

The Pseudoproline Approach to Peptide Cyclization*
Katrina A. JolliffeSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia. Email: kate.jolliffe@sydney.edu.au
Australian Journal of Chemistry 71(10) 723-730 https://doi.org/10.1071/CH18292
Submitted: 19 June 2018 Accepted: 5 July 2018 Published: 13 August 2018
Abstract
The development of efficient methods for the synthesis of cyclic peptides is of interest because of the many potential applications of this class of molecule. Pseudoprolines are derived from serine, threonine, and cysteine and can be used as traceless turn-inducers to facilitate the cyclization of a wide range of linear peptide precursors. The incorporation of a pseudoproline into the peptide to be cyclized generally results in a cyclization reaction that proceeds more quickly and with higher yield than that of an analogous sequence without the pseudoproline. Installation of a pseudoproline at the C-terminal position of a linear peptide sequence has also been shown to eliminate any epimerization of this residue during the reaction. Following pseudoproline-mediated cyclization, these turn-inducers can be removed on treatment with acid in a similar manner to other protecting groups to provide the native peptide sequence, and in the case of cysteine-derived pseudoprolines, the resulting cysteine can be readily converted into alanine through desulfurization. These traceless turn-inducers have been successfully used in the synthesis of cyclic peptides containing either serine, threonine, cysteine or alanine residues.
Introduction
Cyclic peptides are of broad interest for applications ranging from medicinal chemistry to use as biomaterials. Naturally occurring cyclic peptides have been isolated from a variety of sources including bacteria, marine organisms, and plants.[1] These natural products display a wide range of biological activities and generally exhibit improved biological properties (e.g. metabolic stability, resistance to proteases) when compared with their linear counterparts.[2] Cyclization can also be employed to constrain a linear peptide in its bioactive conformation, which can reduce entropy costs associated with a binding event, leading to improved binding potency and/or specificity if the constraint is correctly chosen.[3] More recently, it has been recognized that cyclic peptides may ‘fill the gap’ between small-molecule drugs and antibodies and find applications in the inhibition of protein–protein interactions, with the cyclic nature of these molecules providing a high level of structural definition, together with sufficient flexibility to bind to dynamic protein targets.[4,5] The well-defined structure that results from conformational constraint is also of use in areas other than drug discovery, with cyclic peptides having found application in supramolecular chemistry[6] and as biomaterials.[7]
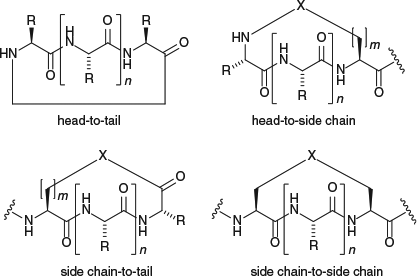
The potential applications of cyclic peptides and related macrocycles have led to significant interest in the development of methods for their synthesis, and these have been summarized in several reviews.[8] There are several ways in which a native peptide sequence can be cyclized: (i) head-to-tail; (ii) head-to-side chain; (iii) side chain to tail; and (iv) side chain to side chain (Fig. 1). Of these, the head-to-tail cyclization reaction is often the most difficult to achieve for small peptide sequences (3–10 amino acids in length), because the preferred transoid conformation of the amide bonds in the linear peptide precursor does not favour a peptide conformation amenable to cyclization.[9] The head-to-tail cyclization reactions of these short peptide sequences are highly sequence-specific and frequently slow and low-yielding. Common side reactions include cyclodimerization or polymerization and epimerization of the C-terminal amino acid (unless this is Gly or Pro).[8]
|
A range of elegant methods have recently been developed to facilitate the synthesis of cyclic peptides and related macrocycles (often labelled cyclic pseudopeptides), many of which introduce modifications into the peptide backbone.[10] Although the resulting macrocyclic structures will find applications as outlined above, their properties are likely to be different to those of a cyclic peptide containing only amide-type linkages[11] and replacement of the amide bond is not always desirable (e.g. in the synthesis of small cyclic peptide natural products). Therefore, the development of methods that facilitate the head-to-tail cyclization of a native peptide sequence remains of interest. Recently developed approaches to the head-to-tail cyclization of a native peptide sequence include the use of native chemical ligation,[12] α-ketoacid–hydroxylamine amide ligation,[13] traceless Staudinger ligation,[14] Ugi peptide ligation,[15] auxiliary-based ring contraction approaches,[16] and the use of traceless turn-inducers.[9,17–31]
Traceless Turn-Inducers to Facilitate Peptide Cyclization
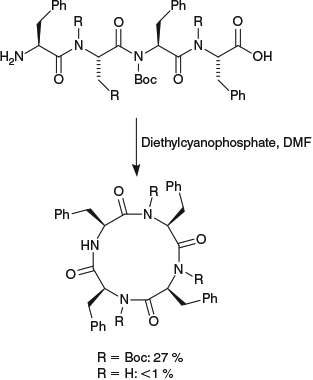
The use of traceless turn-inducers provides a versatile strategy for peptide cyclization. Turn-inducers facilitate head-to-tail peptide cyclization through increasing the population of linear peptide conformers in which the N- and C-termini of the peptide are placed in close proximity, and therefore likely to react. The traceless turn-inducer approach to peptide cyclization was developed following observations that the presence of known turn-inducing elements (e.g. Gly, Pro, N-alkylated or d-amino acids) improved the head-to-tail cyclization reaction of linear peptide sequences.[32] This was attributed to a lowering of the energy barrier for trans–cis isomerization of the amide bonds and first exploited by Cavelier et al.[9] who introduced removable tert-butyloxycarbonyl (Boc) protecting groups onto the main chain nitrogen atoms of a linear peptide sequence, thereby providing, after cyclization, a cyclic peptide that was not accessible by direct methods (Scheme 1). Subsequently, a variety of removable turn-inducers have been employed for peptide cyclization, including 2,4-dimethoxybenzyl (Dmb) protecting groups,[17] 2-hydroxy-6-nitrobenzyl substituents,[18] dehydrophenylalanine,[19] and pseudoprolines.[20–31] A related approach, which uses a removable tether to bring the N- and C-termini of a cyclic peptide into close proximity has also been reported.[33] The present account will focus on the use of pseudoprolines as traceless turn-inducers to facilitate the head-to-tail cyclization of peptides.
Pseudoprolines as Traceless Turn-Inducers
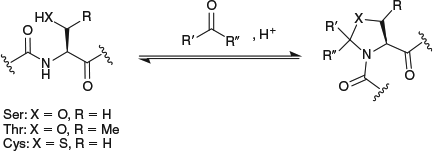
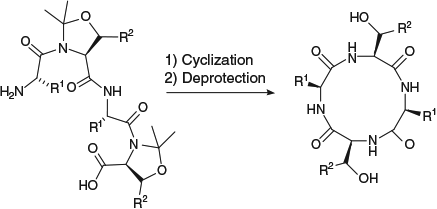
Pseudoprolines (ΨR,R′pros) are five-membered ring analogues of proline, derived from serine, threonine, and cysteine residues on cyclocondensation with an aldehyde or ketone (Fig. 2).[34] They were originally developed as tools to improve yields in solid-phase peptide synthesis by preventing aggregation of growing peptide chains,[35] and are readily incorporated during solid-phase peptide synthesis through coupling of the fluorenylmethoxycarbonyl (Fmoc)-protected dipeptide building blocks, which are commercially available. The introduction of these modified amino acids into a peptide sequence has been found to favour a cisoid conformation of the N-substituted amide bond, with the cis : trans ratio dependent on both the pseudoproline substituents and the nature of the heteroatom in the ring (O vs S).[34,36,37] Theoretical calculations on the cis : trans ratio of CysΨR,Rpro-containing dipeptides were in agreement with experimental results.[37]
|
The high cisoid-amide bond character of peptides containing gem-dimethyl pseudoprolines suggested that these should be ideal turn-inducers to facilitate peptide cyclization. Indeed, early studies showed that replacing the proline residue in the pentapeptide Lys(Dde)–Pro–Gly–Lys(Dde)–Gly [Dde = N-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl)] with a pseudoproline (CysΨMe,Mepro) led to a dramatic increase in the ratio of cyclomonomer to cyclodimer (from 49 : 51 to 100 : 0) obtained on cyclization.[20] Similarly, the cyclic tripeptide cyclo-[Pro–Thr(ΨMe,Mepro)–Pro] was prepared in high yield from the corresponding linear tripeptide.[21] However, both of the above studies focussed on the cyclization reaction itself and in neither case was the pseudoproline treated as a removable turn-inducer.
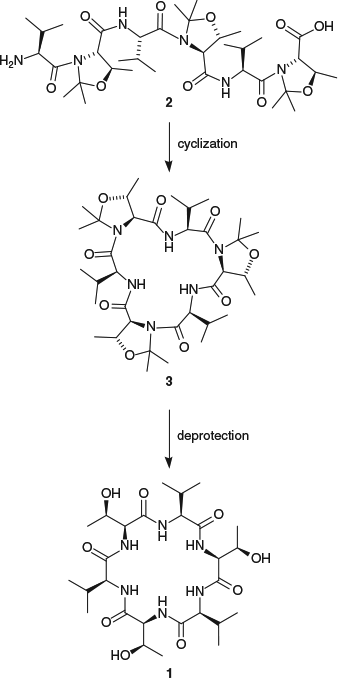
We subsequently employed a Thr-derived pseudoproline as a removable turn-inducer in the synthesis of the cyclic hexapeptide cyclo(Val–Thr–Val–Thr–Val–Thr) 1 (Scheme 2).[22] This is a particularly challenging target, as it comprises all β-branched l-amino acids, and it had been previously reported that attempts to cyclize the tert-butyldimethylsilyl (TBS)-protected linear precursor under a wide range of conditions were unsuccessful.[38] If all three of the TBS protecting groups on the Thr residues were replaced with ΨMe,Mepro groups as in linear peptide 2, we observed that consistently high yields of cyclic peptide 3 (84–99 %) were obtained, irrespective of the coupling reagent used to effect cyclization.[22] Removal of the ΨMe,Mepro groups was then achieved on treatment with either 1 M HCl in dioxane or trifluoroacetic acid (5 % v/v in dichloromethane) to provide the native cyclic hexapeptide 1 in good overall yield. When only one or two of the Thr(TBS) residues were replaced by Thr(ΨMe,Mepro) residues, cyclization yields ranging from 5 to 40 % were obtained. However, following deprotection, it became clear that the cyclic products isolated when a Thr(TBS) residue was at the C-terminus of the linear peptide had all suffered from epimerization of the C-terminal amino acid during the cyclization reaction. Indeed, only the products from cyclization of the C-terminal epimerized precursor were obtained in these cases. This is one of the side reactions common in peptide cyclization and, given that only a single cyclized product was obtained in each case and that the epimerization could only be detected through comparison with an authentic sample, suggests that this may often proceed undetected. In contrast, cyclization of the peptides containing a ΨMe,Mepro residue at the C-terminus proceeded without any epimerization. This highlights an additional benefit of the use of ΨMe,Mepros in peptide cyclization reactions, in that when employed as the C-terminal amino acid, they prevent epimerization and so expand the range of suitable ring-closure sites available.
|
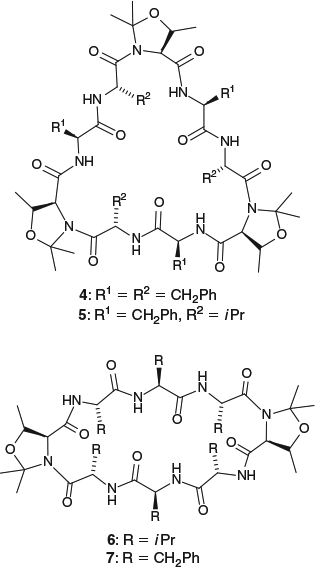
In subsequent studies,[23] we observed that similarly high cyclization yields could be obtained for sterically congested hexapeptides containing either ThrΨMe,Mepro or SerΨMe,Mepro residues at every second amino acid (maintaining a ΨMe,Mepro at the C-terminus to prevent epimerization during the cyclization reactions). Spacing the ΨMe,Mepros further apart (at every third amino acid) in a hexapeptide chain resulted in decreased cyclization yields, suggesting that for demanding hexapeptide cyclization reactions, the amount of turn induced by two ΨMe,Mepros is insufficient to allow efficient cyclization. However, if the ΨMe,Mepro spacing was maintained at every third amino acid, but the peptide was lengthened to nine amino acids, good yields of the cyclic nonapeptides 4 and 5 (Fig. 3) were obtained. In contrast, if the peptide chain was extended to eight amino acids, placing a ΨMe,Mepro at every second amino acid did not give any cyclic products. This was attributed to these peptides adopting a helical conformation, where the turn-inducing effect of the pseudoprolines has effectively ‘gone too far’. In this case, reducing the number of pseudoproline residues to two (at the C-terminus and (n + 4) positions) resulted in moderate to good yields of cyclic octapeptides 6 and 7, with the lowest yields (35 %) obtained for cyclization of the sterically bulky H2N–Val–Val–Val–ThrΨMe,Mepro–Val–Val–Val–ThrΨMe,Mepro–OH precursor.
|
The head-to-tail cyclization of all-l tetrapeptides is particularly challenging as a result of their highly constrained 12-membered ring structure.[9,17] Pleasingly, the incorporation of two ΨMe,Mepro residues (derived from either Thr or Ser) was found to facilitate the cyclization of a range of sterically congested all-l tetrapeptides (containing Leu, Ile, Phe, and Val residues), and subsequent deprotection provided the native Ser- and Thr-containing cyclic peptides in yields of up to 60 % (Scheme 3).[24]
|
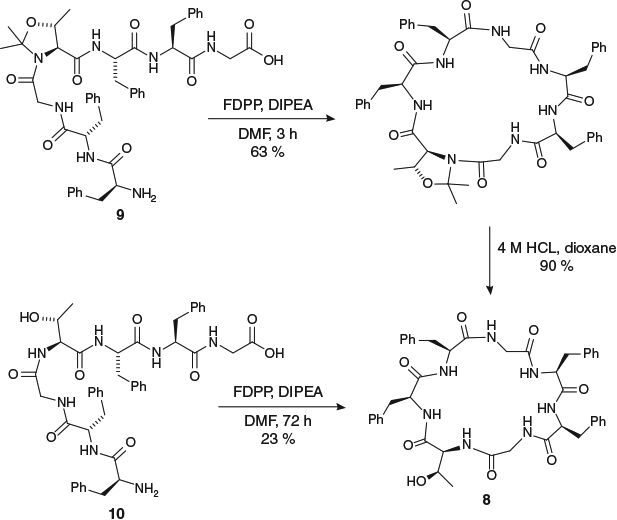
The pseudoproline-mediated traceless turn-inducer approach to peptide cyclization has also been applied to the synthesis of several small cyclic peptide natural products containing Thr residues. In the synthesis of mahafacyclin B (8), a naturally occurring cyclic heptapeptide with antimalarial activity, the use of a ThrΨMe,Mepro residue at the centre of the linear peptide precursor 9 led to cyclization yields more than double those of the native sequence 10 (Scheme 4).[25] Importantly, the rate of cyclization for the pseudoproline-containing peptide was considerably faster than that of the native sequence (3 versus 72 h to completion as determined by consumption of the starting material by HPLC), illustrating the enhanced reaction kinetics that can be obtained through forcing the linear precursor to adopt a conformation that places the reactive N- and C-peptide termini in close proximity.
|
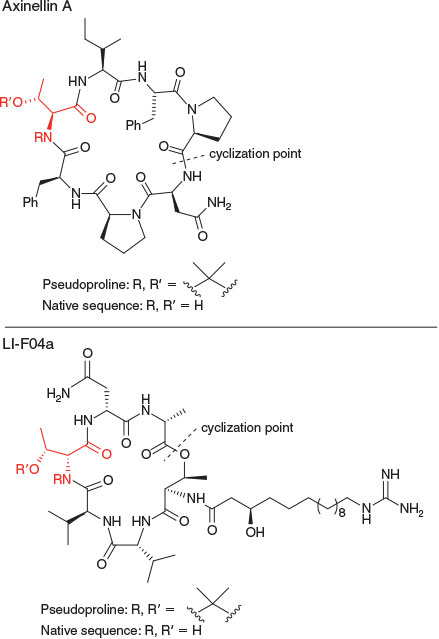
Although a clear improvement in cyclization yield was observed in the synthesis of the cyclic heptapeptide mahafacyclin B, use of the pseudoproline-mediated cyclization in the synthesis of the proline-rich cyclic heptapeptide axinellin A (Fig. 4) gave less spectacular results, with similar yields obtained for the cyclization of the ThrΨMe,Mepro-containing linear precursor and the tBu-protected Thr derivative.[26] Similarly, attempts to use a ThrΨMe,Mepro to improve the yield of the macrolactonization reaction in the synthesis of the antimicrobial cyclic depsipeptide LI-F04a (Fig. 4) were unsuccessful.[27] These examples illustrate that if the turn-inducer does not create a conformation in which the N- and C-termini of the peptide are in close proximity, then cyclization is not assisted by its incorporation, partly explaining the frequently observed sequence specificity of head-to-tail peptide cyclization reactions.
|
Having shown that, in most cases, SerΨMe,Mepro and ThrΨMe,Mepro residues can be employed as traceless turn-inducers to improve the synthesis of peptides that contain either Ser or Thr in their native sequence, we were interested in expanding the traceless turn-inducer methodology to the synthesis of peptides that did not contain either of these residues. CysΨMe,Mepros are ideal for this purpose, because on deprotection to give the free Cys residue, a desulfurization can then be achieved using one of the several methods developed to facilitate the synthesis of non-Cys containing peptides by native ligation.[39]
The first example of the use of a pseudoproline residue in the synthesis of a peptide that does not contain a Ser, Thr or Cys residue in its native sequence was in the synthesis of the cyclic octapeptide cyclogossine B (Scheme 5).[28] Replacement of an Ala residue by a CysΨMe,Mepro in the linear peptide precursor 11 resulted in quantitative cyclization yield, a significant improvement on the 60 % yield obtained for the native sequence under identical cyclization conditions. Although it had previously been reported that CysΨMe,Mepro residues could be removed from linear peptides on treatment with trifluoroacetic acid (TFA) for 32 h,[34a] the pseudoproline in cyclic peptide 12 was resistant to removal under these conditions, leading us to explore alternative options. We found that trifluoromethanesulfonic acid (TFMSA), which is routinely used in Boc solid-phase peptide synthesis,[40] was highly effective for the deprotection of CysΨMe,Mepro residues in cyclic peptides, with treatment of 12 with TFMSA for 15 min at 0°C effecting removal of both the ΨMe,Mepro and Boc protecting groups. Subsequent desulfurization was readily achieved on treatment of 13 with nickel boride to provide the natural product 14 in excellent yield over the three-step sequence (cyclization, deprotection, desulfurization).
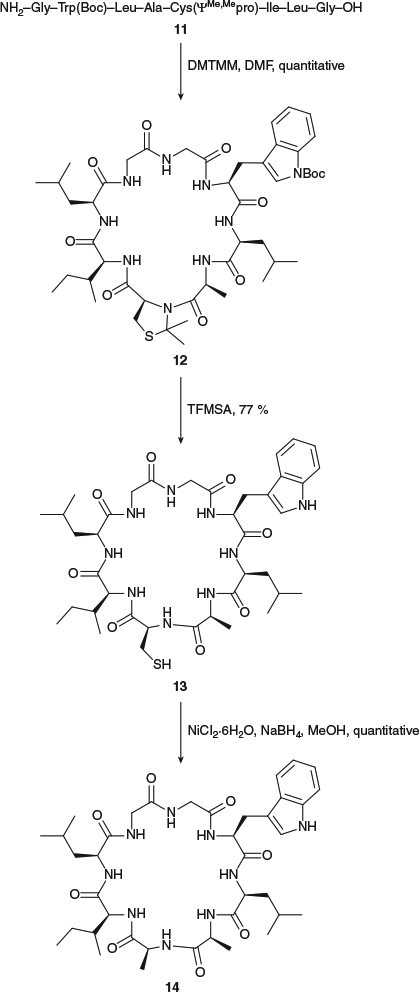
|
In subsequent studies, Postma and Albericio employed CysΨMe,Mepro residues to facilitate the on-resin side chain to side chain cyclization of two conotoxin analogues.[29] They observed that the incorporation of a CysΨMe,Mepro residue greatly enhanced both the efficiency and rate of the cyclization reaction and found that the deprotection reaction was highly sequence-specific in a range of model linear peptides, with near-quantitative deprotection of peptides containing the Ser–CysΨMe,Mepro linkage in under 1 h on treatment with TFA. For the cyclic conotoxin analogues, cleavage from the resin using a TFA/triisopropylsilane/H2O (95 : 2.5 : 2.5) deprotection cocktail effected partial pseudoproline deprotection.
We have also employed CysΨR,Rpro residues in the total synthesis of the naturally occurring cyclic pentapeptides segetalins B and G (Fig. 5).[30] Neither of these cyclic peptides contains a Ser, Thr or Cys residue, but both contain at least one Ala. Replacement of Ala by a CysΨMe,Mepro residue in the linear peptide precursor led to high cyclization yields for both sequences and eliminated the formation of cyclodimers in the cyclization reaction. Subsequent deprotection and desulfurization gave the natural products in good overall yields across the three-step sequence (cyclization, deprotection, desulfurization). However, if CysΨH,Hpro residues, which also favour a cisoid amide bond albeit not as strongly as the CysΨMe,Mepro residue, were employed as the turn-inducers in the cyclization reactions, both cyclomonomers and cyclodimers were obtained and the overall yields of cyclic products were lower. The time required for cyclization of the CysΨH,Hpro-containing linear precursors (96 h for complete consumption of starting material) was also significantly longer than that of the CysΨMe,Mepro derivatives (0.5 h). This highlights the importance of the linear peptide conformation in the cyclization reaction and suggests that slow cis–trans isomerization around the amide bonds may be the rate-limiting step.
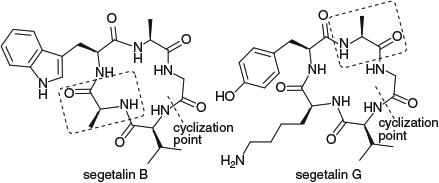
|
Numerous β-thiolated amino acids have now been reported for use in native peptide ligation reactions[41] and we envisaged that these could also be converted into pseudoprolines and used as turn-inducers to facilitate peptide cyclization, thus significantly expanding the scope of the traceless-turn inducer approach to peptide cyclization. As proof-of-principle, we investigated the use of a penicillamine-derived pseudoproline (PenΨMe,Mepro) in the synthesis of the naturally occurring cyclic hexapeptide dichotomin A (Scheme 6).[31] Two linear precursors containing the PenΨMe,Mepro residue were cyclized in excellent yields in only 3 h (87 and 88 % respectively), whereas cyclization of the native linear precursors containing Val residues in place of the PenΨMe,Mepro occurred in only 36 and 33 % yields and took 72 h to reach completion. This is consistent with the incorporation of the turn-inducer increasing both the efficiency and rate of reaction, as was also observed for the incorporation of the CysΨMe,Mepro derivative above. However, deprotection of the PenΨMe,Mepro residue in 15 was found to be slower than that of CysΨMe,Mepro-containing cyclic peptides, requiring treatment with TFMSA for 4 h. Under these conditions, some dehydration of the Thr residue in 15 was also observed, resulting in a decrease in the overall yield across the two-step cyclization–deprotection sequence. Desulfurization of the isolated cyclopeptide 16 to give the natural product 17 was achieved on treatment with nickel boride, illustrating that the pseudoproline approach can be expanded to incorporate β-thiolated amino acids. However, further work is required to optimize the deprotection and desulfurization conditions for these modified amino acids.
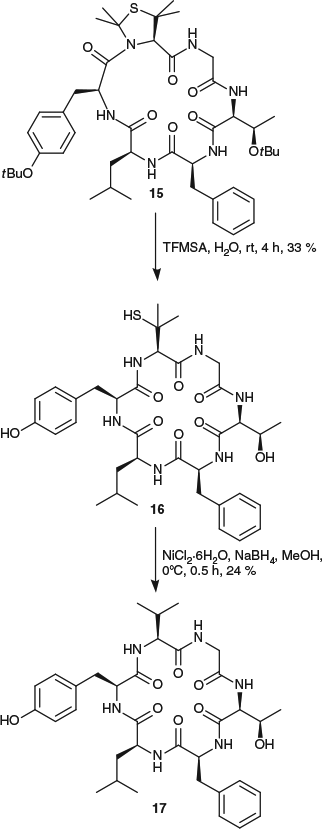
|
Conclusion
The incorporation of gem-dimethyl pseudoprolines derived from Ser, Thr, Cys, and Pen into linear peptides has been found to increase both head-to-tail cyclization yields and the rate of the cyclization reaction for peptides ranging from four to nine amino acids in length. In general, placing a pseudoproline close to the centre of the linear peptide is the most effective strategy to increase cyclization yield, but including a pseudoproline as the C-terminal amino acid has the added benefit of significantly supressing epimerization of this residue, thereby overcoming a problematic side reaction during peptide cyclization. Pseudoproline residues have also been shown to facilitate side chain-to-side chain cyclization reactions of larger macrocycles.
For the cyclization of sterically congested peptides, more than one pseudoproline can be incorporated into the linear precursor and the optimal positioning of these with respect to each other depends on the length of the peptide to be synthesized. For cyclic tetra- and hexapeptides, incorporation of a pseudoproline at every second amino acid provides the highest cyclization yields, whereas for the cyclization of octa- and nonapeptides, additional residues to space the pseudoprolines further apart are required to prevent the peptide from adopting a helical conformation in which the reactive ends cannot be brought together.
For the synthesis of peptides that do not contain Ser, Thr or Cys residues, Cys-derived pseudoprolines provide a means of incorporating a traceless turn-inducer as, after deprotection of the pseudoproline group, the resulting Cys residues can be subjected to desulfurization to give the corresponding Ala-containing cyclic peptides in good overall yields across the three-step sequence (cyclization, deprotection, desulfurization). This approach has been successfully employed in the synthesis of several naturally occurring cyclic peptides such as cyclogossine B and segetalins B and G. We have also shown that a penicillamine-derived pseudoproline can be used to increase cyclization yield, but that in this case, removal of the pseudoproline residue requires harsh conditions, which may result in undesired side reactions (e.g. dehydration) at sensitive side chains. Further investigations on the use of pseudoprolines formed from other β-thiolated amino acids, which will be less sterically hindered than the Pen derivative (containing secondary rather than tertiary thiols), may overcome this problem.
Overall, the incorporation of ΨMe,Mepro residues into a linear peptide precursor provides an effective method for increasing peptide cyclization yields and, in the worst cases, similar yields in comparison with those of the native peptide sequence are obtained. As dipeptides containing these simple side chain protecting groups are commercially available and easily incorporated into a growing peptide chain during solid-phase peptide synthesis, their inclusion in any linear peptide that is to be subject to a cyclization reaction is highly recommended.
Conflicts of Interest
The author declares no conflicts of interest.
Acknowledgements
I thank all of my coworkers, past and present, at the University of Sydney (their names appear in the references). I thank the Australian Research Council (grant no. DP0208266) and the University of Sydney for funding.
References
[1] S. R. Adusumalli, A. K. Yudin, V. Rai, in Natural Lactones and Lactams: Synthesis, Occurrence and Biological Activity (Ed. T. Janecki) 2014, Ch. 8, pp. 321–369 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).[2] (a) D. P. Fairlie, J. D. A. Tyndall, R. C. Reid, A. K. Wong, G. Abbenante, M. J. Scanlon, D. R. March, D. A. Bergman, C. L. L. Chai, B. A. Burkett, J. Med. Chem. 2000, 43, 1271.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. Wang, W. Liao, P. S. Arora, Angew. Chem. Int. Ed. 2005, 44, 6525.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) P. Ermert, K. Moehle, D. Obrecht, in Macrocycles in Drug Discovery (Ed. J. Levin) 2015, Ch. 8, pp. 283–338 (Royal Society of Chemistry: London).
(b) J. Mallinson, I. Collins, Future Med. Chem. 2012, 4, 1409.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. M. J. Liskamp, D. T. S. Rijkers, S. A. Bakker, in Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis (Eds F. Diederich, P. J. Stang, R. R. Tykwinski) 2008, Ch. 1, pp. 1–27 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).
[4] (a) T. A. Hill, N. E. Shepherd, F. Diness, D. P. Fairlie, Angew. Chem. Int. Ed. 2014, 53, 13020.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. K. Yudin, Chem. Sci. 2015, 6, 30.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. Leenheer, P. ten Dijke, C. J. Hipolito, Pept. Sci. 2016, 106, 889.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Zaretsky, A. K. Yudin, in Peptide-Based Drug Discovery: Challenges and New Therapeutics (Ed. V. Srivastava) 2017, Ch. 5, pp. 141–171 (The Royal Society of Chemistry: London).
(e) A. Zorzi, K. Deyle, C. Heinis, Curr. Opin. Chem. Biol. 2017, 38, 24.
| Crossref | GoogleScholarGoogle Scholar |
[5] F. Giordanetto, J. Kihlberg, J. Med. Chem. 2014, 57, 278.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) R. B. P. Elmes, K. A. Jolliffe, Chem. Commun. 2015, 4951.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. A. Jolliffe, Supramol. Chem. 2005, 17, 81.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Kubik, in Artificial Receptors for Chemical Sensors (Eds V. M. Mirsky, A. Yatsimirsky) 2010, Ch. 5, pp. 135–167 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).
[7] (a) R. Chapman, K. A. Jolliffe, S. Perrier, Adv. Mater. 2013, 25, 1170.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. Chapman, M. Danial, M. L. Koh, K. A. Jolliffe, S. Perrier, Chem. Soc. Rev. 2012, 41, 6023.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Montenegro, M. R. Ghadiri, J. R. Granja, Acc. Chem. Res. 2013, 46, 2955.
| Crossref | GoogleScholarGoogle Scholar |
(d) R. J. Brea, C. Reiriz, J. R. Granja, Chem. Soc. Rev. 2010, 39, 1448.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) J. N. Lambert, J. P. Mitchell, K. D. Roberts, J. Chem. Soc., Perkin Trans. 1 2001, 471.
| Crossref | GoogleScholarGoogle Scholar |
(b) P. Li, P. P. Roller, J. Xu, Curr. Org. Chem. 2002, 6, 411.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. S. Davies, J. Pept. Sci. 2003, 9, 471.
| Crossref | GoogleScholarGoogle Scholar |
(d) Y. Hamada, T. Shioiri, Chem. Rev. 2005, 105, 4441.
| Crossref | GoogleScholarGoogle Scholar |
(e) N.-H. Tan, J. Zhou, Chem. Rev. 2006, 106, 840.
| Crossref | GoogleScholarGoogle Scholar |
(f) A. F. Spatola, P. Romanovskis, in The Amide Linkage: Structural Aspects in Chemistry, Biochemistry, and Materials Science (Eds A. Greenberg, C. M. Breneman, J. F. Liebman) 2003, Ch. 16, pp. 519–564 (John Wiley and Sons, Inc.: Hoboken, NJ).
(g) S. Jiang, Z. Li, K. Ding, P. P. Roller, Curr. Org. Chem. 2008, 12, 1502.
| Crossref | GoogleScholarGoogle Scholar |
(h) C. J. White, A. K. Yudin, Nat. Chem. 2011, 3, 509.
| Crossref | GoogleScholarGoogle Scholar |
(i) L. M. De Leon Rodriguez, A. J. Weidkamp, M. A. Brimble, Org. Biomol. Chem. 2015, 13, 6906.
| Crossref | GoogleScholarGoogle Scholar |
(j) Y. S. Ong, L. Gao, K. A. Kalesh, Z. Yu, J. Wang, C. Liu, Y. Li, H. Sun, S. S. Lee, Curr. Top. Med. Chem. 2017, 17, 2302.
[9] F. Cavelier-Frontin, S. Achmad, J. Verducci, R. Jacquier, G. Pèpe, J. Mol. Struct. THEOCHEM 1993, 286, 125.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) For selected examples see: R. A. Turner, A. G. Oliver, S. R. Lokey, Org. Lett. 2007, 9, 5011.
| Crossref | GoogleScholarGoogle Scholar |
(b) V. D. Bock, D. Speijer, H. Hiemstra, J. H. van Maarseveen, Org. Biomol. Chem. 2007, 5, 971.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. Hili, V. Rai, A. K. Yudin, J. Am. Chem. Soc. 2010, 132, 2889.
| Crossref | GoogleScholarGoogle Scholar |
(d) B. H. Rotstein, V. Rai, R. Hili, A. K. Yudin, Nat. Protoc. 2010, 5, 1813.
| Crossref | GoogleScholarGoogle Scholar |
(e) C. J. White, A. K. Yudin, Org. Lett. 2012, 14, 2898.
| Crossref | GoogleScholarGoogle Scholar |
(f) J. M. Smith, J. R. Frost, R. Fasan, J. Org. Chem. 2013, 78, 3525.
| Crossref | GoogleScholarGoogle Scholar |
(g) B. K. W. Chung, C. J. White, A. K. Yudin, Nat. Protoc. 2017, 12, 1277.
| Crossref | GoogleScholarGoogle Scholar |
[11] S. Zaretsky, A. K. Yudin, Drug Discov. Today. Technol. 2017, 26, 3.
| Crossref | GoogleScholarGoogle Scholar |
[12] T. Fukuzumi, L. Ju, J. W. Bode, Org. Biomol. Chem. 2012, 10, 5837.
| Crossref | GoogleScholarGoogle Scholar |
[13] F. Rohrbacher, G. Deniau, A. Luther, J. W. Bode, Chem. Sci. 2015, 6, 4889.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) O. David, W. J. N. Meester, H. Bieräugel, H. E. Schoemaker, H. Hiemstra, J. H. van Marrseveen, Angew. Chem. Int. Ed. 2003, 42, 4373.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. Kleineweischede, C. P. R. Hackenberger, Angew. Chem. Int. Ed. 2008, 47, 5984.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. Sasaki, D. Crich, Org. Lett. 2010, 12, 3254.
| Crossref | GoogleScholarGoogle Scholar |
[15] A. R. Puentes, M. C. Morejón, D. G. Rivera, L. A. Wessjohann, Org. Lett. 2017, 19, 4022.
| Crossref | GoogleScholarGoogle Scholar |
[16] (a) C. T. T. Wong, H. Y. Lam, T. Song, G. Chen, X. Li, Angew. Chem. Int. Ed. 2013, 52, 10212.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. P. A. Rutters, Y. Verdonk, R. de Vries, S. Ingemann, H. Hiemstra, V. Levacherb, J. H. van Maarseveen, Chem. Commun. 2012, 8084.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. A. Horton, G. T. Bourne, J. Coughlan, S. M. Kaiser, C. M. Jacobs, A. Jones, A. Ruhmann, J. Y. Tuner, M. L. Smythe, Org. Biomol. Chem. 2008, 6, 1386.
| Crossref | GoogleScholarGoogle Scholar |
[17] M. El Haddadi, F. Cavelier, E. Vives, A. Azmani, J. Verducci, J. Martinez, J. Pept. Sci. 2000, 6, 560.
| Crossref | GoogleScholarGoogle Scholar |
[18] W. D. F. Meutermans, G. T. Bourne, S. W. Golding, D. A. Horton, M. R. Campitelli, D. Craik, M. Scanlon, M. L. Smythe, Org. Lett. 2003, 5, 2711.
| Crossref | GoogleScholarGoogle Scholar |
[19] D. N. Le, J. Riedel, N. Kozlyuk, R. W. Martin, V. M. Dong, Org. Lett. 2017, 19, 114.
| Crossref | GoogleScholarGoogle Scholar |
[20] C. Sager, M. Mutter, P. Dumy, Tetrahedron Lett. 1999, 40, 7987.
| Crossref | GoogleScholarGoogle Scholar |
[21] T. Ruckle, P. de Lavallaz, M. Keller, P. Dumy, M. Mutter, Tetrahedron 1999, 55, 11281.
| Crossref | GoogleScholarGoogle Scholar |
[22] D. Skropeta, K. A. Jolliffe, P. Turner, J. Org. Chem. 2004, 69, 8804.
| Crossref | GoogleScholarGoogle Scholar |
[23] N. Sayyadi, D. Taleski, S. Leesch, K. A. Jolliffe, Tetrahedron 2014, 70, 7700.
| Crossref | GoogleScholarGoogle Scholar |
[24] K. A. Fairweather, N. Sayyadi, I. J. Luck, J. K. Clegg, K. A. Jolliffe, Org. Lett. 2010, 12, 3136.
| Crossref | GoogleScholarGoogle Scholar |
[25] N. Sayyadi, D. Skropeta, K. A. Jolliffe, Org. Lett. 2005, 7, 5497.
[26] K. A. Fairweather, N. Sayyadi, C. Roussakis, K. A. Jolliffe, Tetrahedron 2010, 66, 935.
| Crossref | GoogleScholarGoogle Scholar |
[27] J. R. Cochrane, D. H. Yoon, C. S. P. McErlean, K. A. Jolliffe, Beilstein J. Org. Chem. 2012, 8, 1344.
| Crossref | GoogleScholarGoogle Scholar |
[28] M. S. Y. Wong, K. A. Jolliffe, Aust. J. Chem. 2010, 63, 797.
| Crossref | GoogleScholarGoogle Scholar |
[29] T. M. Postma, F. Albericio, Org. Lett. 2014, 16, 1772.
| Crossref | GoogleScholarGoogle Scholar |
[30] M. S. Y. Wong, K. A. Jolliffe, Pept. Sci. 2018, 110, e24042.
| Crossref | GoogleScholarGoogle Scholar |
[31] M. S. Y. Wong, D. Taleski, K. A. Jolliffe, Aust. J. Chem. 2015, 68, 627.
| Crossref | GoogleScholarGoogle Scholar |
[32] (a) K. D. Kopple, J. Pharm. Sci. 1972, 61, 1345.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. Titlestad, Acta Chem. Scand. 1997, B31, 641.
(c) A. Ehrlich, H.-U. Heyne, R. Winter, M. Beyermann, H. Haber, L. A. Carpino, M. Bienert, J. Org. Chem. 1996, 61, 8831.
| Crossref | GoogleScholarGoogle Scholar |
[33] J. Illesinghe, C. X. Guo, R. Garland, A. Ahmed, B. van Lierop, J. Elaridi, W. R. Jackson, A. J. Robinson, Chem. Commun. 2009, 295.
| Crossref | GoogleScholarGoogle Scholar |
[34] (a) T. Wöhr, F. Wahl, A. Hefzi, B. Rohwedder, T. Sato, X. Sun, M. Mutter, J. Am. Chem. Soc. 1996, 118, 9218.
| Crossref | GoogleScholarGoogle Scholar |
(b) P. Dumy, M. Keller, D. E. Ryan, B. Rohwedder, T. Wöhr, M. Mutter, J. Am. Chem. Soc. 1997, 119, 918.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Keller, C. Sager, P. Dumy, M. Schutkowski, G. S. Fischer, M. Mutter, J. Am. Chem. Soc. 1998, 120, 2714.
| Crossref | GoogleScholarGoogle Scholar |
[35] (a) M. Mutter, A. Nefzi, T. Sato, X. Sun, F. Wahl, T. Wöhr, Pept. Res. 1995, 8, 145.
(b) W. R. Sampson, H. Patsiouras, N. J. Ede, J. Pept. Sci. 1999, 5, 403.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Mutter, Chimia 2013, 67, 868.
| Crossref | GoogleScholarGoogle Scholar |
[36] (a) J. K. Clegg, J. R. Cochrane, N. Sayyadi, D. Skropeta, P. Turner, K. A. Jolliffe, Aust. J. Chem. 2009, 62, 711.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. Zhang, J. Gong, Y. Yang, S. Dong, J. Pept. Sci. 2011, 17, 601.
| Crossref | GoogleScholarGoogle Scholar |
[37] H. Jamet, M. Jourdan, P. Dumy, J. Phys. Chem. B 2008, 112, 9975.
| Crossref | GoogleScholarGoogle Scholar |
[38] P. Wipf, C. P. Miller, J. Am. Chem. Soc. 1992, 114, 10975.
| Crossref | GoogleScholarGoogle Scholar |
[39] (a) L. Z. Yan, P. E. Dawson, J. Am. Chem. Soc. 2001, 123, 526.
| Crossref | GoogleScholarGoogle Scholar |
(b) Q. Wan, S. J. Danishefsky, Angew. Chem. Int. Ed. Engl. 2007, 46, 9248.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. Jin, T. Li, H. Y. Chow, H. Liu, X. P. B. Li, Angew. Chem. Int. Ed. 2017, 56, 14607.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. S. Kulkarni, J. Sayers, B. Premdjee, R. J. Payne, Nat. Rev. Chem. 2018, 2, 0122.
| Crossref | GoogleScholarGoogle Scholar |
[40] A. L. Doherty-Kirby, G. A. Lajoie, in Solid-Phase Peptide Synthesis: A Practical Guide (Eds S. A. Kates, F. Albericio) 2000, Ch. 4, pp. 129–196 (Marcel Dekker Inc.: New York, NY).
[41] (a) For reviews see: C. T. C. Wong, C. L. Tung, X. Li, Mol. Biosyst. 2013, 9, 826.
| Crossref | GoogleScholarGoogle Scholar |
(b) L. R. Malins, N. J. Mitchell, R. J. Payne, J. Pept. Sci. 2014, 20, 64.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Rohde, O. Seitz, Biopolymers 2010, 94, 551.
| Crossref | GoogleScholarGoogle Scholar |
(d) P. E. Dawson, Isr. J. Chem. 2011, 51, 862.
| Crossref | GoogleScholarGoogle Scholar |
* Professor Kate Jolliffe was awarded the 2017 Arthur J. Birch Medal.