

A Scalable, Combined-Batch, and Continuous-Flow Synthesis of a Bio-Inspired UV-B Absorber
Mark York A C , Karen E. Jarvis A , Jamie A. Freemont A , John H. Ryan A , G. Paul Savage A , Stephanie A. Logan A and Larissa Bright BA CSIRO Manufacturing, CSIRO, Clayton, Vic. 3168, Australia.
B Coral Sunscreen Pty Ltd, Aitkenvale, Qld 4814, Australia.
C Corresponding author. Email: mark.york@csiro.au
Australian Journal of Chemistry 72(11) 860-866 https://doi.org/10.1071/CH19252
Submitted: 3 June 2019 Accepted: 5 July 2019 Published: 24 July 2019
Abstract
A new, chromatography-free synthesis for the preparation of an experimental UV-B absorber is reported. A key step of the process is a one-pot partial reduction of a symmetrical imide with a sequential dehydration step. The synthesis uses several continuous-flow steps to increase sample throughput and was used to prepare sufficient material to support further testing activities in >99 % purity.
Introduction
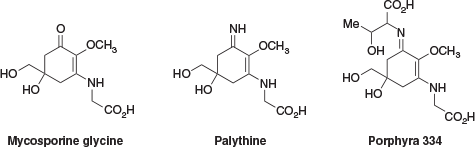
Despite prolonged exposure to the damaging ultraviolet (UV) radiation of the Australian climate, the shallow-water coral polyps of the Great Barrier Reef demonstrate resilience to extended periods of solar irradiation. This resilience is due in part to the presence of UV-absorbing compounds within the corals.[1] The presence of UV-absorbing compounds in the extracts of shallow-water coral species has been known since they were reported by Shibata in 1969.[2] These compounds were identified as a family of mycosporine-like amino acids (MAAs, Fig. 1). Since their identification, there has been interest in developing these compounds for use as commercial sunscreens.[3,4] These efforts have been hampered by a lack of readily available sustainable sources of material and, more importantly, a lack of hydrolytic and oxidative stability of the parent MAAs.[5,6]
|
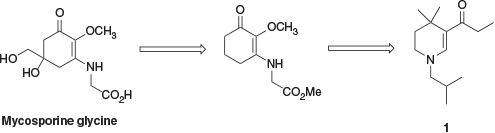
In an attempt to address these shortfalls and improve synthetic accessibility, a collaboration between groups at the Australian Institute of Marine Science (AIMS) and Imperial Chemical Industries (ICI) developed a series of bio-inspired UV-absorbing compounds. These retained the enaminone chromophore of the MAAs but with a simplified structure, movement of the enaminone nitrogen from an exocyclic to an endocyclic position to improve hydrolytic stability, and the addition of a geminal dimethyl substituent to improve oxidative stability.[3] The evolution of these compounds can be seen in Fig. 2.
|
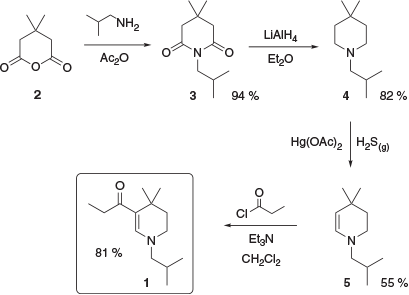
This series of compounds is typified by 1, a tetrahydropyridine derivative. This is a UV-B absorber with a reported absorbance maximum in methanol of 307 nm and a molar extinction coefficient of 29300 M−1 cm−1.[7] Several synthetic pathways to absorber 1 have been reported.[7,8] One reported synthesis begins with commercially available 3,3-dimethylglutaric anhydride, 2, which was treated with isobutylamine followed by acetic anhydride to yield the corresponding imide 3. This was then exhaustively reduced with lithium aluminium hydride to give piperidine 4, which was oxidised with mercuric acetate gave the corresponding enamine 5. Finally, acylation of 5 gave UV absorber 1 (Scheme 1).
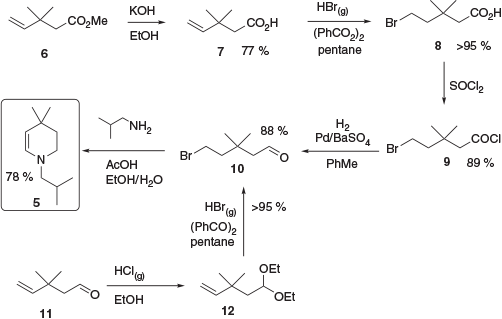
Alternative approaches to advanced enamine intermediate 5 have also been reported commencing with either methyl 3,3-dimethylpent-4-enoate 6 or 3,3-dimethylpent-4-enal 11 and following a radical bromination approach to yield common intermediate 10. This intermediate was then cyclised in the presence of isobutylamine to give 5 (Scheme 2).[7]
|
A concise approach to enamine 5 from pyranone 13 has also been reported (Scheme 3).[8] This involved condensation of 13 with isobutylamine to form enaminone 14, followed by reduction of 14 to the desired advanced intermediate 5.
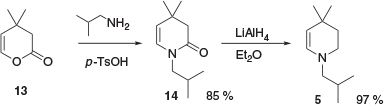
|
Results and Discussion
In order to further explore the properties of UV absorber 1, we required access to >100 g of material and so undertook an assessment of the published methods for suitability of use at this (and potentially larger) scales. The synthesis described in Scheme 1 was discounted owing to the use of excess amounts of mercury reagents in the key oxidation step and the high risk of contamination of final products with mercury. The two related syntheses reported in Scheme 2 were initially judged to be more suitable; however, several chromatographic separations and distillations were required to provide pure 1. Owing to the scale of the work proposed, these operations were not desirable. Additionally, we experienced considerable difficulty replicating the reported yields on anything but a very small scale for both the radical bromination and Rosenmund reduction processes. The reported reaction of aldehyde 10 with isobutylamine to give enamine 5 also proved troublesome to perform in a reproducible fashion owing to the instability of both 5 and 10.
We were initially drawn to the synthesis outlined in Scheme 3 owing to the brevity and high yields reported. However, although pyranone starting material 13 was reported to be readily commercially available at the time of the initial publication,[8] this was no longer the case for us, with the material being available only from a limited range of suppliers at prohibitively high cost from a manufacturing point of view.[9] Syntheses of 13 have been reported but these protocols were judged to be unsuitable for the desired application. In one report, a Wacker oxidation process requiring high temperatures and pressures of oxygen was used.[10] Another synthesis used more accessible reaction conditions but was a four-step, low-yielding synthesis from a starting material that was not commercially available.[11] Nevertheless, for completeness we obtained a small sample of pyranone 13 to examine the published synthesis (Scheme 3), but in our hands, the reported procedures did not give adequate yields of intermediate 14.
Given the unsuitability of the previously reported syntheses for production of material at the desired scale, a new approach was devised (Scheme 4).[12] Commercially available 3,3-dimethylglutaric acid, 15, was converted to the corresponding anhydride 2 in quantitative yield (2 is also commercially available but expensive on scale). Conversion of the anhydride to the corresponding glutarimide 3 was achieved in 92 % yield followed by a partial reduction to enamide 14. This was then further reduced to enamine 5 and acylated with propionyl chloride to yield 1.
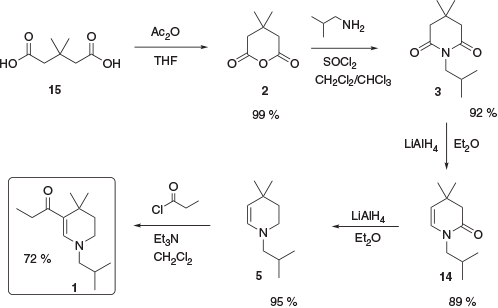
|
Formation of Anhydride 2
The transformation of 3,3-dimethylglutaric acid, 15, to the corresponding anhydride 2 is a known reaction that has been reported using either oxalyl chloride in 70 % yield,[13] an 8-fold excess of acetic anhydride in 79 % yield,[14] or an excess of thionyl chloride in 96 % yield.[15] In an attempt to avoid the large excess of acetic anhydride used in the published procedure and the resulting lengthy distillation and recrystallisation, a continuous-flow approach was developed. The performance of synthetic transformations under continuous-flow conditions is an area of intense current interest. This interest can be explained, at least in part, by the array of potential advantages that continuous-flow processing can have over traditional batch processing. Such advantages include high reproducibility, ease of scale-up, rapid mixing and heat transfer, and inherently higher safety owing to smaller reactor volumes and the containment of hazardous reaction intermediates.[16] Thus, a continuous-flow approach to anhydride 2 was developed using 1.2 equiv. of acetic anhydride to effect the transformation over the course of a 4-min reaction (Scheme 5). Anhydride 2 was isolated in near-quantitative yield after removal of volatiles. By utilising a series of four 10-mL reactor coils, the reaction pumping rate was able to be increased 4-fold over that used with one coil while maintaining the same residence time. This allowed the production of 165 g of the product in 1 h.
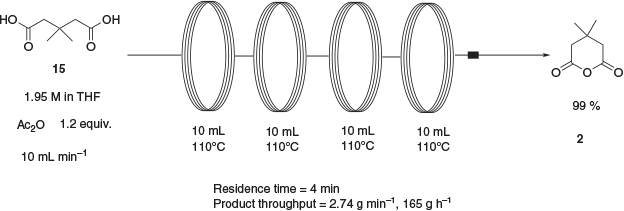
|
Formation of Imide 3
The conversion of 2 to 3 has been reported previously by a one-pot process in 94 % yield whereby 2 is treated with isobutylamine followed by an excess of acetic anhydride.[7] The addition of isobutylamine to 2 under solvent-free conditions as reported proved to be an extremely exothermic process that was deemed unsuitable for large-scale application. Additionally, the reported procedure gave only minor amounts of the desired product in our hands, with the bulk of the material being a mixture of intermediate amide 16 and the corresponding mixed acetic anhydride. With this in mind, a two-step approach to 3 was developed involving initial synthesis of the intermediate amide 16 over a 3-min reaction time (Scheme 6). The product was isolated in near-quantitative yield after an aqueous workup. As the cyclisation of 16 to 3 in the presence of acetic anhydride had already been found to give only minimal amounts of product, cyclisation in the presence of thionyl chloride was attempted. This gave the product in 97 % yield after a reaction time of 10 min at 95°C (Scheme 7) This process allowed the preparation of 3 in sufficient quantities to complete the synthesis of absorber 1. In all instances of the continuous-flow reactions, flow rate was calculated in conjunction with total reactor volume in order to give the desired reaction and residence time.

|

|
A telescoped synthesis of 3 directly from anhydride 2 was also demonstrated whereby a solution of anhydride 2 was reacted with isobutylamine at 50°C and the resulting solution of amide 16 further treated with thionyl chloride and reacted at 100°C to give 92 % of the desired product after aqueous workup and a total reaction time of 10 min (Scheme 8).

|
Formation of Enamide 14
Previously reported methods for the conversion of imide 3 to enamine building block 5 involved the complete reduction of 3 to substituted piperidine 4 followed by oxidation with mercuric acetate.[7] In order to avoid the need for such a low-yielding and hazardous oxidation process, a reduction approach was developed whereby 3 was partially reduced to enamide 14 via hemiaminal intermediate 17 (Scheme 9). Initial attempts to form 17 through a sodium borohydride reduction were mostly unsuccessful, with the major isolated product being ring-opened compound 18. Ring opening could be avoided by performing the reaction in HCl/EtOH but a large excess of borohydride was required (8 equiv.) and the reaction did not proceed to completion, necessitating chromatographic purification. Reduction using sodium bis(2-methoxyethoxy)aluminium hydride (1 equiv.) gave ~50 % conversion to hemiaminal 17 when performed in both continuous-flow and batch modes. An attempt to increase conversion by increased reaction temperature or equivalents of reducing agent resulted in over-reduction to piperidine 4.
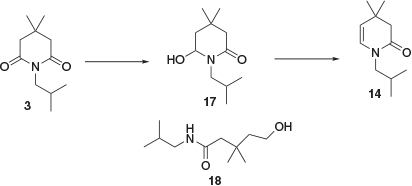
|
In an attempt to avoid over-reduction of 3 to piperidine 4, reduction with lithium aluminium hydride was attempted in diethyl ether at low temperature, with the rationale being that the lower solubility of organoaluminates derived from LiAlH4 would cause the product of the initial reduction to form a precipitate and disfavour over-reduction. This was borne out, with 40 % of 17 being isolated by column chromatography after reduction with LiAlH4. In order to avoid an additional step in the synthesis, dehydration to 14 was performed in situ by acidification of the reaction mixture. Thus, an 89 % yield of 14 could be obtained from the reduction of 3 with LiAlH4 followed by an acidic workup (Scheme 4).
Formation of Enamine 5
The reduction of 14 to enamine 5 has been previously reported to proceed with 97 % yield in the presence of LiAlH4.[8] In the present work, a slightly modified version of this procedure was used to provide 5 in 95 % yield. Enamine 5 was found to be particularly unstable with respect to hydrolysis and oxidation and so the reduction reaction was quenched with sodium sulfate decahydrate in place of water. In the majority of cases, the material was used directly on isolation. Samples of enamine 5 could be stored in a freezer for up to 3 months if treated with 1 wt-% dibutylhydroxytoluene (BHT) before removal of solvent.
Formation of UV absorber 1
Acylation of enamine 5 with propionyl chloride was reported to result in the isolation of 1 in 81 % yield after purification by column chromatography.[7] In our hands, it was possible to isolate 1 in 75–88 % yield but the material remained significantly coloured even after chromatographic purification (in its pure state, 1 is an almost colourless oil). It was found that the final product 1 could be isolated from the reaction mixture in >99 % HPLC purity (254 nm) and 72 % yield without recourse to chromatography by formation of the hydrochloride salt, washing with ethyl acetate and conversion to the free base. Additionally, it was found that the HCl salt provided a highly stable form of 1, which was suitable for extended storage at ambient temperature.
Conclusions
An efficient and scalable synthesis of 1, a bio-inspired UV absorber, has been developed to enable further testing and evaluation. Using a combination of batch and continuous-flow steps, the process allowed the chromatography-free preparation of ~0.5 kg of 1 over several batches in >99 % HPLC purity (254 nm). The overall yield of the process was 55 %.
Experimental
Methods and Materials
Continuous-flow chemistry was performed on a Vapourtec R-4 flow reactor heater with connected R2+ and R2 pumping modules. Reagents were used as received from commercial suppliers. 1H and 13C NMR scans were performed on Bruker Advance 400 MHz NMR spectrometer at 400 and 100 MHz respectively. NMR scans were performed with the sample held at 25 ± 0.1°C. Chemical shifts for all experiments are referenced using the Unified Scale relative to 0.3 % tetramethylsilane in deuteriochloform. Samples for NMR spectroscopy were prepared by dissolving the analyte in CDCl3. Spectroscopic data are reported using the following format: (1) chemical shift (ppm); (2) multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad); (3) J, coupling constant (Hz); (4) integration. Melting points were determined using a Büchi melting point B-545 apparatus and are uncorrected. Mass spectrometric analyses were performed on a Thermo Scientific Q Exactive mass spectrometer fitted with an atmospheric solids analysis probe (ASAP) ion source (M&M Mass Spec Consulting).[17] The design and method of ionisation have been described previously.[18,19] Positive and negative ions were recorded in an appropriate mass range at 140000 mass resolution. The atmospheric pressure chemical ionisation (APCI) probe was used without flow of solvent. The nitrogen nebulising and desolvation gas used for vaporisation was heated to 350°C in these experiments. The sheath gas flow rate was set to 25, the auxiliary gas flow rate to 5, and the sweep gas flow rate to 2 (all arbitrary units). The discharge current was 4 mA and the capillary temperature was 320°C. HPLC analysis was performed on an Alliance system comprising a Waters 2695 Separations module and a 2996 photodiode array detector (scanning from 190 to 700nm). The analysis was performed on a 150 × 4.6 mm Alltima C18 column using isocratic 65 % acetonitrile/25 % water at 1.0 mL min−1 as mobile phase. The chromatograms were recorded using a wavelength of 254 nm. Data collection and processing were performed in Waters Empower 3 software.
4,4-Dimethyldihydro-2H-pyran-2,6(3H)-dione 2
Acetic anhydride (265 mL, 2.81 mol) was added to a solution of 3,3-dimethylglutaric acid, 15, (375 g, 2.34 mol) in THF (500 mL) and diluted to a total volume of 1200 mL with THF. The solution was then pumped at a rate of 10 mL min−1 through a series of four 10-mL reactor coils (perfluoroalkoxy alkane (PFA) tubing, 1-mm ID) heated to 110°C and fitted with an 8-bar (800 kPa) acid-resistant backpressure regulator. The combined eluents were evaporated under vacuum, toluene added (100 mL), and the solvent evaporated to give the title compound as a colourless solid (335.0 g, 99 %). Spectroscopic data corresponded to those reported previously.[14] δH 2.62 (s, 4H), 1.17 (s, 6H).
5-(Isobutylamino)-3,3-dimethyl-5-oxopentanoic acid 16
A solution of 4,4-dimethyldihydro-2H-pyran-2,6(3H)-dione, 2, (1.3 M in CH2Cl2, 1791 mL, 2.33 mol) pumped at a rate of 10 mL min−1 was mixed at ambient temperature with a solution of isobutylamine (5 M in CH2Cl2, 278 mL, 2.79 mol) pumped at a rate of 3.12 mL min−1 via a T-piece and passed through a series of four 10-mL reactor coils (PFA tubing, 1-mm ID) heated to 50°C and fitted with an 8-bar acid-resistant backpressure regulator. The combined eluents were then washed with dilute HCl solution (2M, 500 mL), dried with magnesium sulfate, and evaporated under vacuum to a yellow oil, which on standing solidified to give the title compound as a cream solid (496.6 g, 99 %). Mp 70–71°C. m/z [M + H]+ 216.1595; C11H22NO3 requires [M + H]+ 216.1594. δH 6.17 (s, br, 1H), 3.18 (t, J 6.3, 2H), 2.45 (s, 2H), 2.33 (s, 2H), 1.90–1.80 (m, 1H), 1.14 (s, 6H), 0.97 (d, J 6.7, 6H). δc (CDCl3, 400 MHz) 173.92, 173.82, 47.51, 47.22, 46.64, 33.79, 29.37, 28.36, 20.24.
1-Isobutyl-4,4-dimethylpiperidine-2,6-dione (3,3-Dimethyl-N-isobutylglutarimide) 3
A solution of 5-(isobutylamino)-3,3-dimethyl-5-oxopentanoic acid, 16, (1.63 M in CHCl3, 935 mL, 1.52 mol) pumped at a rate of 2.96 mL min−1 was mixed at ambient temperature with a solution of thionyl chloride (6.85 M in CHCl3, 167 mL, 2.29 mol) pumped at a rate of 1.04 mL min−1 via a T-piece and passed through a series of four 10-mL reactor coils (PFA tubing, 1-mm ID) heated to 95°C and fitted with two 8-bar acid-resistant backpressure regulators. The combined eluents were evaporated under vacuum and the residue was dissolved in diethyl ether (1000 mL), and washed with water (2 × 500 mL) and aqueous Na2CO3 solution (10 % w/w, 500 mL). The ethereal solution was dried with magnesium sulfate and evaporated under vacuum to an orange oil, which on standing solidified to give the title compound as a pale orange solid (291.3 g, 97 %). Mp 49–50°C. m/z 198.1490 [M + H]+; C11H20NO2 requires [M + H]+ 198.1489. δH 3.63 (d, J 7.4, 2H), 2.52 (s, 4H), 2.04–1.95 (m, 1H), 1.10 (s, 6H), 0.88 (d, J 6.7, 6H). δc 172.3, 46.6, 29.1, 27.9, 27.3, 20.4.
One-Pot Preparation of 1-Isobutyl-4,4-dimethylpiperidine-2,6-dione (3,3-Dimethyl-N-isobutylglutarimide) 3
A solution of 4,4-dimethyldihydro-2H-pyran-2,6(3H)-dione, 2, (1.3 M in CHCl3, 7.7 mL, 10 mmol) pumped at a rate of 2.58 mL min−1 was mixed at ambient temperature with a solution of isobutylamine (5 M in CHCl3, 2 mL, 10 mmol) pumped at a rate of 0.67 mL min−1 via a T-piece and passed through a 10-mL reactor coil (PFA tubing, 1-mm ID) heated to 50°C. The resulting solution was then mixed at ambient temperature with a solution of thionyl chloride (6.85 M in CHCl3, 167 mL, 15 mmol) pumped at a rate of 0.74 mL min−1 via a T-piece and passed through a series of three 10-mL reactor coils (PFA tubing, 1-mm ID) heated to 100°C and fitted with two 8-bar acid-resistant backpressure regulators. The combined eluents were then evaporated under vacuum, and the residue dissolved in diethyl ether (50 mL) and washed with water (2 × 50 mL) and aqueous Na2CO3 solution (10 % w/w, 50 mL). The ethereal solution was then dried with magnesium sulfate and evaporated under vacuum to an orange oil, which on standing solidified to give the title compound as a pale orange solid (1.8 g, 92 %).
6-Hydroxy-1-isobutyl-4,4-dimethylpiperidin-2-one 17
A solution of 1-isobutyl-4,4-dimethylpiperidine-2,6-dione, 3, (0.50 g, 2.53 mmol) in tetrahydrofuran (25 mL) was cooled on an ice bath and treated dropwise with lithium aluminium hydride (1 M in THF, 2.53 mL, 2.53 mmol) under a nitrogen atmosphere at a rate appropriate to keep the temperature below 20°C. Once addition was complete, a large mass of precipitate was formed that inhibited stirring. The mixture was stirred for 10 min and quenched by addition of sodium sulfate decahydrate (1 g, 30 mmol H2O). The cooling bath was removed and the mixture was stirred for 10 min. The mixture was filtered and the filter cake was washed with three portions of toluene. The filtrates were combined and evaporated under vacuum. The residue was purified by column chromatography, eluting with 0–100 % v/v light petroleum/ethyl acetate, to give the title compound as a pale yellow oil, which solidified on standing to a low-melting solid (0.21 g, 40 %). m/z 200.1645 [M + H]+; C11H22NO2 requires [M + H]+ 200.1645. δH 4.98–4.91 (m, 1H), 3.61–3.58 (m, 1H), 3.12–3.06 (m, 1H), 2.38–1.98 (m, 5H), 1.61–1.54 (m, 1H), 1.07 (s, 3H), 1.01 (s, 3H), 0.91 (d, 3H), 0.85 (d, 3H). δc 170.4, 79.4, 49.3, 46.2, 45.0, 30.2, 29.4, 26.9, 26.6, 20.8, 20.5.
5-Hydroxy-N-isobutyl-3,3-dimethylpentanamide 18
A solution of 1-isobutyl-4,4-dimethylpiperidine-2,6-dione, 3, (0.20 g, 1.01 mmol) in EtOH (5 mL) was cooled to 0°C, under a nitrogen atmosphere and treated portion-wise with sodium borohydride (0.077 g, 2.03 mmol). Once addition was complete, the reaction mixture was allowed to warm to room temperature and stirred for 4 h. Analysis showed incomplete reaction and so the mixture was diluted with EtOH (4 mL) and treated periodically with sodium borohydride (0.35 g, 9.3 mmol) over a period of 15 h. The reaction mixture was then cooled to 0°C, quenched with 1 M HCl and water (5 mL), and the mixture stirred for 5 min. The mixture was extracted with ethyl acetate (3 × 5 mL), the combined organic extracts were washed with a saturated brine solution, dried over phase separation paper, and evaporated under vacuum to give the title compound as a colourless oil (0.17 g, 85 %). m/z 202.1801 [M + H]+; C11H24NO2 requires [M + H]+ 202.1802. δH 6.00 (s, br, 1H), 3.78 (t, J 5.6, 2H), 3.08 (t, J 6.2, 2H), 2.25 (s, 2H), 1.84–1.73 (m, 1H), 1.67 (t, J 5.8, 2H), 1.02 (s, 6H), 0.91 (d, J 6.7, 6H). δc 172.7, 59.5, 48.2, 47.1, 42.5, 32.8, 29.2, 28.6, 20.3.
One-Pot Preparation of 1-Isobutyl-4,4-dimethyl-3,4-dihydropyridin-2(1H)-one 14
A solution of 1-isobutyl-4,4-dimethylpiperidine-2,6-dione, 3, (118 g, 544 mmol) in diethyl ether (590 mL) was cooled on an ice bath and treated dropwise with lithium aluminium hydride (1 M in diethyl ether, 283 mL, 283 mmol) under a nitrogen atmosphere at a rate suitable to keep the temperature below 30°C. Once addition was complete (~20 min), the mixture was stirred for 10 min and quenched by addition of dilute HCl solution (2 M, 40 mL) followed by addition of further HCl solution (4 M, 450 mL) until a clear biphasic mixture was obtained. The cooling bath was removed and the mixture was stirred for 25 min. The aqueous phase was discarded. The organic phase was dried with magnesium sulfate and evaporated under vacuum to give the title compound as a pale orange oil (87.6 g, 89 %). Spectroscopic data corresponded to those reported previously.[8] δH 5.90 (d, J 7.8, 1H), 4.95 (d, J 7.8, 1H), 3.28 (d, J 7.4, 2H), 2.36 (s, 2H), 2.04–1.91 (m, 1H), 1.08 (s, 6H), 0.91 (d, J 6.7, 6H).
1-Isobutyl-4,4-dimethyl-1,2,3,4-tetrahydropyridine 5
Lithium aluminium hydride pellets (14.24 g, 375 mmol) were added to diethyl ether (375 mL) and the resulting mixture was stirred at ambient temperature for 20 min under a nitrogen atmosphere. The resulting suspension was then treated dropwise with a solution of 1-isobutyl-4,4-dimethyl-3,4-dihydropyridin-2(1H)-one, 14, (68 g, 375 mmol) in diethyl ether (290 mL) at a rate suitable to maintain a gentle reflux. Once addition was complete (~20 min), the mixture was heated at reflux for 1 h. The mixture was then quenched by portion-wise addition of sodium sulfate decahydrate (25.6 g, 795 mmol). The resulting suspension was stirred for 20 min, treated with anhydrous sodium sulfate (10 g), and stirred for a further 10 min. The mixture was filtered into a flask containing BHT (0.63 g, 1 wt-% assuming 100 % yield). The filter pad was washed with diethyl ether (2 × 100 mL) and the combined filtrate was evaporated under vacuum to give the title compound as a pale yellow liquid (59.5 g, 95 %). Spectroscopic data corresponded to those reported previously.[6] δH 5.78 (d, J 7.9, 1H), 4.10 (d, J 7.9, 1H), 2.92 (t, J 5.7, 2H), 2.61 (d, J 7.3, 2H), 1.90–1.83 (m, 1H), 1.60 (t, J 5.5, 2H), 1.02 (s, 6H), 0.88 (d, J 6.6, 6H).
1-(1-Isobutyl-4,4-dimethyl-1,4,5,6-tetrahydropyridin-3-yl)propan-1-one 1
A solution of 1-isobutyl-4,4-dimethyl-1,2,3,4-tetrahydropyridine, 5 (40.0 g, 239 mmol) and triethylamine (33.3 mL, 239 mmol) in CH2Cl2 (300 mL) was treated with BHT (0.4 g, 1 wt-% with respect to starting enamine), cooled on a salt/ice bath, and treated dropwise with a solution of propionyl chloride (20.89 mL, 239 mmol) in CH2Cl2 (200 mL) at a rate suitable to keep the temperature of the solution below 0°C. Once addition was complete (~45 min), the mixture was stirred for a further 90 min before quenching with water (300 mL) and stirring vigorously for a further 10 min. The organic phase was washed with sodium carbonate solution (10 % w/w, 200 mL) and dried with sodium sulfate. Evaporation under vacuum gave a pale orange oil (57.9 g), which was dissolved in diethyl ether (300 mL) and treated dropwise with a solution of hydrogen chloride in diethyl ether (2 M, 200 mL, 400 mmol) while stirring. The mixture was evaporated under vacuum and the residual yellow gum treated with ethyl acetate (300 mL), and the mixture heated at reflux until a yellow solid had formed. The mixture was cooled to room temperature, the solid was crushed to uniform size, and the mixture was filtered. The solid was suspended in ethyl acetate (500 mL) and the suspension was heated at reflux with stirring for 30 min. The yellow liquor was removed by filtration and the solid residue was resuspended in ethyl acetate (500 mL) and heated at reflux for 30 min. The almost colourless liquor was removed by filtration, and the off-white solid was suspended between light petroleum (500 mL) and sodium carbonate solution (10 % w/w, 500 mL). The biphasic mixture was shaken until the solid dissolved. The organic phase was dried with magnesium sulfate and evaporated under vacuum to give product 1 as an almost colourless oil (38.4 g, 72 %). HPLC purity was measured at >99 %. Spectroscopic data corresponded to those reported previously.[7] δH 7.15 (s, 1H), 3.12 (t, J 5.8, 2H), 2.96 (d, J 7.4, 2H), 2.46 (q, J 7.5, 2H), 2.00–1.91 (m, 1H), 1.62 (t, J 5.9, 2H), 1.29 (s, 6H), 1.10 (t, J 7.5, 3H), 0.92 (d, J 6.7, 6H). δc 196.3, 147.9, 114.7, 64.3, 43.5, 39.4, 30.2, 29.9, 28.2, 27.6, 19.9, 10.5.
Supplementary Material
1H NMR spectra of all synthesised compounds and 13C NMR spectra of novel compounds (3, 16–18) are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported, in part, by Enterprise Connect’s Researcher in Business program.
References
[1] W. C. Dunlap, J. M. Shick, Y. Yamamoto, Redox Rep. 1999, 4, 301.| Crossref | GoogleScholarGoogle Scholar | 10772069PubMed |
[2] K. Shibata, Plant Cell Physiol. 1969, 10, 325.
[3] W. C. Dunlap, B. E. Chalker, W. M. Bandaranayake, J. J. Wu Won, Int. J. Cosmet. Sci. 1998, 20, 41.
| Crossref | GoogleScholarGoogle Scholar | 18505488PubMed |
[4] R. Losantos, I. Funes-Ardoiz, J. Aguilera, E. Herrera-Ceballos, C. Garcia-Iriepa, P. J. Campos, D. Sampedro, Angew. Chem. Int. Ed. 2017, 56, 2632.
| Crossref | GoogleScholarGoogle Scholar |
[5] I. Tsujino, K. Yabe, I. Sekikawa, N. Hamanaka, Tetrahedron Lett. 1978, 19, 1401.
| Crossref | GoogleScholarGoogle Scholar |
[6] S. Takano, D. Uemara, Y. Hirata, Tetrahedron Lett. 1978, 19, 2299.
| Crossref | GoogleScholarGoogle Scholar |
[7] G. Bird, P. J. Chalmers, N. Fitzmaurice, D. J. Rigg, S. H. Thang, U.S. Patent 5 637 718 1997.
[8] S. H. Thang, D. J. Rigg, Synth. Commun. 1993, 23, 2355.
| Crossref | GoogleScholarGoogle Scholar |
[9] Scifinder commercial sources search April 2019. Available at https://scifinder.cas.org
[10] M. Tanaka, H. Urata, T. Fuchikami, Tetrahedron Lett. 1986, 27, 3165.
| Crossref | GoogleScholarGoogle Scholar |
[11] K. Kondo, T. Takashima, M. Suda, U.S. Patent 4 235 780 1980.
[12] J. Ryan, M. York, Australian Patent 2013351912 2013.
[13] G. Kantin, E. Chupakhin, D. Dar’in, M. Krasavin, Tetrahedron Lett. 2017, 58, 3160.
| Crossref | GoogleScholarGoogle Scholar |
[14] J. Picha, V. Vanek, M. Budesinsky, J. Mladkova, T. A. Garrow, Eur. J. Med. Chem. 2013, 65, 256.
| Crossref | GoogleScholarGoogle Scholar | 23727536PubMed |
[15] H. Flink, T. Putkonen, A. Sipos, R. Jokela, Tetrahedron 2010, 66, 887.
| Crossref | GoogleScholarGoogle Scholar |
[16] For a recent review, see: M. B. Plutschack, B. Pieber, K. Gilmore, P. H. Seeberger, Chem. Rev. 2017, 117, 11796.
| Crossref | GoogleScholarGoogle Scholar | 28570059PubMed |
[17] Atmospheric Solids Analysis Probe (M&M Mass Spec Consulting LLC: Harbeson, DE). Available at http://www.asap-ms.com/ (accessed 8 July 2019)
[18] C. Petucci, J. Diffenda, J. Mass Spectrom. 2008, 43, 1565.
| Crossref | GoogleScholarGoogle Scholar | 18470958PubMed |
[19] A. D. Ray, J. Hammond, H. Major, Eur. J. Mass Spectrom. 2010, 16, 169.
| Crossref | GoogleScholarGoogle Scholar |