. L. Stern, R. J. Thomson, Org. Lett. 2010, 12, 3010.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmvFaktLs%3D&md5=8a6c3581468e1948c7cb60d930c0064fCAS | 20521835PubMed |
[68] E. J. Corey, A. G. Myers, J. Am. Chem. Soc. 1985, 107, 5574.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXlslyntbo%3D&md5=774397451fa9314b595f3343b7077323CAS |
[69] C. Zheng, I. Dubovyk, K. E. Lazarski, R. J. Thomson, J. Am. Chem. Soc. 2014, 136, 17750.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXitFans7nJ&md5=eef967a16f9074f1d66bc29b24eb0c94CAS | 25495370PubMed |
[70] K. C. Nicolaou, C. J. N. Mathison, T. Montagnon, Angew. Chem. Int. Ed. 2003, 42, 4077.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnslSlsbw%3D&md5=7c9b9dd5839402fef842f4ecc86b4d37CAS |
[71] K. C. Nicolaou, C. J. N. Mathison, T. Montagnon, J. Am. Chem. Soc. 2004, 126, 5192.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXisl2itr8%3D&md5=e9794c776bd87705e03032d8bad79ff3CAS | 15099102PubMed |
[72] C. G. Newton, S. L. Drew, A. L. Lawrence, A. C. Willis, M. N. Paddon-Row, M. S. Sherburn, Nat. Chem. 2014, 7, 82.
| Crossref | GoogleScholarGoogle Scholar | 25515894PubMed |
[73] W. Adam, M. Makosza, C. R. Saha-Möller, C. -G. Zhao, Synlett 1998, 1335.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXot1Kq&md5=ca5f2cbeaf5352130c6be36f0f591de2CAS |
[74] C. G. Newton, M. S. Sherburn, Nat. Prod. Rep. 2015, 32, 865.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXms1GqtL4%3D&md5=24c2479e9864c764ff78a2b907de19b0CAS | 25882677PubMed |
[75] M. J. Narcis, D. J. Sprague, B. Captain, N. Takenaka, Org. Biomol. Chem. 2012, 10, 9134.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1equ7nP&md5=8616f5400fc2492c798fc995531f4d1dCAS | 23104427PubMed |
[76] Z.-J. Jia, Q. Zhou, Q.-Q. Zhou, P.-Q. Chen, Y.-C. Chen, Angew. Chem. Int. Ed. 2011, 50, 8638.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptFOjt7o%3D&md5=8dfc056c2965edbcd2708340b5269c59CAS |
[77] J.-G. Fu, Y.-F. Shan, W.-B. Sun, G. -Q. Lin, B. -F. Sun, Org. Biomol. Chem. 2016, 14, 5229.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XotF2qs7w%3D&md5=0623b69489e7bc01f240157707a9541bCAS | 27219468PubMed |
[78] E. J. Corey, T. Ravindranathan, S. Terashima, J. Am. Chem. Soc. 1971, 93, 4326.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXltV2qs7Y%3D&md5=61a200f6e607e4730b4c765a0da7ece0CAS |
[79] C. S. Marvel, J. Dec, H. G. Cooke, J. C. Cowan, J. Am. Chem. Soc. 1940, 62, 3495.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaH3MXmsVGq&md5=a02c2b0c83f1cb73d499f09d3bc985a9CAS |
[80] E. J. Corey, N. Imai, S. Pikul, Tetrahedron Lett. 1991, 32, 7517.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhtVGqs74%3D&md5=00c1d015b7f0a7655bc5c9090067487aCAS |
[81] W. Oppolzer, C. Chapuis, D. Dupuis, M. Guo, Helv. Chim. Acta 1985, 68, 2100.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXhslKltb8%3D&md5=6516cd6edb07fb58286676a58014b00bCAS |
[82] C. Ebner, E. M. Carreira, Angew. Chem. Int. Ed. 2015, 54, 11227.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXht1KitbjE&md5=cc02f81aac524e9949a7e4ec3f6908afCAS |
[83] R. L. Snowden, S. M. Linder, M. Wüst, Helv. Chim. Acta 1989, 72, 892.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXhsFOksLs%3D&md5=d6b02a6bba86115b035b0d794b069dd4CAS |
[84] P. Li, J. N. Payette, H. Yamamoto, J. Am. Chem. Soc. 2007, 129, 9534.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvVSiu78%3D&md5=eb97f94668c1e7263af35192492f06e9CAS | 17630748PubMed |
[85] J. N. Payette, H. Yamamoto, J. Am. Chem. Soc. 2007, 129, 9536.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXns1Ckurk%3D&md5=79a0aa07c0ec91852f1672ffe4df5835CAS | 17630749PubMed |
[86] J. N. Payette, H. Yamamoto, J. Am. Chem. Soc. 2008, 130, 12276.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVChsL%2FM&md5=bf266ca0dc63dbf0211991a11d4603dcCAS | 18722431PubMed |
[87] K. C. Nicolaou, A. Li, D. J. Edmonds, Angew. Chem. Int. Ed. 2006, 45, 7086.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1ans7bL&md5=228841dfa3cb7dbffdce594f3e3f6c74CAS |
[88] M. Olbrich, P. Mayer, D. Trauner, Org. Biomol. Chem. 2014, 12, 108.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVKjt73O&md5=3ac4c4e2a8e6b7a698957080ae35024fCAS | 24217004PubMed |
[89] S. Mukherjee, A. P. Scopton, E. J. Corey, Org. Lett. 2010, 12, 1836.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvVeju7k%3D&md5=109827cf03d67082cec0fc09594f2cccCAS | 20337419PubMed |
[90] P. Metz, M. Fleischer, R. Fröhlich, Tetrahedron 1995, 51, 711.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjsVGqtL0%3D&md5=177ed5a0f2faf91bf8710d481855187bCAS |
[91] S. Doye, T. Hotopp, E. Winterfeldt, Chem. Commun. 1997, 1491.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlvV2ntrs%3D&md5=28a3397c6ff809124ceba5699cc12dcdCAS |
[92] M. Brüggemann, A. I. McDonald, L. E. Overman, M. D. Rosen, L. Schwink, J. P. Scott, J. Am. Chem. Soc. 2003, 125, 15284.
| Crossref | GoogleScholarGoogle Scholar | 14664560PubMed |
[93] N. P. Pavri, M. L. Trudell, Tetrahedron Lett. 1997, 38, 7993.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXnsV2hu7w%3D&md5=166bdca5b1c516d9814b19e4cdf0d765CAS |
[94] S. R. Fletcher, R. Baker, M. S. Chambers, S. C. Hobbs, P. J. Mitchell, J. Chem. Soc. Chem. Commun. 1993, 1216.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhtVOnsbs%3D&md5=f18a99538bdd2a456607e1ea7bdd3ee0CAS |
[95] H. H. Wasserman, B. H. Lipshutz, Tetrahedron Lett. 1975, 16, 4611.
| Crossref | GoogleScholarGoogle Scholar |
[96] B. M. Trost, Y. Tamaru, J. Am. Chem. Soc. 1975, 97, 3528.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXkslOkurg%3D&md5=7a45abf0bfaaf84d9f1b34704d329713CAS |
[97] T. Sasaki, Y. Ishibashi, M. Ohno, Tetrahedron Lett. 1982, 23, 1693.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XltV2ktL4%3D&md5=6684699091d2ad195fb55be7e25aff1dCAS |
[98] V. K. Aggarwal, D. E. Jones, A. M. Martin-Castro, Eur. J. Org. Chem. 2000, 2939.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmtV2lsL4%3D&md5=fe38940e25bd4812c7c433249162779eCAS |
[99] T. G. Back, D. Wehrli, Synlett 1995, 1123.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXpslCrtL0%3D&md5=265e1e21ae36fbfc04a37c5710b13482CAS |
[100] E. Wenkert, C. Vial, F. Näf, Chimia 1992, 46, 95.
| 1:CAS:528:DyaK38Xks12ntbY%3D&md5=0a8d8ee6a5981dca97f37910a6c0fb74CAS |
[101] D. A. Singleton, J. P. Martinez, J. Am. Chem. Soc. 1990, 112, 7423.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXlsFamsLk%3D&md5=35e746157d38e8bfb199ab880ca25fc2CAS |
[102] D. A. Singleton, J. P. Martinez, J. V. Watson, Tetrahedron Lett. 1992, 33, 1017.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhvFekt7g%3D&md5=fa677c75e7700e7731c38df44ed390faCAS |
[103] D. A. Singleton, J. P. Martinez, J. V. Watson, G. M. Ndip, Tetrahedron 1992, 48, 5831.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XmtFamt7s%3D&md5=d669e9659c039e8f5803b162d7883bc9CAS |
[104] D. A. Singleton, J. P. Martinez, Tetrahedron Lett. 1991, 32, 7365.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38Xht12jsr0%3D&md5=d1b6feda714a739a24814fad8c3ac21bCAS |