

Introduction to the chemistry and pharmacology of psychedelic drugs
Scott R. Walker A , Glenn A. Pullella B , Matthew J. Piggott B and Peter J. Duggan A C *
A , Glenn A. Pullella B , Matthew J. Piggott B and Peter J. Duggan A C *
A CSIRO Manufacturing, Research Way, Clayton, Vic. 3168, Australia.
B Chemistry, School of Molecular Sciences, University of Western Australia, Crawley, WA 6009, Australia. Email: matthew.piggott@uwa.edu.au
C College of Science and Engineering, Flinders University, Adelaide, SA 5042, Australia.
Abstract
The science of psychedelics is an intriguing, multi-disciplinary field that has recently been the subject of heightened public interest. This has mainly resulted from publicity associated with a number of high-profile investigations into psychedelic-assisted therapy for a range of difficult-to-treat mental health conditions. With many psychedelic substances known, including natural, semi-synthetic and fully synthetic, and a rangeof receptors, enzymes and transporters implicated in their modes of action, although very interesting, the field can appear daunting to newcomers to the area. This Primer Review is designed to give an overview of the chemistry and pharmacology of psychedelics. It is hoped that it will provide a useful resource for science undergraduates, postgraduates and their instructors, and experienced scientists who require a comprehensive and up-to-date summary of the field. The Review begins with a summary of the important classes of psychedelics and then goes on to summarise the known history of their traditional human use, dating back to prehistoric times. Following that, important classes of psychedelics are examined in more detail, namely the ergolines, such as lysergic acid diethylamide (LSD), tryptamines like psilocybin and N,N-dimethyltryptamine, phenethylamines typified by mescaline and 3,4-methylenedioxymethamphetamine (MDMA), arylcyclohexylamines including ketamine and phenylcyclohexylpiperidine (PCP), and a group of naturally occurring drugs that do not belong to any of these three classes, examples being muscimol and salvinorin A. The contributions made by early pioneers like Albert Hofmann and Alexander Shulgin are briefly summarised. References to primary literature and more specialised reviews are provided throughout.
Keywords: arylcyclohexylamines, ergolines, hallucinogens, natural products chemistry, phenethylamines, psychedelics, receptors, tryptamines.
Introduction
The term ‘psychedelic’ was coined by the British psychiatrist, Humphry Osmond in 1957[1] by combining the Greek ‘psyche’, meaning mind or soul, and ‘dēleín’ or ‘deloun’ meaning to manifest or reveal. It is implied by this word construction that psychedelic drugs unlock certain otherwise inaccessible aspects of the mind. The common effect of psychedelics is their ability to induce an altered state of consciousness, including a changed perception of the environment, and can include dream-like and mystical experiences. Users also often have an increased belief in the consciousness of a range of living and non-living things.[2] Entheogens are the sub-class of psychedelics that are used in traditional cultural and religious practices. The effects of psychedelic drugs can be profound and long-lasting.[3] Recently-developed theories suggest that psychedelics lead to neuroplasticity and increased neuronal connectivity within the brain.[4]
In recent years, there has been a surge in interest in the use of psychedelic drugs in clinical settings, mainly because of the promise they show as therapies for difficult-to-treat mental health conditions such as post-traumatic stress disorder (PTSD), treatment-resistant depression, addiction, anorexia nervosa and various anxiety states including those experienced near the end of life. This interest has been bolstered by recent seminal publications from United States researchers showing positive clinical effects in depressed cancer patients. A wide variety of natural substances, purified natural products and synthetic compounds are classified as psychedelics, each with intriguing chemistry and pharmacology. As with cannabis, the worldwide prohibition of most psychedelic substances in the early 1970s has greatly hindered their scientific investigation and as a result, there is still much to be learnt about their effects on humans and their modes of action. The purpose of this Primer Review is to provide a succinct overview of the current knowledge of the chemistry and pharmacology of these fascinating bioactive compounds.
This review classifies psychedelic compounds into four broad classes, selected examples of which are shown in Fig 1. The first and largest class of compounds bears the tryptamine substructure and includes compounds like psilocybin (1), derived from certain varieties of mushrooms, dimethyltryptamine (DMT, 2), which is present in many plants, and lysergic acid diethylamide (LSD, 3), a synthetic derivative of the ergoline alkaloids found in ergot fungus and morning glory seeds. These are usually termed ‘classic psychedelics’. The second class is the phenethylamines and includes the cactus-derived mescaline (4), also considered a classic psychedelic, and entactogens like the synthetic 3,4-methylenedioxymethamphetamine (MDMA, 5). The third class is the arylcylohexylamines, which are often described as dissociative hallucinogens and includes drugs like ketamine (6) and phenylcyclohexylpiperidine (PCP, 7). The fourth class encompasses naturally derived hallucinogens that do not fit neatly into any of the above groupings and includes muscimol (8), derived from Amanita muscaria mushrooms; salvinorin A (9), a rare type of non-alkaloid psychedelic, which is derived from the sage Salvia divinorum; and ibogaine (10), an alkaloid found in the West African iboga plant. Some psychoactive components of Cannabis, most notably Δ9-tetrahydrocannabinol (Δ9-THC) and its analogues, are also sometimes considered psychedelics; however they are not covered here, having been extensively reviewed in another primer.[5] There is also a class of psychoactive compounds that cause hallucinations together with delirium, known as deliriants, including the tropane alkaloids scopolamine, atropine and hyoscyamine, that are not usually considered to be psychedelics and hence are not included in this review.
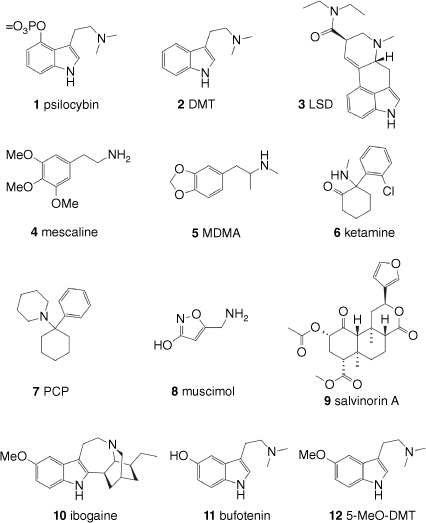
History of human use of psychedelics
It has been surmised that the consumption of psychedelic substances such as muscimol (8), present in fly agaric mushrooms (Amanita muscaria), distinguished by their iconic white-spotted orange-red caps,[6] originated among the Finno-Ugric people of northern Europe and Asia in prehistoric times.[7] These mushrooms, like many plants and fungi that produce psychedelic substances, are associated with shamanism, a religious practice in which participants profess to interact with the spirit world through altered states of consciousness. It seems likely that such rituals spread eastward from northern Europe across Siberia and into North America with the waves of migration that led to the human habitation of the Americas.[7] As such, the use of psilocybin (1)-containing mushrooms of the genera Psilocybe, Stropharia and Panaeolus became prominent in Mesoamerica, the region that extends from present-day central Mexico to Central America. Stone carvings found in the highlands of Central America suggest that these mushrooms were used in religious practices at least 8000 years ago.[7] There is also archaeological evidence for the ancient use of A. muscaria mushrooms in North Africa,[6] and both Amanita and Psilocybe mushrooms by the Ancient Egyptians.[8]
During their expansionary phases in the Americas, early cultures appear to have also found alternative sources of psychedelic substances in the plants that they encountered. Mescaline (4) is present in a number of cactus species including peyote (Lophophora williamsii), native to present-day southwest Texas and Mexico, Peruvian torch cactus (Echinopsis peruviana, syn. Trichocereus peruvianus) and San Pedro cactus (E. pachanoi, syn. T. pachanoi), which occur naturally in the Andes Mountains. There is archaeological evidence of the use of these plants by humans dating back to over 6000 years ago,[9] and given their mescaline content, it is assumed that they were consumed for their psychedelic effects, as has been the case in more recent times. On the other side of the Andes, the indigenous tribes of the Amazon Basin have long used a psychedelic decoction known as ayahuasca. It is prepared from a combination of rainforest plants – usually the stem and bark of the liana Banisteriopsis caapi, and the leaves of the shrub Psychotria viridis. The leaves of the latter plant contain high concentrations of DMT (2), which, although strongly psychedelic, is essentially inactive if taken orally, as its metabolism is greatly accelerated by monoamine oxidase enzymes in the liver. The indigenous people of the Amazon have developed an ingenious way of extending the DMT (2) time of action by incorporating into the decoction parts of plants such as B. caapi, which contain β-carboline alkaloids that reversibly inhibit monoamine oxidase enzymes.[10] An alternative strategy discovered by early cultures is to administer the psychoactive substances in the form of snuffs, thus avoiding the ‘first-pass metabolism’ that occurs in the liver if taken orally. Although widespread across South America and the Caribbean, this practice is thought to originate from continental South America. Seeds of two Anadenanthera species, A. peregrina and A. colubrina, and the resin of a number of Virola species, were used for this purpose and have been shown to contain DMT (2), a range of DMT analogues and β-carbolines.[7] Bufotenin (11), in particular, is found in seeds of certain species of Anadenanthera and in the latex of a sub-species of the north-eastern South American tree Brosimum acutifolium used by indigenous shamans.[11] In addition to plant sources, bufotenin (11) is also found in the skin secretions and eggs of several toads, particularly the Colorado River toad (Incilius alvarius). The evidence that bufotenin is actually psychoactive is weak, however, and these toad secretions contain several other tryptamines including more powerful psychedelics such as 5-MeO-DMT (12, Fig. 1).[12] Representations of the Colorado River toad are a recurring motif in Mesoamerican art, which has been interpreted as a sign of indigenous knowledge of the psychedelic properties of this toad.[13] Toad skin extracts have also been used in traditional Chinese medicine for centuries.[14]
Salvinorin A (9), an unusual non-alkaloid psychoactive, is present in Salvia divinorum, its scientific name meaning ‘sage of the diviners’. A plant from the mint family, it was used by inhabitants of a mountainous region known today as Oaxaca, Mexico, when psychoactive mushrooms were not available.[15]
The root bark of the African shrub Tabernanthe iboga has been traditionally used by West African forest-dwelling people of present-day Gabon and Cameroon.[16] It is used at low doses to alleviate fatigue, hunger and thirst on long hunting expeditions. At higher doses, T. iboga bark is hallucinogenic and this effect is integral to initiation ceremonies and rituals of the Bwiti religion.[17] The practice may have been learnt from the Pygmy peoples of central Africa, who are believed to have used iboga bark from very early times.[18] A key bioactive component of iboga root bark is 10-methoxyibogamine or ibogaine (10). Although this compound is a tryptamine derivative, its subjective effects and pharmacology are quite different to other psychedelic tryptamines, and it is usually considered distinct from that class.
Lysergic acid derivatives structurally related to LSD (3) and members of the class of natural products known as ergoline or ergot alkaloids are present in the seeds of certain ‘sacred’ morning glory plants of the Convolvulaceae family, from the Ipomoea, Rivea and Turbina genera. These are found in present-day Mexico and Central America and although not as potent as LSD (3), ‘sacred’ morning glory seeds are believed to have been used by the indigenous inhabitants of these regions for their psychoactive effects.[7] The seeds of another morning glory, commonly known as Hawaiian baby wood rose, Argyreia nervosa, also contain ergoline alkaloids.[19] Despite the common name, this plant originates from the Indian sub-continent and is used in traditional ayurvedic medicine. Better known are the ergoline alkaloids produced by Claviceps purpurea (ergot), a fungus that can infest grain crops such as barley, wheat and rye, and have been responsible for mass poisonings, particularly in medieval Europe. Ingestion of grains contaminated with ergot can lead to ‘ergotism’, a disturbing cluster of symptoms that can take two main forms: ergotismus gangrenosus and ergotismus convulsivus,[20] the latter often associated with hallucinations. There is evidence to suggest that the ancient Greek ceremonial drink kykeon contained ergot and by inference, ergoline alkaloids.[21] Interestingly, the ergoline alkaloids found in some morning glory seeds are most likely produced by endophytic clavicipitaceous fungi and not the plant itself.[22,23] The presence in perennial rye grass of another genus of endophytic fungus, Epichloë, which has been shown to produce the ergoline alkaloid ergovaline (13a, Fig. 2), has been associated with ‘rye grass staggers’ and ergot poisoning in New Zealand sheep.[24]
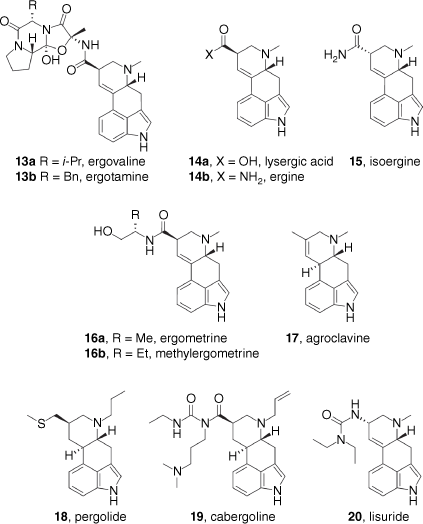
Ergolines
The ergoline alkaloids are a major class of the classic psychedelics, best exemplified by the semi-synthetic and potent psychedelic LSD (3). As mentioned above, the ergot alkaloids are natural products produced by fungi of the genus Claviceps, parasitic on grasses and grains. During the reproductive process, Claviceps species produce hardened tuber-like bodies termed sclerotium, which can contain significant quantities of ergot alkaloids, typically 0.2–0.3% of dry weight in Australian examples.[25] The primary ergot alkaloid found in sclerotium is ergotamine (13b), which bears the tetracyclic ergot alkaloid structure found in lysergic acid (14a), linked to a cyclised tripeptide motif. Results of the analysis of Claviceps extracts suggest that they can contain up to 67 different ergot alkaloids, with many yet to be fully identified.[26] Also as mentioned above, ergot alkaloids are found in some plants, notably members of the Convolvulaceae family. For example, 0.5–0.9% of the dry weight of A. nervosa seeds are indole alkaloids, with the simple lysergic acid primary amide ergine (14b) and its epimer isoergine (15) being the major constituents.[27] Ergot alkaloid-producing plants are quite rare and are distributed across unrelated taxa. Evidence that the alkaloids are actually produced by associated fungi comes from the observation that the treatment of some ergoline-producing plant species with broad-spectrum fungicides eliminates ergoline production.[28] Attempts to culture the associated fungal species have so far been unsuccessful; however, molecular phylogeny studies indicate the presence of a species related to the ergot-producing fungi C. purpurea,[29] and DNA sequencing indicates that this fungal species has genes responsible for ergot alkaloid biosynthesis.[30] There seems little doubt that these fungi are ultimately responsible for the biosynthesis of ergot alkaloids in the Convolvulaceae family.[23]
LSD (3) is the most prominent member of a broad class of lysergic acid amides that consists of both naturally occurring and semi-synthetic compounds. The psychedelic properties of LSD were uncovered by Albert Hofmann in 1943 as part of a wider research program into a semi-synthetic lysergic acid derivatives at Sandoz Ltd., with his experiences later published in book form.[31] During the synthesis of a sample of LSD, Hofmann was struck with ‘remarkable restlessness, combined with a slight dizziness … [later I] sank into a not unpleasant intoxicated-like condition, characterised by an extremely stimulated imagination’. Puzzled by such a vivid occurrence, Hofmann hypothesised that he had been accidently exposed to trace amounts of LSD during its synthesis. To investigate the occurrence further, he decided to deliberately take some of the substance, and mindful of the high toxicity of a number of ergot derivatives, ingested a small, 0.25 mg sample of LSD as the tartrate salt. It is now known that LSD is incredibly potent and that amount equated to a 10-fold higher dose than the minimally recognisable human oral dose of 25 μg.[32] Within an hour, he began to experience effects, and within two a ‘most severe crisis… everything in my field of vision wavered and was distorted as if seen in a curved mirror. I also had the sensation of being unable to move from the spot’.
Hofmann had begun the modern era of psychedelic research. He would later go on to isolate the active principles psilocybin (1) and psilocin (24) from the traditionally used fungi teonanacatl (Psilocybe mexicana), using human ingestion to guide separation after mice were found to be unresponsive.[33] He would also discover the ergot nature of active principles in the traditionally used plant ololiuhqui (Ipomoea corymbose), first disclosed in Sydney at the 1960 International Union of Pure and Applied Chemistry (IUPAC) Natural Products Conference.[31] In addition, he attempted unsuccessfully to isolate the active principle from the traditionally used plant ska María Pastora, returning from Mexico with an undescribed species later named Salvia divinorum, from which would eventually be isolated salvinorin A (9).[34]
Despite the common lysergic acid amide substructure, these compounds have a variety of biological effects, and only some members are potent psychedelics. The simplest possible lysergamide, ergine (14b) has been isolated from both fungal and, especially, plant sources.[23] A medically important naturally-occuring lysergamide is ergometrine (16a), the lysergic acid amide of 2-aminopropanol, which is used to treat post-partum bleeding.[35] Extension of the alkyl sidechain of ergometrine by one carbon unit gives the synthetic drug methylergometrine (16b), also used in the treatment of post-partum bleeding, as well as migrane.[35] Neither of these compounds (16a or 16b) are psychedelic at their clinical dose, although they both display psychedelic properties at high doses.[36] In addition to naturally occurring lysergamides, a wide variety of LSD analogues has been prepared where the amide substituents have been varied, and their pharmacological properties investigated (see below).[37]
The naturally occurring ergovaline (13a) and ergotamine (13b) are members of larger group of natural products termed the ergopeptines, bearing the ergot alkaloid core conjugated to a tripeptide-derived motif. The peptide-derived portion is formed from proline and two hydrophobic amino acids, typically alanine, valine, leucine or phenylalanine, giving a variety of permutations. The high commercial demand for ergot alkaloids for medicinal purposes (see below) is currently met by semi-synthesis from ergopeptines isolated from fungal extracts, with ~7 tonnes of ergopeptines isolated annually from industrial fermentation and extraction of field-sourced fungal material.[38] The ergopeptines have a variety of biological effects, including useful vasoconstrictive properties that, as with the ergometrines, have led to applications in the treatment of migraines and post-partum haemorrhage. Despite the presence of the core ergoline motif, however, the ergopeptines do not have a substantial psychedelic effect.[35]
In addition to the lysergic acid derivatives, there is also the related clavine group of natural products containing the ergoline skeleton with a methyl substituent in the place of the carboxyl group. These alkaloids are non-psychedelic; however, some, such as agroclavine (17), are intermediates in the biosynthesis of lysergamides.[39]
The tetracyclic ergoline scaffold is present in several semi-synthetic therapeutics, such as pergolide (18), cabergoline (19) and lisuride (20). These compounds are generally not considered to be psychedelic, although isolated cases of hallucinations have been reported for patients treated with pergolide (18).[40] Pergolide, cabergoline and lisuride are dopamine D2 receptor agonists and have been, and are still in some countries, used to treat Parkinson’s disease. The use of pergolide and cabergoline has fallen out of favour owing to increased risk of valvular heart disease, due to agonism of the 5-HT2B receptor, and a host of other side-effects.[41] Cabergoline is still used to treat abnormally high prolactin levels and prolactinomas (tumours producing high levels of prolactin), and pergolide is used in veterinary medicine for the treatment of Cushing’s disease in horses.
Ergoline pharmacology
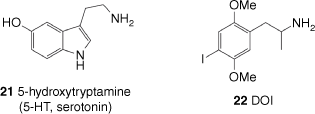
LSD (3) has affinity for a wide range of receptors in the human brain. These receptors’ usual function is to bind neurotransmitters such as serotonin, dopamine and histamine as part of signal transmission in the nervous system.[32] As an extremely potent psychedelic, active doses of LSD in humans are typically 50–200 µg, giving peak plasma concentrations of 3–9 nM.[42] LSD binds to a number of different types of receptors with affinities ranging from micromolar to sub-nanomolar, showing particular affinity for members of the 5-hydroxytrytamine (5-HT) and dopamine receptor family. Although a number of targets could be responsible for its psychedelic effects, the activity of LSD (3) at extremely low concentrations limits the potential candidates to receptors with very high affinity for LSD. These receptors are all members of the 5-HT family, whose primary ligand is 5-HT or serotonin (21, Fig. 3).
The chemical structure of 5-hydroxytrytamine, also known as serotonin, and the synthetic psychedelic DOI (2,5-dimethoxy-4-iodoamphetamine).
Partial agonism of the 5-HT2A receptor is thought to be the primary cause of the psychedelic effects of LSD. The 5-HT2A receptor is a cell-surface protein expressed widely throughout the central nervous system (CNS), but particularly in the areas of the neocortex involved in higher-order brain function such as cognition and perception. Like many receptors in the 5-HT family, the 5-HT2A receptor is a G-protein coupled receptor (GPCR), a type of transmembrane protein that allows signals to traverse cell membranes by conversion of extracellular binding events into intracellular responses. The result is the activation of bound G-protein complexes. Like all proteins, GPCRs exist in a variety of interconverting conformational states, and these can favour or hinder the interaction of the GPCR with other signalling proteins within the cell. Binding of a ligand – usually a low-molecular-weight organic molecule – to the extracellular side of the GPCR stabilises certain conformations of the receptor, shifting the population of protein conformations away from the baseline population that occurs in the absence of a ligand. These ligands can be endogenous substances such as serotonin (21), or exogenous substances such as pharmaceuticals. This change in the conformational population of the GPCR results in a different balance of interactions with downstream signalling proteins compared with that observed in the absence of ligand, and is ultimately responsible for the transmission of the binding signal into the cell.
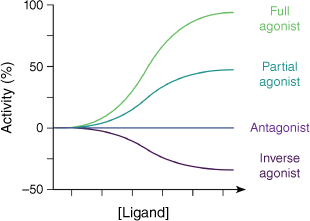
Ligands signalling through GPCRs can be broadly divided into four main classes. Agonists are molecules that, on binding, cause receptor activation by stabilising activated conformations of the receptor. Full agonists achieve the same levels of receptor activation as would be achieved by high concentrations of the endogenous ligand (i.e. maximum activation, Fig. 4). Partial agonists can only achieve partial GPCR activation, even at very high concentrations. These differences in GPCR activation at saturating concentrations reflect the differing stabilisations of the active GPCR state depending on the chemical structure of the agonist. Antagonists block the action of agonists, by binding in their place (orthosteric) or in a position remote from the agonist binding site (allosteric), thereby preventing stabilisation of the active GPCR conformations. Antagonists binding in this fashion can reduce the signalling of the GPCR to the baseline level observed in the absence of agonists. However, even in the absence of any agonists, a small fraction of the GPCR conformational population is in an activated state. Inverse agonists are chemicals that act in the opposite way to agonists, and stabilise inactive conformations of the receptor, thereby reducing signalling below baseline levels.
Modulation of the activity of GPCRs by different types of ligands. Increasing concentration of the ligand is left to right along the x-axis and the level of signalling activity of the GPCR is along the y-axis. Activity is expressed as a percentage of the maximum activity in the presence of saturating concentrations of the endogenous ligand with baseline activity in the absence of ligand set as zero. A full agonist is capable of increasing signalling to the maximum rate, whereas a partial agonist increases signalling to a lower level even at high concentrations. An antagonist binds without affecting the signalling rate from its baseline level, whereas an inverse agonist can lower signalling below baseline.
As mentioned, GPCRs transmit signals into the cell via their interaction with intracellular protein partners, which themselves trigger further signalling pathways. The originally considered interaction partners of GPCRs were thought to be G-protein complexes, which on dissociation from the activated GPCR activate a variety of downstream signals such as those elicited by phosphorylation processes catalysed by phospholipase C. However, subsequent research has uncovered additional signal transduction pathways, both via differing G protein partners and via other interaction partners such as the arrestins.[43]
A GPCR may transmit different signals into the cell depending on which downstream signalling pathways are activated by ligand binding. Binding of a ligand to the GPCR can activate multiple pathways, either equally or unequally depending on the chemical structure of the ligand and how it stabilises particular GPCR conformations.[44] Unequal activation of signalling downstream of the GPCR is termed biased agonism. This biased signalling is an important factor in the biological activity of psychedelics and can lead to different outcomes for otherwise comparable compounds. For example, the natural product ergotamine (13b) and semi-synthetic ergolines LSD (3) and lisuride (20) are all high-affinity (nM to sub-nM) agonists of the 5-HT2A receptor, yet only LSD is psychedelic; the difference in pharmacological effects elicited by these alkaloids is ascribed to biased signalling at the 5-HT2A receptor.[43]
LSD (3) is a partial agonist of the 5-HT2A receptor and binds with extraordinary affinity, displaying an inhibition constant, Ki, of 0.48 nM for displacement of radiolabelled ketanserin.[45] A large contribution to this tight binding is the slow rate of dissociation of the LSD-5-HT2A receptor complex, with a residence time2 for 5-HT2A receptor-bound LSD of 221 min at 37°C.[46] As mentioned, LSD is also a biased agonist, like other psychedelics such as 2,5-dimethoxy-4-iodoamphetamine (DOI, 22), activating a subset of G-protein-dependent signalling mediated by the 5-HT2A receptor. This produces a specific set of downstream phosphorylation changes unique to psychedelic 5-HT2A agonists such as LSD (3) that is not produced by non-psychedelic agonists such as ergotamine (13b) and lisuride (20). In addition to the originally defined GPCR signalling pathways that are all G-protein mediated, there are other non-G protein-mediated pathways activated by LSD that are important to its biological activity, such as the β-arrestin2 signalling pathway, also downstream of 5-HT2A. Not all psychedelic 5-HT2A receptor agonists activate this pathway however; DOI’s activity, for example, is independent of β-arrestin2.[47]
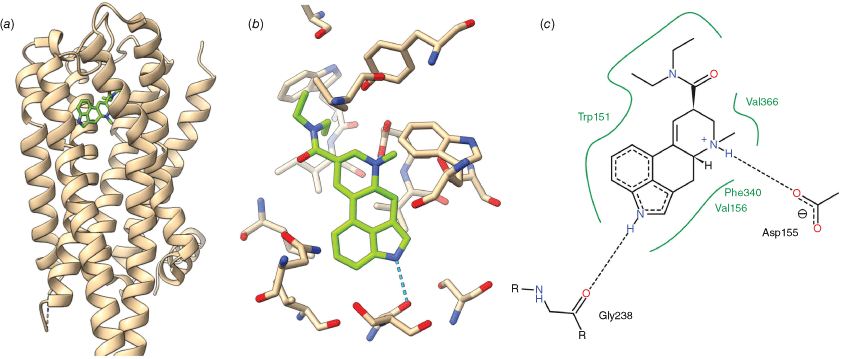
The structural biology of the LSD-5-HT2A receptor complex has been revealed by X-ray crystallography of LSD complexed with 5-HT receptors, initially with models based on the 5-HT2B receptor, and more recently on the X-ray crystallography of the LSD-5-HT2A receptor complex itself.[46,48,49] In the structure reported by Cao et al.,[49] the potent psychedelic 3 binds to the 5-HT2A receptor in a cavity within the bundle of transmembrane helices that comprise the main structure of the GPCR. This cavity is the binding site for a variety of agonists, antagonists and inverse agonists of the 5-HT2A receptor including the endogenous ligand serotonin (21) and is thus termed the orthosteric pocket. The precise details of the interactions between the amino acid residues of the receptor and the chemical structure of the ligands are ultimately responsible for the remarkable spectrum of effects that ligand binding at this one site can cause. In the case of LSD, binding occurs between transmembrane helices II, IV and V, after which a loop encloses the ligand, blocking ready exit from the receptor (Fig. 5a). Occlusion of the binding site by this loop is thought to be responsible for the above-mentioned slow off-rate of LSD from the 5-HT2A receptor.[46] Key features of the LSD-5-HT2A receptor complex include a hydrogen bond between the indole NH of LSD and Gly238 and a salt bridge between the protonated ergoline amino group and Asp 155. (Fig. 5b, c) Phe340 sits above the indole of LSD in an edge-to-face geometry, allowing interaction with the electron-rich pyrrole ring. In addition, the surface of the LSD binding pocket is lined with other hydrophobic residues (Val366, Val156, Phe339, Trp151) that promote favourable interactions with hydrophobic portions of LSD. In an earlier structure by Kim et al.,[48] the core of LSD is in a similar position, but a slight shift out of plane results in the indole NH being closer to Ser242 than Gly238. The function and relative importance of these two possible interactions remain unclear, and while removing this interaction by mutation of Ser242 to alanine does not significantly change the affinity of LSD for the 5-HT2A receptor, it does increase the rate of dissociation of LSD from the receptor.[48] In humans, most other members of the 5-HT receptor family naturally have alanine at this position in the sequence and cannot form this interaction, including receptors with comparable affinity for LSD, such as the 5-HT2B receptor.[50] Importantly, the diethylamide of LSD is a key motif in LSD’s psychedelic effect, with even minor changes resulting in order-of-magnitude losses in potency.[37] The diethylamide group adopts a trans orientation with respect to the terminal methyl groups, which participate in Van der Waals interactions with Val 366 and Trp151. Studies of conformationally restricted LSD analogues have shown this trans arrangement to be an important component of both β-arrestin2 recruitment[46] and in vivo bioactivity in rats.[37] Hinting at the complexity of the pharmacology of these compounds, the non-psychedelic agonist lisuride (20) makes largely similar interactions with the 5-HT2A receptor, suggesting that differences in biological activity hinge on subtle changes within the extended binding pocket.
The binding of LSD to the human 5-HT2A receptor as revealed by X-ray crystallography: (a) location of the LSD (green) in relation to the transmembrane helices of the human 5-HT2A receptor (tan), with the extracellular face of the receptor orientated towards the top. The loop blocking rapid dissociation of LSD from the receptor sits above LSD towards the extracellular face. (b) Detail of the binding site interactions, with (LSD green). (c) Key hydrogen bonds (dotted) and hydrophobic interactions (green). Drawn from the PDB structure 7wc6[49] with the aid of USCF ChimeraX[51] and Poseview.[52]
In addition to changes around the key amide motif of LSD, modification of the N6-methyl group of LSD is known to produce potent psychedelics. In rats trained to distinguish LSD from saline, removal of the methyl group results in loss of activity whereas longer alkyl groups can be active. Sequentially extending the N6-alkyl group gives a spread of potencies relative to LSD: N6-ethyl (approximately 1.6× potency), N6-propyl (approximately equipotent) and N6-butyl (0.1× potency). Increasing the bulk of this substituent is deleterious (N6-isopropyl, 0.4× potency). Introduction of small unsaturated substituents improves activity (N6-allyl, 2.0× potency) whereas larger unsaturated groups are inactive (N6-2-phenethyl).[53] Anecdotal reports of experiences in humans loosely parallel these results, with compounds with N6-allyl and N6-ethyl substituents active at similar concentrations to LSD, whereas N6-propyl-substituted compounds were less active than LSD and N6-2-phenethyl compounds were inactive at 0.5 mg.[54]
Tryptamines
The tryptamine class of psychedelics contain a pendant aminoethyl group extending from the 3-position of an indole core. This substructure is also found in endogenous neurotransmitters such as serotonin (21) and its biosynthetic precursor, the amino acid tryptophan, and in a conformationally restricted form in the ergoline alkaloids. In the latter case, the pendant aminoethyl group has been cyclised to form additional rings. The prototypical tryptamine psychedelic is DMT (2), which is a natural product found in a wide variety of plant species, often alongside close analogues. Endogenously generated DMT is also detectable in trace amounts in biological fluids from several animal species including humans.[55] In both cases, the biosynthetic pathway involves the enzyme-catalysed decarboxylation of tryptophan followed by methylation promoted by aromatic l-amino acid decarboxylase and indole-N-methyltransferase (INMT) respectively. Intriguingly, high levels of INMT expression are detectable in human lung and adrenal tissue, with results from antibody labelling studies suggesting that INMT is also present in the CNS.[55,56] In situ measurements in living rats show extracellular DMT levels in the cortex are in the low nanomolar range, comparable with more typical neurotransmitters such as serotonin (Fig. 3).[56]
DMT (2) is a potent psychedelic, with typical human doses of 4–30 mg (intravenous) or 20–60 mg (inhalation). Despite its potent activity when administered by intravenous, intramuscular or inhaled routes, DMT (2) is essentially inactive when dosed orally, with high oral doses (350 mg) producing no effect in humans.[54] The poor oral bioavailability of DMT (2) is attributed to rapid first-pass metabolism in the liver promoted by the enzyme monoamine oxidase A. However, co-dosing of a monoamine oxidase inhibitor sufficiently slows the metabolism of DMT (2) such that oral doses are effective. As mentioned above, the combination of a DMT-containing plant such as Psychotria viridis with the plant Banisteriopsis caapi, which contains monoamine oxidase-inhibiting harmala alkaloids, is the basis of the traditional South American psychoactive drink ayahuasca.[57] Plants contain other tryptamine natural products in addition to DMT (2), an important sub-set of these being the oxygenated derivatives such as bufotenin (5-hydroxy-DMT, 11) and 5-methoxy-DMT (12). These compounds bear an oxy substituent at C5, in common with the human neurotransmitters serotonin (21) and melatonin (23) (Fig. 6).
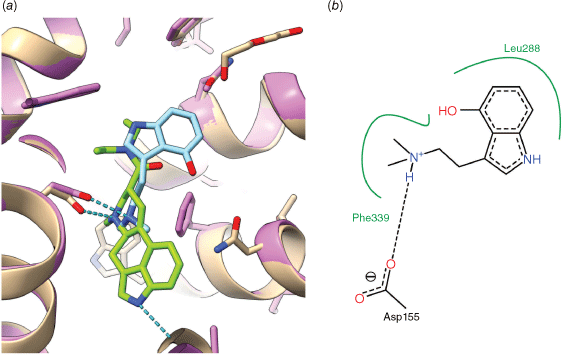
(a) Overlay of LSD (green) and psilocin (light blue) bound to the human 5-HT2A receptor (tan, magenta) as determined by X-ray crystallography. Despite an indole core in both molecules, and a common interaction of the basic side chain with Asp155, the molecules appear to bind in different locations, with LSD in the orthosteric binding pocket and psilocin in the extended binding pocket. (b) Annotated detail of the interaction of psilocin with surrounding amino acids, hydrogen bonds (dashed) and hydrophobic regions (green) are indicated. Drawn from the PDB structures 7wc5, 7wc653 with the aid of USCF ChimeraX[51] and Poseview.[52]
In addition to psychedelic tryptamines from plants and animals, there are also psychedelic tryptamines in the fungal kingdom. Several species of fungi contain a variety of tryptamines, some of which are powerful psychedelics. The major fungal-derived tryptamine psychedelic is psilocin (24), which is derived from psilocybin (1), which bears a 4-phosphate substitution on the core tryptamine structure. Psilocybin-containing fungi are largely within the genus Psilocybe, although other fungi[58] and possibly a lichen[59] have acquired psilocybin biosynthesis by horizontal gene transfer. Psilocybin-containing fungi also typically contain varying levels of the dephosphorylated form psilocin (24), and on human dosing, psilocybin (1) is rapidly dephosphorylated to the active form psilocin (24). This hydroxylated DMT analogue is a powerful psychedelic in humans, and unlike DMT (2), is orally bioavailable, with monoamine oxidases making only a minor contribution to its overall metabolism. Psilocybin (1) is thus a natural prodrug of psilocin (24). In Psilocybe fungi, tissue damage induces enzymatic dephosphorylation of psilocybin and subsequent enzymatic oxidative polymerisation of psilocin generates a characteristic blue quinoid polymer.[60] This polymerisation is thought to be a defence against insect predation, and could explain why some Psilocybe species accumulate very high levels of psilocybin, in some cases up to 2% of dry weight.[61] In addition to psilocybin (1) and psilocin (24), Psilocybe species can contain other minor tryptamines. Variation in the methylation state of the exocyclic amino group gives the secondary amine baeocystin (25), the primary amine norbaeocystin (26) and quaternary amine aeruginascin (27), and its dephosphorylated analogue of baeocystin, norpsilocin (28). It is unclear to what extent these minor tryptamines are psychedelic. Although a 4 mg dose of baeocystin (25) has been reported to be mildly psychedelic in humans,[62] results from more recent in vitro binding and rodent head-twitch assay studies suggest that the tertiary amine group of psilocin is required for the psychedelic effect.[63]
In addition to naturally occurring tryptamines, a variety of synthetic tryptamines have been developed for varied medical purposes. The triptan class of pharmaceuticals (30–33, Fig. 7) are 5-HT1B and 5-HT1D receptor agonists used in the treatment of migraine, and have largely supplanted synthetic ergotamine derivatives for this purpose. Triptans retain the tryptamine core of DMT (2) incorporating larger substituents at the 5-position, although modification to the alkylamine substituent is also possible. These modifications render the triptans non-psychedelic at typical doses.
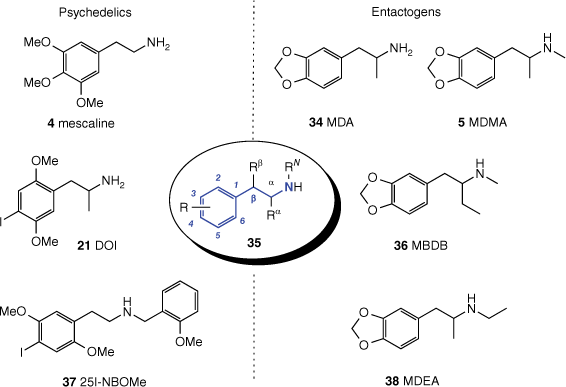
General chemical structure of a substituted phenethylamine 35, with the phenethylamine core highlighted in blue, surrounded by structures of noteworthy phenethylamines segregated on the basis of psychoactivity.
Along with the pharmaceutical optimisation of tryptamines for anti-migraine medications, attempts have been made to explore the psychedelic properties of tryptamines. Notable is the pioneering work of Shulgin, who described the synthesis and psychedelic properties of 55 tryptamines.[54] A wide variety of substitutions were explored in this work, revealing interesting structure–activity relationships (SARs). In contrast to the fairly broad range of substitution in the triptan class of pharmaceuticals, relatively few modifications to the tryptamine scaffold are tolerated to allow retention of psychedelic activity.[37] Modification of one or more of the N-substituents to ethyl, propyl, isopropyl or allyl is tolerated, as is incorporation of small groups on the benzene moiety such as 5-methoxy. Replacement of the phosphate ester of psilocybin with an acetate ester gives psilacetin (29), which retains psychedelic properties in humans. Although the labile acetyl group is likely to be cleaved in vivo to give psilocin (24), mimicking the dephosphorylation process observed with psilocybin (1), psilacetin (29) may also have psychedelic effects in its own right.[63] Although all of these compounds can be broadly classed as psychedelic, users report a variety of differing subjective effects from compound to compound, which may reflect subtle differences in target binding affinity and biased pharmacology.[54]
Tryptamine pharmacology
Like LSD, tryptamines have affinity for a number of CNS receptors. The close structural similarity between tryptamine psychedelics and the endogenous neurotransmitter serotonin is suggestive of a role for the serotoninergic nervous system, and indeed the primary cause of the psychedelic activity of tryptamines is thought to be agonism of the 5-HT2A receptor, similarly to LSD (3). Like LSD, both DMT (2) and psilocin (24) are potent partial agonists of the 5-HT2A receptor (Ki 3–5 and 8 nM respectively),[64] although unlike LSD, they have much lower affinity for adrenergic receptors and no significant affinity for dopamine receptors. Despite the overall different receptor profiles for ergotamines and tryptamines, it is thought to be their common high-affinity partial agonism of the 5-HT2A receptor that causes psychedelic effects in vivo. However, as discussed above for the ergolines, within this simple model, a paradox emerges – whereas the ergotamine and tryptamine psychedelics are all 5-HT2A partial agonists, not all 5-HT2A agonists are psychedelic. This paradox extends to the endogenous 5-HT2A ligand serotonin, which despite its structural similarity to tryptamine psychedelics is not psychedelic. This is again thought to be due to biased signalling at the 5-HT2A receptor, with the relative modulation of downstream biological pathways that occurs on ligand binding being dependent on how each ligand stabilises different receptor conformations. Recent results have raised an intriguing alternative possibility, where signalling from the 5-HT2A receptor may be occurring not only from the expected extracellular-facing 5-HT2A receptor but also from intracellular populations of 5-HT2A receptor. The differing biological effects induced by structural changes to tryptamines may not only reflect differing affinities and biased signalling at the 5-HT2A receptor but also differences in membrane permeability and access to intracellular 5-HT2A receptors. In neurons engineered to transport the otherwise membrane-impermeable serotonin into the cell, addition of serotonin produces changes in neuron growth normally only associated with psychedelics such as DMT. In mice whose cortical neurons have been engineered to transport serotonin intracellularly, administration of a serotonin-releasing agent produces head-twitch responses usually associated with psychedelic compounds, along with antidepressant activity. This intracellular signalling mechanism may be physiologically relevant for endogenously produced membrane-permeable psychedelics such as DMT.[65] Interestingly, in contrast to LSD (3), signalling pathways triggered downstream of tryptamine psychedelics binding to 5-HT2A do not appear to involve β-arrestin2.[66]
The molecular details of the interaction of psilocin (24) with the 5-HT2A receptor have recently been determined by X-ray crystallography, along with LSD (3), serotonin (21) and the non-psychedelic lisuride (20).[49] Like LSD, psilocin (24) binds to the 5-HT2A receptor in the orthosteric site. Whereas the positively charged ammonium groups of LSD and psilocin overlay, the indole ring system of both psilocin and serotonin does not overlap with that of LSD (Fig. 8). Instead, for both psilocin and serotonin, the indole moiety binds nearer to the extracellular face of the receptor, in the same location as the key diethylamide group of LSD. This region of the hydrophobic cleft has been termed the extended binding pocket. The 5-HT2A receptor complexes contrast with the structures of serotonin bound to the 5-HT1A and 5-HT1D receptors,[67] which show a common binding location for the indole of serotonin and LSD, in the orthosteric binding pocket. The reasons for these differences in binding poses and their implications remain to be fully elucidated.
Phenethylamines
The chemical literature, both scientific and clandestine, is replete with psychedelic substances belonging to the family of compounds known as phenethylamines. These psychedelics share a common molecular scaffold (highlighted blue in Fig. 9) consisting of a primary or secondary amine that is linked by a two-carbon unit to a substituted phenyl group. It is no coincidence that the neurotransmitters dopamine and noradrenaline share this scaffold, although they are not considered psychedelics. The amphetamine stimulants are also phenethylamines.
The simple phenethylamine scaffold lends itself to modification, and the combination of substituents at the amino nitrogen, the α- and β-positions, and at any of the five positions on the phenyl group can potentially generate a psychoactive phenethylamine, leading to a vast number of possible psychedelics. Accordingly, synthetic phenethylamines dominate the compound class, with one of the best-known psychoactive phenethylamines, MDMA (5), first synthesised by chemists at Merck in 1912, albeit for reasons entirely unrelated to psychedelia.[68] Among the few naturally occurring psychedelic phenethylamines,[69] mescaline (4) is the most significant, with its rich history of human use, as mentioned above. Although phenethylamines like mescaline and more potent synthetic analogues are often called ‘classic hallucinogens’, they are better described as ‘pseudohallucinogens’, as the sensory disturbances they cause are recognised as being subjective and unreal. True hallucinations are indistinguishable from reality.
There are many reports in which a psychoactive phenethylamine has been subjected to subtle chemical modification, with the resulting analogue found to produce markedly different subjective effects. The occurrence of such dramatically varying SARs hints at the complex pharmacology underlying the activity of these compounds. The common illicit entactogenic phenethylamine MDMA (5), for example, is thought to exert its psychoactivity primarily through increasing extracellular levels of the monoamine neurotransmitters serotonin (21), dopamine and norepinephrine in the brain, but is influenced by a raft of interactions with other CNS receptors and transporters.[70] The foundations of this pharmacological knowledge were gathered as MDMA (5) rose to prominence as a party drug in the 1980s. Now, decades later, MDMA (5) is being intensely investigated as a potential therapeutic agent for a range of mental illnesses,[71–73] thus placing increasing importance on understanding its mechanisms of action.
Mescaline
The history of psychedelic phenethylamines, as noted earlier, is ancient and begins with mescaline (4), which has been consumed by humans in preparations of the peyote (L. williamsii) and wachuma (E. pachanoi, E. peruvianus or E. lageniformis) cacti throughout the Americas for millennia.[9] Mescaline (4) occurs in small amounts in many other cacti,[74] though L. williamsii, E. pachanoi, E. peruvianus and E. lageniformis are the only species that contain enough for a full psychedelic experience.[9] The mescaline (4) content of L. williamsii is approximately 2–6% of the dry weight of the cactus, and the majority of this is concentrated in the above-ground head or crown.[75] The heads are harvested and dried to give discs, referred to as peyote buttons, which are usually chewed or used to prepare a tea.[76]
The traditional Native American usage of peyote and wachuma did not draw the attention of scientists until the late 19th century, whereupon chemical investigation of peyote led to isolation of the alkaloid primarily responsible for its psychoactivity.[9] More than two decades later, in 1919, the identity of this compound was confirmed via synthesis as 3,4,5-trimethoxyphenethylamine (4).[77] With synthetic access to the drug established, mescaline (4) began to appear in Western culture, notably influencing philosopher and novelist Jean-Paul Sartre’s 1938 novel La Nausée, among other of his works,[78] and prompting author Aldous Huxley to pen an account detailing his experience with the drug, entitled The Doors of Perception.[79] In 1955, mescaline (4) even gained the attention of British politician Christopher Mayhew, who participated in an ‘experiment’ in which he was recorded after being administered mescaline (4). The footage of Mayhew was to be televised on the BBC programme Panorama; however, the broadcaster decided against airing it. Mayhew later described the experiment as ‘the most interesting thing [he has] ever done’ and ‘profoundly thought-provoking’.[80]
Mescaline (4) produces changes in colour and visual perception, followed by visual (pseudo)hallucinations,[81] which are often described as kaleidoscopic in nature and can persist for 10–12 h.[76] Distortion of time or space may also be experienced, as well as an array of emotional effects including, feelings of euphoria, heightened anxiety and ego dissolution.[82] In the years following its synthesis, the psychedelic effects of mescaline (4) drew the attention of psychiatrists, who used the drug for varying lines of psychological investigation.[79,82] For example, some drew parallels between mescaline-induced (pseudo)hallucinations and psychosis, and thus made attempts to use mescaline (4) as a tool to study schizophrenic psychosis[83] and as a potential treatment for schizophrenia.[84] The 1960s, however, brought a series of ever-tightening restrictions on research use of mescaline (4) – and of psychedelics more broadly – and it was eventually made illegal in many parts of the world, including in Australia. The drug was internationally outlawed in 1971 by the Convention on Psychotropic Substances.
Relative to other classic psychedelics, LSD (3) and psilocin (24), pharmacological study of mescaline (4) is limited. This may be owing to the low potency of mescaline (4), which requires human oral dosage of approximately 200 mg,[85] some 4000-fold greater than LSD (3). Furthermore, much more potent synthetic phenethylamine psychedelics (see below) can be easily accessed,[85] making mescaline a less attractive target for research. The studies that have been conducted indicate that, like LSD (3) and psilocin (24), mescaline (4) is a serotonergic psychedelic that acts mainly through agonism of the 5-HT2A receptor.[9,82] Mescaline (4) binds 5-HT2A with an affinity (Ki) of 6.3 μM and is also a 5-HT2C receptor agonist with slightly lower affinity, as well as a low-micromolar antagonist of the adrenergic receptor α2A.[86] These interactions are thought to largely account for the effects elicited by mescaline (4), though its receptor interaction profile contains a raft of other targets. Mescaline (4) modestly inhibits the norepinephrine (NET), dopamine (DAT) and serotonin (SERT) transporters,[86] and binds other serotonergic 5-HT1A/2B receptors and the dopaminergic D1/2/3 receptors.[82] The trace amine-associated receptor 1 (TAAR1) regulates monoamine (norepinephrine, dopamine and serotonin) neurotransmission,[87] and mescaline (4) has also been found to bind rat TAAR1,[86] although a recent study questions the relevance of this interaction to the activity of mescaline (4) in humans.[88]
The therapeutic potential of mescaline (4) has been explored in a similarly piecemeal manner to the study of its mode of action. Early studies showed that mescaline did not have potential as a treatment for schizophrenia,[84] while much more recent cross-sectional studies have suggested therapeutic benefits of mescaline (4) for depression, PTSD and substance abuse disorders.[89,90] A recent review from Vamvakopoulou et al.[82] collates preclinical and clinical mescaline (4) research and illustrates the sporadic appearance of such studies. At the time of writing, there are over 450 clinical trials pertaining to psychedelics or hallucinogens registered on clinicaltrials.gov, and only five of these specifically involve mescaline (4). The current state of mescaline (4) research is perhaps attributable to a combination of its poor potency in vivo, its long duration of action (10–12 h) and its persisting status as a prohibited substance in many parts of the world.
Synthetic phenethylamine psychedelics
Prior to its use being criminalised, mescaline (4) also played a significant role in the development of the synthetic phenethylamines by inspiring the investigations of medicinal chemist Alexander Shulgin. Shulgin synthesised at least 179 phenethylamines, which he then tested either alone on himself or along with his wife and close friends, referred to as the ‘research group’.[85] This work established some structure–psychoactivity relationships for phenethylamines in humans and resulted in the discovery of a number of substituted phenethylamines that remain important, such as the ‘classic hallucinogens’ DOM (2,5-dimethoxy-4-methylamphetamine; 40) and 2C-B (2,5-dimethoxy-4-bromophenethylamine; 44, Fig. 10), and entactogens MDEA (3,4-methylenedioxy-N-ethylamphetamine; 38) and MBDB (N-methyl-1-(benzodioxol-5-yl)butan-2-amine; 36, Fig. 9). Alexander (Sasha) and Ann Shulgin’s book Phenethylamines I have known and loved (PiHKAL)[85] is a seminal work that contains frank and detailed descriptions of the subjective effects of phenethylamines and remains extremely valuable to current phenethylamine research.
Structure–activity relationships for 2,4,5-trisubstituted phenethylamine (pseudo)hallucinogens, as assessed by 5-HT2A binding affinity (Ki) and effective oral dose in humans. For TMA-2 (39), DOM (40) and R-(–)-DOM (R-40), reported affinities are for rat 5-HT2A labelled with [3H]ketanserin;[91] for 2C-1 (41), 25I-NBOMe (42), 43 and 2C-B (44) reported affinities are for rat 5-HT2A labelled with [125I]DOI.[92–94]
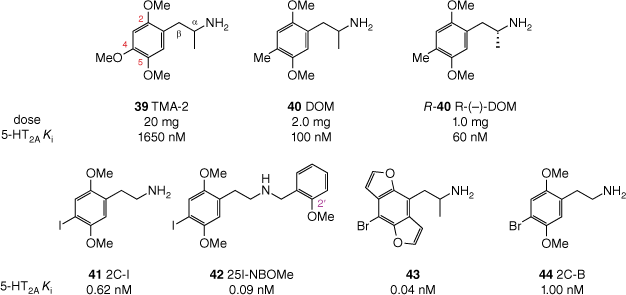
Shulgin identified that, when comparing isomeric phenethylamines with different ring substitution patterns, the 2,4,5-trisubstituted isomers consistently proved the most active.[85] Furthermore, optimal psychoactivity and receptor affinity are seen in analogues with methoxy substituents at both the 2- and 5-positions.[95] Even subtle changes at either of these positions, such as replacing the 2-oxygen atom with a sulfur atom, can lead to a significant drop in potency.[96] Modifications at the 4-position have been extensively explored, establishing that a fairly lipophilic group at this position produces the most active phenethylamines, exemplified by comparison of 2,4,5-trimethoxyamphetamine (TMA-2, 39) and DOM (40, Fig. 10).
Compared with ring substitution, there are fewer options for modulation of activity through the α-position. There are numerous (pseudo)hallucinogenic phenethylamines that either lack an α-substituent or bear an α-methyl group. However, if the group is made any larger (e.g. α-ethyl), essentially all activity is lost.[97] Notably, of the two enantiomers generated by the presence of an α-methyl group, the R-enantiomer ((R)-40) is more potent (Fig. 10).[91]
Separate synthetic studies by Shulgin and Shulgin[85] and Glennon et al.[94] showed that many N-alkyl substituents (e.g. methyl, ethyl, propyl, cyclopropyl, N,N-dimethyl) either reduce the potency of the resulting analogue compared with the parent primary amine, or render it completely inactive. Thus, it was long held that for phenethylamines, a primary amino group was optimal for psychedelic activity. This notion was later refuted by work from Glennon et al., who identified the N-benzyl group as conferring a dramatically increased affinity for the 5-HT2A receptor, with such compounds being potent agonists with high efficacy.[94] The introduction of an N-benzyl substituent provides yet another phenyl group that may be decorated with substituents. Subsequent investigations of SARs around N-benzyl substituents revealed that hydrogen bond acceptors, such as hydroxy or methoxy groups, in the 2′- or 3′-position(s) produce compounds with incredibly potent 5-HT2A receptor affinity.[93,98,99] For example, the affinity of 4-iodo-2,5-dimethoxyphenethylamine (2C-1, 41) for the 5-HT2A receptor, already subnanomolar at 0.62 nM, was increased seven-fold by addition of the N-(2′-methoxybenzyl) substituent (see Fig. 10).[93] The resulting analogue 42 may be abbreviated as 25I-NBOMe, and other phenethylamines sharing the N-(2′-methoxybenzyl) group collectively form the subclass referred to as NBOMes, colloquially pronounced ‘N-bombs’.
The SAR studies spanning the (pseudo)hallucinogenic phenethylamines and leading to the genesis of the NBOMes have been more comprehensively reviewed elsewhere.[95,100,101] A summary of these SARs is illustrated in Fig. 11. The figure highlights many, but not all, structural modifications to the phenethylamine backbone that produce potent 5-HT2A receptor agonists. Indeed, the generalisations made in Fig. 11 do not cover a small family of structurally constrained phenethylamines that exhibit affinity for the 5-HT2A receptor rivalling that of the NBOMes, exemplified by benzodifuran 43 (Fig. 10).[102]
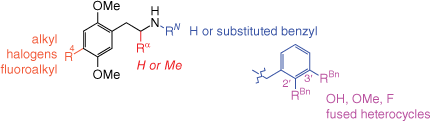
The NBOMes and other phenethylamines (e.g. DOM (40), 2C-1 (41)) exert their psychoactivity as ligands for 5-HT2 receptors, and the majority of their psychedelic effects are attributed to activation of the 5-HT2A receptor.[103–105] The subnanomolar 5-HT2A receptor affinity exhibited by some NBOMes translates into effective doses for humans as low as 0.10 mg.[106] Anecdotally, NBOMes are said to produce visual and auditory psuedohallucinations akin to those elicited by LSD (3), which has provided motivation for their recreational use.[107] These sensory disturbances are frequently accompanied by a range of adverse physiological responses that include hyperthermia, tachycardia, agitation and seizures.[100] The adverse effects, coupled with the low effective dose, make NBOMes a significant health risk, and numerous fatalities have been attributed to 25I-NBOMe (42) toxicity alone.[100] Accordingly, 25I-NBOMe and other NBOMes have been banned in a number of jurisdictions internationally. Many of the comparatively less potent phenethylamines have also been criminalised; DOM, for example, is currently a Schedule I illicit substance in the USA, and similarly Schedule 9 in Australia.[108]
It seems that past synthetic studies for phenethylamines were conducted in the pursuit of 5-HT2A ligands with ever-increasing potency, perhaps driven by the desire to study this interaction as a mechanism of (pseudo)hallucinogenesis. With renewed interest in the therapeutic potential of psychedelics, it is possible that some of the earlier synthetic phenethylamine psychedelics that were discarded may be revisited. The analogues discussed above are a small fraction of those synthesised and documented by Shulgin alone.[85]
MDMA and the entactogens
Shulgin is largely credited with the rediscovery of MDMA (5), colloquially known as ‘ecstasy’ or ‘Molly’, earning him the title ‘the stepfather of MDMA’. The compound had received almost no attention since its production as a synthetic intermediate at Merck in 1912. In 1976, Shulgin presented the effects of MDMA (5) in humans at a scientific conference,[109] and together with medicinal chemist David Nichols, was the first to publish on this topic.[110] Perhaps more influentially, Shulgin introduced MDMA (5) to retired psychotherapist Leo Zeff in 1977. Zeff had previously been interested in the therapeutic potential of LSD (3) and MDA (34), and was impressed by the effects of MDMA (5), so much so that he came out of retirement.[111] Over the 15 years that followed, Zeff propagated the use of MDMA as an adjunct to psychotherapy to approximately 150 therapists and 4000 patients.[111] Psychotherapists adopted the use of MDMA as it facilitates more intimate patient–therapist communication, achieved by enhancing empathetic feelings, reducing defensiveness and assuaging the fear of emotional injury.[112] MDMA also induces intense euphoria and the ‘rush’ associated with classic pyschostimulants,[70] which both likely contributed to its uptake by recreational users.
As word of its pleasurable effects spread, the demand for MDMA (5) became so great that it was mass-produced and overtly marketed; one could even buy the newly commercialised ‘ecstasy’ openly at US bars. Unsurprisingly, the US Drug Enforcement Agency (DEA) stepped in, recommending that the drug be assigned a US Schedule I status, and thus MDMA was outlawed in the USA in 1985. Australia followed suit, declaring MDMA illegal in 1987. Despite being an illicit substance, MDMA remains extremely popular, and in 2019, approximately 630 000 Australians reported using MDMA within the previous 12 months, making it the third most-commonly abused illicit substance after cannabis and cocaine.[113] MDMA remains a Schedule 9 (prohibited) substance in Australia, although, along with psilocybin, this will change on 1 July 2023 for two very specific indications and treatment regimens (see below).
‘Ecstasy’ (MDMA, 5) users generally report an experience distinct from that induced by classic psychedelics such as mescaline (4) or LSD (3). In Shulgin’s words, MDMA induces ‘an easily controlled altered state of consciousness with emotional and sensual overtones.’[110] Although high doses can cause alterations to visual perception, MDMA apparently does not cause true hallucinations.[70] In addition to its unique subjective effects, Nichols invoked structure–psychoactivity relationships for MDMA to argue that it should not be classed as a psychedelic.[114] Nichols’ argument hinged on three major observations. The first is a stereochemical consideration: for the (pseudo)hallucinogenic amphetamines (e.g. DOM (40)), the R-enantiomer is more psychoactive (see Fig. 10), whereas the opposite is true for MDMA. This incongruity hints at key differences in biological target profiles of the two drugs. The second observation is that, for the psychedelic phenethylamines, extending the α-substituent to an ethyl group – consider α-ethyl-DOM (‘ariadne’, 45) compared with DOM (Fig. 12) – results in essentially a complete loss of psychoactivity.[97,115] In contrast, both MDMA and its homologue MBDB (47) are psychedelic. Notably, the stereodisposition of MBDB is consistent with MDMA (i.e. S-MBDB is the more active enantiomer). Last, N-methylation of the ‘classic hallucinogenic’ phenethylamines causes a dramatic attenuation of their activity, illustrated by the approximately 10-fold drop in potency when comparing DOM and N-methyl-DOM (46) (Fig. 12).[85,91] Again, MDMA, being the N-methylated analogue of MDA, does not follow this trend and is active with potency similar to the primary amphetamine (MDA, 34).[116]
Structure–activity relationships differentiating the phenethylamine psychedelics (top row) from the entactogens (bottom row),[114] as illustrated by the effective human oral doses.[85,116,117]
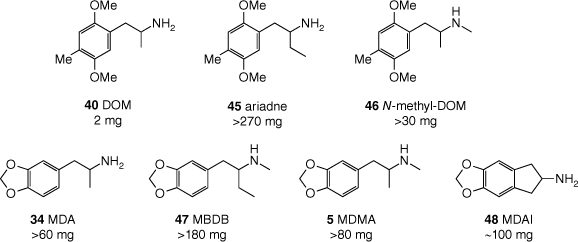
Two structural features – the α-ethyl and N-methyl groups – that are known to suppress pseudohallucinogenic activity are present in MBDB (47). Indeed, MBDB does not induce pseudohallucinations; however, it is psychoactive, and reportedly generates effects very similar to those of MDMA (5).[85] Thus, these compounds and their effects are now widely considered as distinct from those of the hallucinogens, and instead are representative of a class of psychedelics that Nichols dubbed the entactogens.[114] The name entactogen derives from the Greek ‘en’ (within), ‘tactus’ (touch) and ‘gen’ (producing), literally ‘producing touching within’. The name empathogens was also considered, as MDMA and MBDB induce a sense of empathy and connectedness with others. This name was rejected by Nichols owing to the negative connotations of ‘pathogens’, but is still used today.
‘Ecstasy’ (MDMA, 5) is racemic and as mentioned above, the enantiomers have different subjective effects. Potential therapeutic applications, pharmacology and synthesis of MDMA enantiomers are reviewed elsewhere in this issue.[118] The N-methyl group of MDMA may be extended to an N-ethyl group, giving 38 (MDEA, Fig. 9), without significant loss of psychoactivity. The subjective effects of MDEA are reportedly slightly shorter in duration, but otherwise difficult to distinguish from those of MDMA.[116] Extension of the α-substituent, however, to anything other than the α-ethyl group of MBDB (47) renders the resulting MDMA analogue non-psychoactive.[85,119] The α-methyl group of MDA (34) may be tethered to the benzene moiety, producing the aminoindane analogue 48 (MDAI, Fig. 12), which retains the entactogenic activity of unconstrained MDMA. The chemistry and pharmacology of this interesting compound and other closely related aminoindanes have been reviewed by Brandt et al.[117]
The most common illicit entactogen, MDMA (5) is thought to act primarily by reversing the monoamine transporters SERT, NET and DAT to release serotonin (21), norepinephrine and dopamine respectively from neurons, thereby increasing the extracellular levels of these neurotransmitters.[70,120] As a first approximation, the entactogenic, stimulant and euphoric effects of MDMA are thought to be due to the reversal of SERT, NET and DAT respectively. The α-ethylated analogue, MBDB (47), for example, retains the SERT-reversing and entactogenic activity of MDMA, but is a poor DAT reverser, and is accordingly much less euphoric.[121,122] Additionally, MDMA interacts with numerous receptors, including the adrenergic α2A and dopaminergic D1/2 receptors,[120] and is a low-potency agonist of the 5-HT2A receptor.[123] TAAR1 has been posited as another target of MDMA on the basis of sub-micromolar activation of the receptor in rodents, though it activates the human receptor with low potency in cellular assays.[123] The drug also exerts hormonal influences in humans, possibly as a result of altered serotonin activity, which are likely responsible for some of its effects.[124] For further information, we direct readers to the excellent ‘Dark Classics’ review by Dunlap et al., which encapsulates current understanding of the mechanisms of action of MDMA.[70]
The recent resurgence of interest in the therapeutic potential of MDMA (5) has been led by the Multidisciplinary Association for Psychedelic Studies (MAPS), who have published the results of one Phase III clinical trial of MDMA-assisted psychotherapy, with another ongoing. The completed trial demonstrated statistically significant improvement in PTSD symptoms after three treatment sessions.[73] Earlier promising results at Phase II led MDMA to be designated a ‘breakthrough therapy’ by the US Food and Drug Administration (FDA) in 2017, and the drug will be further evaluated for mainstream use in PTSD in 2023.[125] Following the approval in 2014 by Switzerland’s Federal Office of Public Health (FOPH) for the limited clinical use of MDMA (5), LSD (3) and psilocybin (1),[126] the Australian Therapeutic Goods Administration (TGA) also recently approved from 1 July 2023, the prescription of MDMA and LSD specifically for the treatment of PTSD and treatment-resistant depression respectively, under highly restricted treatment regimens.[127]
Although MDMA (5) has the potential to improve the treatment of PTSD and possibly other psychiatric conditions, it not an ideal drug. With an average duration of action of 4–6 h and significant impairment of motor skills, treatment sessions are lengthy and complex, and therefore very expensive. A shorter-acting MDMA analogue could have clinical benefits. The intense euphoria that MDMA induces, probably due to its ability to elevate extracellular brain dopamine levels, is an abuse liability. It remains to be seen whether the ‘high’ that comes with MDMA use is essential or at least beneficial in psychotherapy, or if a less-euphoric but still effective MDMA analogue can be developed. Repeated recreational use of MDMA has been associated with depleted brain serotonin levels,[128] although these potentially neurotoxic effects of MDMA remain contentious.[70] As a noradrenaline releaser, MDMA is also a stimulant, causing increases in heart rate and blood pressure,[129] which are problematic in individuals with underlying cardiovascular disease, and properties not conducive to psychotherapy. Although most of these issues can be managed in the controlled environment of a clinical setting, there is room for improvement.
Arylcyclohexylamines
Ketamine (6) and PCP (7) are examples of synthetic dissociative hallucinogens of the arylcyclohexylamine class of compounds. Phencyclidine (7) was first synthesised by accident in 1956 by Victor Maddox as part of Parke-Davis’s anaesthetic development program.[130] It was initially considered to be a highly promising new anaesthetic and analgesic as it was highly efficacious while not causing respiratory depression.[131] Phencyclidine (7) was marketed under the name Sernyl but was withdrawn from human use in 1967 owing to a range of troubling side effects. The drug can cause schizophrenic-like psychosis, agitation, aggressive behaviour, catatonia, a range of adverse physiological effects and neurotoxicity.[131] Its use as an anaesthetic for non-human primates continued until 1978 but was withdrawn because by the mid- to late 1970s, PCP (7) had become a major illicit drug, especially in the United States.[131] Commonly known as “angel dust”, it became one of the first fully synthetic drugs of abuse. Following numerous media reports describing users engaging in a range of disturbing behaviours including extreme violence, self-mutilation, cannibalism and suicide, its popularity waned.[132]
More recently, there has been a resurgence in illicit PCP (7) use, while it is also being used to adulterate many other illicit drugs.[131] A number of PCP analogues have also found their way into the illicit market and result from chemical modification of either the amine or aromatic portion of the molecule.[132] These derivatives are favoured for their hallucinogenic and euphoric effects. Phencyclidine (7) readily penetrates the blood–brain barrier and, owing to its lipophilic nature, also enters the adipose tissue (body fat), where it can remain for long periods, being slowly released over time.
The pharmacology of PCP (7) is extremely complex, but one very important property appears to be its N-methyl-D-aspartate (NMDA) receptor antagonism, while it also inhibits dopamine and serotonin uptake.[133,134] Its ability to induce schizophrenic-like symptoms has been instrumental in understanding the molecular basis of schizophrenia and in the development of antipsychotic drugs.[131]
Ketamine (6) was first synthesised in 1962 by Wayne State University professor Calvin Stevens while consulting for Parke-Davis, as a part of efforts to develop a safer alternative to PCP (7).[135] The human trials that began in 1964 showed ketamine to be an effective anaesthetic and much safer than PCP (7), and it was subsequently brought on to the market in 1969. Ketamine (6) is often colloquially referred to as a ‘horse tranquilliser’ but it is now widely used in human medicine, especially for its anaesthetic and analgesic properties.[136] Ketamine is currently being investigated as an antidepressant, with its rapid onset following intravenous administration concomitant with a reduction in suicidal ideation being considered particularly promising. At sub-anaesthetic levels, patients can often experience hallucinations or dream-like experiences and an ‘out of body’ feeling,[137] hence its categorisation as a ‘dissociative hallucinogen’. Such effects have led to its illicit use, despite in some cases leading to extreme fear, anxiety and near-death experiences.[138]
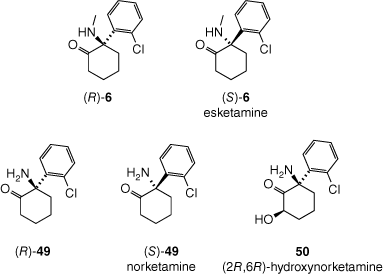
Like PCP (7), ketamine (6) readily crosses the blood–brain barrier[139] and has been shown to be an NMDA receptor antagonist, which appears to a key mechanism by which it induces anaesthesia and analgesia.[136] Ketamine (6) is administered as a racemate, but the (S)-enantiomer (esketamine, Fig. 13) is the more potent NMDA receptor antagonist and analgesic, and is marketed as Ketanest for anaethesia, and Spravato for depression. Ketamine (6) undergoes metabolism to a wide array of metabolites, mainly catalysed by cytochrome P450 enzymes in the liver. Many of these metabolites also have biological effects, thus complicating understanding the drug’s modes of action. One key metabolite appears to be norketamine, generated through demethylation of the secondary amine. The (S)-enantiomer of norketamine (S-49) is also a more potent NMDA receptor antagonist than its (R)-counterpart (R-49). It has been suggested that NMDA receptor antagonism does not fully explain the antidepressant effects of ketamine (6), and that activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, especially by a metabolite of (S)-ketamine (S-6), (2R,6R)-hydroxynorketamine (50), may be central to the antidepressant effect.[140] Although NMDA receptor antagonism is thought to play a central role in the inducement of hallucinations, ketamine (6),[141] like PCP (7) and its metabolites display complex polypharmacology, affecting a range of neuronal receptors and transporters involved in perception.[136]
Other naturally occurring psychedelics
Fly agaric mushrooms (A. muscaria) are native to the temperate and cold temperate forests of the Northern Hemisphere and are believed to have originated from the Siberian–western North American region more than 3 million years ago.[6] They are now widespread in temperate forests of South America, New Zealand and southern Australia.[142] Apart from their use by shamans in northern Europe and Asia, mentioned above, there is evidence of their consumption in northern Africa, as they have appeared in cave paintings in present-day Algeria dating back 5500 years.[6] These iconic mushrooms are ectomycorrhizal fungi, meaning that they form symbiotic relationships with the roots of trees.
Fly agaric mushrooms have been shown to contain a range of bioactives and toxins, and are bioaccumulators of vanadium and other toxic metals.[6] Even when one sets aside the dangers of misidentification of apparently similar but deadly mushrooms, their consumption is fraught with danger and frequently produces a wide range of negative effects. The main constituent thought to be responsible the hallucinogenic activity of A. muscaria is muscimol (8), which is derived from ibotenic acid (51, Fig. 14) through a decarboxylation reaction. Ibotenic acid is a moderate neurotoxin and, like muscimol (8), can cross the blood–brain barrier.[143] Intracranial injections of ibotenic acid can cause brain damage, leading to similar symptoms to Alzheimer’s disease. In fact, ibotenic acid (51) injections are used to generate Alzheimer’s-like rodent models for use in dementia drug discovery and development studies.[6] The hallucinogenic effects of muscimol (8), although not particularly well understood, appear to result from very different pharmacology to any of the other psychedelic substances described above. Muscimol (8) is structurally related to the neurotransmitter γ-aminobutyric acid (GABA, 52) and is an agonist of both GABAA and GABAC receptors.[144] There have been several reports of patients experiencing hallucinations following the administration of the anti-insomnia drug zolpidem,[145] which is also a GABA receptor agonist, and it is thought that muscimol (8) may induce hallucinations in a similar way.
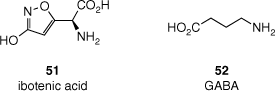
Salvinorin A (9) is present in the leaves of Salvia divinorum, a plant that originates from a high-altitude region of present-day southern Mexico. The short-acting psychedelic effect of salvinorin A (9) and ease of cultivation of S. divinorum have led to the expansion of its use worldwide.[15] In 2002, Australia was the first country to make the possession and sale of S. divinorum and salvinorin A illegal, and several countries have since followed suit. Salvinorin A is rapidly metabolised to inactive products in the gastrointestinal tract and liver and hence users prefer to either hold S. divinorum leaves in the mouth while chewing, employing the oral mucosal route of delivery, or to smoke them.[15] These methods provide more rapid delivery to the brain and, especially in the case of inhalation, avoid first-pass metabolism. The onset of psychedelic effects can be very rapid and they usually do not last any longer than an hour. Being a diterpenoid rather than an alkaloid, salvinorin A (9) and certain phytocannabinoids are the only classes of psychedelic that do not contain nitrogen. Salvinorin A (9) also has a unique mode of action, being a potent and selective κ-opioid receptor agonist.[146] It has been established that other κ-opioid receptor agonists induce perceptual distortions and it is now assumed that salvinorin A (9) causes psychedelic effects through its activation of κ-opioid receptors.[146]
The ingestion of T. iboga bark is said to induce a dream-like state in which adherents to the Bwiti religion are thought to connect with their ancestors and also become more socially connected to their fellow tribal members.[18] The practice can apparently also induce near-death experiences,[147] an effect that is sometimes encountered by users of classic psychedelics.[148] The T. iboga plant produces a large number of alkaloids, with ibogaine (10) being dominant among these. Ibogaine (10), its analogues and metabolites are considered to be mainly responsible for the pharmacological effects of T. iboga bark. The drug is fairly rapidly metabolised in humans, with a half-life of approximately 7 h, and yet its effects can be felt for 1–3 days after administration.[17] A major metabolite of ibogaine (10) is 12-hydroxyibogaine (53, Fig. 15), commonly known as noribogaine, and it is very likely to be an important contributor to the pharmacological effects observed following the ingestion of the parent drug. These two alkaloids (10 and 53) have been assayed against a wide range of receptor and transporter proteins, and despite their structural similarity to the tryptamines, do not induce serotonin receptor activation.[17] Instead, the subjective effects most likely result from the combination of fairly weak interactions with a number of CNS receptors and transporters. The psychedelic and dissociative effects of 10 and 53 may result from partial agonism of the κ-opioid receptor, similar to but weaker than salivinorin A (9), and the inhibition of the NMDA receptor, akin to ketamine (6) and PCP (7). Ibogaine came to prominence in the early 1960s when a young New Yorker, Howard Lotsof, claimed to have been cured of his heroin addiction following experimentation with the drug. Apart from experiencing vivid hallucinations, he reported experiencing a detailed ‘life review’ combined with rationalisations of important life events.[149] The latter appeared to significantly contribute to the overcoming of his opioid addiction. Pharmacologically, the non-competitive antagonism of several nicotinic acetylcholine receptors (nAChRs) is thought to make important contributions to the antiaddictive effects of ibogaine, although it has been argued that simultaneous actions at multiple receptors are likely to be very important to the observed behavioural effects.[17] Lotsof’s advocacy led to sporadic efforts to develop ibogaine as a treatment for addiction; however, the compound is neurotoxic and cardiotoxic, the latter most likely stemming from inhibition of potassium ion channel KV11.1 (the product of the human ether-a-go-go-related gene (hERG)), which has been implicated in the adverse cardiac effects of certain non-steroidal anti-inflammatory drugs.[150] Ibogaine use has been associated with a number of deaths, many of which appear to result from cardiac arrest.[17] This has prompted efforts to develop less toxic ibogaine analogues, as well as structurally unrelated nAChR inhibitors for the treatment of addiction.[151]
Summary
Psychedelics are substances that induce an altered state of consciousness. This can include a dream-like state together with a changed perception of the environment, and can sometimes lead to an increased belief in the consciousness of a range of living and non-living things. Four main classes of psychedelics can be identified: classic psychedelics such as the ergoline LSD (3) and tryptamines like psilocybin (1) and DMT (2); phenethylamines like mescaline (4) and the entactogen MDMA (5); dissociative hallucinogens like ketamine (6); and a group of natural hallucinogens that do not neatly fit into the other three categories, such as muscimol (8) and salvinorin A (9). Hallucinogenic deliriants like scopolamine are generally not considered to be psychedelic. There is a history of traditional use of natural psychedelics dating back to prehistoric times, evidence of which can be found in Europe, Asia, North and South America and Africa, and some of these cultural practices continue today.
LSD (3) is a semi-synthetic compound derived from a class of fungal metabolites known as ergot alkaloids or ergolines and is incredibly potent, with an effective human dose in the tens of micrograms. Psilocybin (1) is found in some species of mushroom and is a prodrug, cleaving to the actual psychoactive compound psilocin (24) in vivo. DMT (2) is also psychedelic and is found in many plant species, but is very rapidly metabolised in the body through a process catalysed by monoamine oxidase enzymes, and is effectively inactive if taken orally. Its duration of action can be extended if co-administered with monoamine oxidase-inhibiting harmala alkaloids, which is the basis of the traditional Amazonian decoction ayahuasca. These psychedelics, as well as mescaline (4), appear to induce their psychedelic effect through biased signalling in a particular serotonin receptor known as the 5HT2A receptor. The synthetic compound MDMA (5) and certain closely related analogues are generally considered separate from psychedelics and are termed entactogens. They are thought to exert their psychoactive effect by reversing the monoamine transporters SERT, NET and DAT, thereby increasing the extracellular levels of serotonin, norepinephrine and dopamine. Lysergic acid diethylamide, psilocybin and MDMA are being investigated clinically for their use with conditions such as treatment-resistant depression, PTSD, anxiety, anorexia nervosa and addiction. These drugs were approved for limited clinical use in Switzerland in 2014, with psilocybin and MDMA due to be available in Australia for very restricted prescription by specially authorised psychiatrists from 1 July 2023.
The synthetic dissociative hallucinogens ketamine (6) and PCP (7) and their metabolites display complex polypharmacology, affecting a range of neuronal receptors and transporters involved in perception, of which antagonism of the NMDA receptor is thought to be quite important. Ketamine is already a registered drug and is used clinically as an anaesthetic and analgesic, but is also now being investigated for its antidepressant properties. Muscimol (8) is present in the iconic fly agaric mushrooms, which are possibly the first species, either fungal or plant, to be consumed by humans for their psychedelic or hallucinogenic effects. Muscimol is thought to induce its hallucinogenic effects through agonism of certain GABA receptors. Salvinorin A (9) is found in the leaves of a species of salvia, S. divinorum, and is assumed to cause its psychedelic effects through activation of κ-opioid receptors. Finally, ibogaine (10), present in the bark of T. iboga is thought to exert its psychoactive effects through a combination of a range of fairly weak interactions in the CNS, with the psychedelic and dissociative effects most likely resulting from partial agonism of the κ-opioid receptor and the inhibition of the NMDA receptor.
Conclusion
Psychedelics are a fascinating class of psychoactive drugs, many of which, owing to their illegal status, have not been available for scientific study or medical use in most parts of the world for approximately five decades. With the results of a number of clinical trials now becoming available, regulators are taking small, hesitant steps towards allowing their registration as approved drugs for a select group of mental health conditions. This has led to a resurgence of interest in the multifaceted scientific investigation of psychedelics, which will no doubt provide a rich vein of endeavour for many years to come.
Conflicts of interest
The CSIRO has undertaken research for several Australian companies whose aim is to commercialise medicinal psychedelics, including Natural MedTech, Psylo, Psychae Therapeutics and Reset Mind Sciences, and in some cases the research is ongoing. MJP and GAP collaborate with, and are sponsored by, Emyria Ltd on MDMA-inspired medicinal chemistry research. PJD and MJP are Guest Editors for the Australian Journal of Chemistry Special Issue in which this article appears, and were blinded from the peer review of this manuscript.
Acknowledgements
GAP and MJP gratefully acknowledge financial, logistical and intellectual support from Emyria Ltd.
References
1 Tanne JH. Humphry Osmond. BMJ 2004; 328: 713.
| Crossref | Google Scholar |
2 Nayak SM, Griffiths RR. A single belief-changing psychedelic experience is associated with increased attribution of consciousness to living and non-living entities. Front Psychol 2022; 13: 852248.
| Crossref | Google Scholar |
3 MacLean KA, Johnson MW, Griffiths RR. Mystical experiences occasioned by the hallucinogen psilocybin lead to increases in the personality domain of openness. J Psychopharmacol 2011; 25: 1453-1461.
| Crossref | Google Scholar |
4 Daws RE, Timmermann C, Giribaldi B, Sexton JD, Wall MB, Erritzoe D, Roseman L, Nutt D, Carhart-Harris R. Increased global integration in the brain after psilocybin therapy for depression. Nat Med 2022; 28: 844-851.
| Crossref | Google Scholar |
5 Duggan PJ. The chemistry of cannabis and cannabinoids. Aust J Chem 2021; 74: 369-387.
| Crossref | Google Scholar |
6 Voynova M, Shkondrov A, Kondeva-Burdina M, Krasteva I. Toxicological and pharmacological profile of Amanita muscaria (L.) Lam. – a new rising opportunity for biomedicine. Pharmacia 2020; 67: 317-323.
| Crossref | Google Scholar |
8 Berlant SR. The entheomycological origin of Egyptian crowns and the esoteric underpinnings of Egyptian religion. J Ethnopharmacol 2005; 102: 275-288.
| Crossref | Google Scholar |
9 Cassels BK, Sáez-Briones P. Dark classics in chemical neuroscience: Mescaline. ACS Chem Neurosci 2018; 9: 2448-2458.
| Crossref | Google Scholar |
10 Brito-da-Costa AM, Dias-da-Silva D, Gomes NGM, Dinis-Oliveira RJ, Madureira-Carvalho Á. Toxicokinetics and toxicodynamics of ayahuasca alkaloids N,N-dimethyltryptamine (DMT), harmine, harmaline and tetrahydroharmine: Clinical and forensic impact. Pharmaceuticals 2020; 13: 334.
| Crossref | Google Scholar |
11 Moretti C, Gaillard Y, Grenand P, Bévalot F, Prévosto J-M. Identification of 5-hydroxy-tryptamine (bufotenine) in takini (Brosimum acutifolium Huber subsp. acutifolium C.C. Berg, Moraceae), a shamanic potion used in the Guiana Plateau. J Ethnopharmacol 2006; 106: 198-202.
| Crossref | Google Scholar |
12 Schwelm HM, Zimmermann N, Scholl T, Penner J, Autret A, Auwärter V, Neukamm MA. Qualitative and quantitative analysis of tryptamines in the poison of Incilius alvarius (Amphibia: Bufonidae). J Anal Toxicol 2022; 46: 540-548.
| Crossref | Google Scholar |
13 Davis W, Weil AT. Identity of a New World psychoactive toad. Anc Mesoam 1992; 3: 51-59.
| Crossref | Google Scholar |
14 Jensen H, Chen KK. The chemical identity of certain basic constituents present in the secretions of various species of toads. J Biol Chem 1936; 116: 87-91.
| Crossref | Google Scholar |
15 Hernández-Alvarado RB, Madariaga-Mazón A, Ortega A, Martinez-Mayorga K. Dark classics in chemical neuroscience: Salvinorin A. ACS Chem Neurosci 2020; 11: 3979-3992.
| Crossref | Google Scholar |
16 Jenks C. Extraction studies of Tabernanthe Iboga and Voacanga Africana. Nat Prod Lett 2002; 16: 71-76.
| Crossref | Google Scholar |
17 Wasko MJ, Witt-Enderby PA, Surratt CK. Dark classics in chemical neuroscience: Ibogaine. ACS Chem Neurosci 2018; 9: 2475-2483.
| Crossref | Google Scholar |
18 Alper KR, Lotsof HS, Kaplan CD. The ibogaine medical subculture. J Ethnopharmacol 2008; 115: 9-24.
| Crossref | Google Scholar |
19 Paulke A, Kremer C, Wunder C, Wurglics M, Schubert-Zsilavecz M, Toennes SW. Studies on the alkaloid composition of the Hawaiian Baby Woodrose Argyreia nervosa, a common legal high. Forensic Sci Int 2015; 249: 281-293.
| Crossref | Google Scholar |
20 Smakosz A, Kurzyna W, Rudko M, Dąsal M. The usage of ergot (Claviceps purpurea (fr.) Tul.) in obstetrics and gynecology: A historical perspective. Toxins 2021; 13: 492.
| Crossref | Google Scholar |
21 Johnson MW, Hendricks PS, Barrett FS, Griffiths RR. Pharmacol Therapeut 2019; 197: 83-102.
| Crossref |
22 Steiner U, Ahimsa-Müller MA, Markert A, Kucht S, Groß J, Kauf N, Kuzma M, Zych M, Lamshöft M, Furmanowa M, Knoop V, Drewke C, Leistner E. Molecular characterization of a seed transmitted clavicipitaceous fungus occurring on dicotyledoneous plants (Convolvulaceae). Planta 2006; 224: 533-544.
| Crossref | Google Scholar |
23 Steiner U, Leistner E. Ergot alkaloids and their hallucinogenic potential in morning glories. Planta Med 2018; 84: 751-758.
| Crossref | Google Scholar |
24 Eady C. The impact of alkaloid-producing Epichloë endophyte on forage ryegrass breeding: A New Zealand perspective. Toxins 2021; 13: 158.
| Crossref | Google Scholar |
25 Blaney BJ, Molloy JB, Brock IJ. Alkaloids in Australian rye ergot (Claviceps purpurea) sclerotia: implications for food and stockfeed regulations. Anim Prod Sci 2009; 49: 975-982.
| Crossref | Google Scholar |
26 Uhlig S, Rangel-Huerta OD, Divon HH, Rolén E, Pauchon K, Sumarah MW, Vrålstad T, Renaud JB. Unraveling the ergot alkaloid and indole diterpenoid metabolome in the Claviceps purpurea species complex using LC–HRMS/MS diagnostic fragmentation filtering. J Agric Food Chem 2021; 69: 7137-7148.
| Crossref | Google Scholar |
27 Chao JM, Der Marderosian AH. Ergoline alkaloidal constituents of Hawaiian Baby Wood Rose, Argyreia nervosa (Burm. f.) Bojer. J Pharm Sci 1973; 62: 588-591.
| Crossref | Google Scholar |
28 Kucht S, Groß J, Hussein Y, Grothe T, Keller U, Basar S, König WA, Steiner U, Leistner E. Elimination of ergoline alkaloids following treatment of Ipomoea asarifolia (Convolvulaceae) with fungicides. Planta 2004; 219: 619-625.
| Crossref | Google Scholar |
29 Li S-M, Unsöld I. Post-genome research on the biosynthesis of ergot alkaloids. Planta Med 2006; 72: 1117-1120.
| Crossref | Google Scholar |
30 Markert A, Steffan N, Ploss K, Hellwig S, Steiner U, Drewke C, Li S-M, Boland W, Leistner E. Biosynthesis and accumulation of ergoline alkaloids in a mutualistic association between Ipomoea asarifolia (Convolvulaceae) and a Clavicipitalean fungus. Plant Physiol 2008; 147: 296-305.
| Crossref | Google Scholar |
32 Passie T, Halpern JH, Stichtenoth DO, Emrich HM, Hintzen A. The pharmacology of lysergic acid diethylamide: A review. CNS Neurosci Ther 2008; 14: 295-314.
| Crossref | Google Scholar |
33 Hofmann A, Heim R, Brack A, Kobel H. Psilocybin, ein psychotroper Wirkstoff aus dem mexikanischen RauschpilzPsilocybe mexicana Heim. Experientia 1958; 14: 107-109.
| Crossref | Google Scholar |
34 Ortega A, Blount JF, Manchand PS. Salvinorin, a new trans-neoclerodane diterpene from Salvia divinorum(Labiatae). J Chem Soc Perkin Trans 1 1982; 2505-2508.
| Crossref | Google Scholar |
35 Schiff Jr PL. Ergot and its alkaloids. Am J Pharm Educ 2006; 70: 98.
| Crossref | Google Scholar |
36 Ott J, Neely P. Entheogenic (hallucinogenic) effects of methylergonovine. J Psychedelic Drugs 1980; 12: 165-166.
| Crossref | Google Scholar |
38 Wong G, Lim LR, Tan YQ, Go MK, Bell DJ, Freemont PS, Yew WS. Reconstituting the complete biosynthesis of D-lysergic acid in yeast. Nat Commun 2022; 13: 712.
| Crossref | Google Scholar |
39 Wallwey C, Li SM. Ergot alkaloids: structure diversity, biosynthetic gene clusters and functional proof of biosynthetic genes. Nat Prod Rep 2011; 28: 496-510.
| Crossref | Google Scholar |
40 Arnold G, Gasser T, Storch A, Lipp A, Kupsch A, Hundemer HP, Schwarz J. High doses of pergolide improve clinical global impression in advanced Parkinson's disease—A preliminary open label study. Arch Gerontol Geriatr 2005; 41: 239-253.
| Crossref | Google Scholar |
41 Tsuboi T, Watanabe H, Katsuno M, Sobue G. Cabergoline in the treatment of Parkinson’s disease. In: Riederer P, Laux G, Mulsant B, Le W, Nagatsu T, editors. NeuroPsychopharmacotherapy. Springer; 2019. pp. 1–10. https://doi.org/10.1007/978-3-319-56015-1_223-1
42 Dolder PC, Schmid Y, Steuer AE, Kraemer T, Rentsch KM, Hammann F, Liechti ME. Pharmacokinetics and pharmacodynamics of lysergic acid diethylamide in healthy subjects. Clin Pharmacokinet 2017; 56: 1219-1230.
| Crossref | Google Scholar |
44 Gurevich VV, Gurevich EV. GPCR signaling regulation: The role of GRKs and arrestins. Front Pharmacol 2019; 10: 125.
| Crossref | Google Scholar |
45 Janowsky A, Eshleman AJ, Johnson RA, Wolfrum KM, Hinrichs DJ, Yang J, Zabriskie TM, Smilkstein MJ, Riscoe MK. Mefloquine and psychotomimetics share neurotransmitter receptor and transporter interactions in vitro. Psychopharmacology 2014; 231: 2771-2783.
| Crossref | Google Scholar |
46 Wacker D, Wang S, McCorvy JD, Betz RM, Venkatakrishnan AJ, Levit A, Lansu K, Schools ZL, Che T, Nichols DE, Shoichet BK, Dror RO, Roth BL. Crystal structure of an LSD-bound human serotonin receptor. Cell 2017; 168: 377-389.e12.
| Crossref | Google Scholar |
47 Karaki S, Becamel C, Murat S, Mannoury la Cour C, Millan MJ, Prézeau L, Bockaert J, Marin P, Vandermoere F. Quantitative phosphoproteomics unravels biased phosphorylation of serotonin 2A receptor at Ser280 by hallucinogenic versus nonhallucinogenic agonists. Mol Cell Proteomics 2014; 13: 1273-1285.
| Crossref | Google Scholar |
48 Kim K, Che T, Panova O, DiBerto JF, Lyu J, Krumm BE, Wacker D, Robertson MJ, Seven AB, Nichols DE, Shoichet BK, Skiniotis G, Roth BL. Structure of a hallucinogen-activated Gq-coupled 5-HT2A serotonin receptor. Cell 2020; 182: 1574-1588.e19.
| Crossref | Google Scholar |
49 Cao D, Yu J, Wang H, Luo Z, Liu X, He L, Qi J, Fan L, Tang L, Chen Z, Li J, Cheng J, Wang S. Structure-based discovery of nonhallucinogenic psychedelic analogs. Science 2022; 375: 403-411.
| Crossref | Google Scholar |
50 Knight AR, Misra A, Quirk K, Benwell K, Revell D, Kennett G, Bickerdike M. Pharmacological characterisation of the agonist radioligand binding site of 5-HT2A, 5-HT2B and 5-HT2C receptors. Naunyn Schmiedebergs Arch Pharmacol 2004; 370: 114-123.
| Crossref | Google Scholar |
51 Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 2021; 30: 70-82.
| Crossref | Google Scholar |
52 Stierand K, Maaß PC, Rarey M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics 2006; 22: 1710-1716.
| Crossref | Google Scholar |
53 Hoffman AJ, Nichols DE. Synthesis and LSD-like discriminative stimulus properties in a series of N(6)-alkyl norlysergic acid N,N-diethylamide derivatives. J Med Chem 1985; 28: 1252-1255.
| Crossref | Google Scholar |
55 Barker SA, McIlhenny EH, Strassman R. A critical review of reports of endogenous psychedelic N, N-dimethyltryptamines in humans: 1955–2010. Drug Test Anal 2012; 4: 617-635.
| Crossref | Google Scholar |
56 Dean JG, Liu T, Huff S, Sheler B, Barker SA, Strassman RJ, Wang MM, Borjigin J. Biosynthesis and extracellular concentrations of N,N-dimethyltryptamine (DMT) in mammalian brain. Sci Rep 2019; 9: 9333.
| Crossref | Google Scholar |
57 Riba J, Valle M, Urbano G, Yritia M, Morte A, Barbanoj MJ. Human pharmacology of ayahuasca: Subjective and cardiovascular effects, monoamine metabolite excretion, and pharmacokinetics. J Pharmacol Exp Ther 2003; 306: 73-83.
| Crossref | Google Scholar |
58 Reynolds HT, Vijayakumar V, Gluck-Thaler E, Korotkin HB, Matheny PB, Slot JC. Horizontal gene cluster transfer increased hallucinogenic mushroom diversity. Evol Lett 2018; 2: 88-101.
| Crossref | Google Scholar |
59 Schmull M, Dal-Forno M, Lücking R, Cao S, Clardy J, Lawrey JD. Dictyonema huaorani (Agaricales: Hygrophoraceae), a new lichenized basidiomycete from Amazonian Ecuador with presumed hallucinogenic properties. Bryologist 2014; 117: 386-394.
| Crossref | Google Scholar |
60 Lenz C, Wick J, Braga D, García-Altares M, Lackner G, Hertweck C, Gressler M, Hoffmeister D. Injury-triggered blueing reactions of Psilocybe “magic” mushrooms. Angew Chem Int Ed 2020; 59: 1450-1454.
| Crossref | Google Scholar |
61 Gotvaldova K, Borovička J, Hájková K, Cihlářová P, Rockefeller A, Kuchař M. Extensive collection of psychotropic mushrooms with determination of their tryptamine alkaloids. Int J Mol Sci 2022; 23: 14068.
| Crossref | Google Scholar |
62 Gartz J. Biotransformation of tryptamine derivatives in mycelial cultures of Psilocybe. J Basic Microbiol 1989; 29: 347-352.
| Crossref | Google Scholar |
63 Glatfelter GC, Pottie E, Partilla JS, Sherwood AM, Kaylo K, Pham DNK, Naeem M, Sammeta VR, DeBoer S, Golen JA, Hulley EB, Stove CP, Chadeayne AR, Manke DR, Baumann MH. Structure–activity relationships for psilocybin, baeocystin, aeruginascin, and related analogues to produce pharmacological effects in mice. ACS Pharmacol Transl Sci 2022; 5: 1181-1196.
| Crossref | Google Scholar |
64 Roth BL, Lopez E, Patel S, Kroeze WK. The multiplicity of serotonin receptors: Uselessly diverse molecules or an embarrassment of riches? Neuroscientist 2000; 6: 252-262.
| Crossref | Google Scholar |
65 Vargas MV, Dunlap LE, Dong C, Carter SJ, Tombari RJ, Jami SA, Cameron LP, Patel SD, Hennessey JJ, Saeger HN, McCorvy JD, Gray JA, Tian L, Olson DE. Psychedelics promote neuroplasticity through the activation of intracellular 5-HT2A receptors. Science 2023; 379: 700-706.
| Crossref | Google Scholar |
66 Schmid CL, Bohn LM. Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a β-arrestin2/Src/Akt signaling complex in vivo. J Neurosci 2010; 30: 13513-13524.
| Crossref | Google Scholar |
67 Xu P, Huang S, Zhang H, Mao C, Zhou XE, Cheng X, Simon IA, Shen DD, Yen HY, Robinson CV, Harpsøe K, Svensson B, Guo J, Jiang H, Gloriam DE, Melcher K, Jiang Y, Zhang Y, Xu HE. Structural insights into the lipid and ligand regulation of serotonin receptors. Nature 2021; 592: 469-473.
| Crossref | Google Scholar |
68 Bernschneider-Reif S, Öxler F, Freudenmann RW. The origin of MDMA (‘ecstasy’) – separating the facts from the myth. Pharmazie 2006; 61: 966-972.
| Google Scholar |
69 Bruhn JG, EI-Seedi HR, Stephanson N, Beck O, Shulgin AT. Ecstasy analogues found in cacti. J Psychoactive Drugs 2008; 40: 219-222.
| Crossref | Google Scholar |
70 Dunlap LE, Andrews AM, Olson DE. Dark classics in chemical neuroscience: 3,4-methylenedioxymethamphetamine. ACS Chem Neurosci 2018; 9: 2408-2427.
| Crossref | Google Scholar |
71 Sessa B. Why MDMA therapy for alcohol use disorder? And why now? Neuropharmacology 2018; 142: 83-88.
| Crossref | Google Scholar |
72 Sartori SB, Singewald N. Novel pharmacological targets in drug development for the treatment of anxiety and anxiety-related disorders. Pharmacol Ther 2019; 204: 107402.
| Crossref | Google Scholar |
73 Mitchell JM, Bogenschutz M, Lilienstein A, Harrison C, Kleiman S, Parker-Guilbert K, Ot’alora G M, Garas W, Paleos C, Gorman I, Nicholas C, Mithoefer M, Carlin S, Poulter B, Mithoefer A, Quevedo S, Wells G, Klaire SS, van der Kolk B, Tzarfaty K, Amiaz R, Worthy R, Shannon S, Woolley JD, Marta C, Gelfand Y, Hapke E, Amar S, Wallach Y, Brown R, Hamilton S, Wang JB, Coker A, Matthews R, de Boer A, Yazar-Klosinski B, Emerson A, Doblin R. MDMA-assisted therapy for severe PTSD: A randomized, double-blind, placebo-controlled phase 3 study. Nat Med 2021; 27: 1025-1033.
| Crossref | Google Scholar |
75 Klein MT, Kalam M, Trout K, Fowler N, Terry M. Mescaline concentrations in three principal tissues of Lophophora williamsii (Cactaceae): Implications for sustainable harvesting practices. Haseltonia 2015; 2015: 34-42.
| Crossref | Google Scholar |
76 Dinis-Oliveira RJ, Pereira CL, da Silva DD. Pharmacokinetic and pharmacodynamic aspects of peyote and mescaline: Clinical and forensic repercussions. Curr Mol Pharmacol 2019; 12: 184-194.
| Crossref | Google Scholar |
77 Späth E. Über dieanhalonium-alkaloide. Monatsh Chem 1919; 40: 129-154.
| Crossref | Google Scholar |
78 Jay M. ‘Sartre’s Bad Trip’, The Paris Review. 2019. Available at https://www.theparisreview.org/blog/2019/08/21/sartres-bad-trip/ [verified 15 February 2023].
80 Last A. The History Hour: Why a British MP was filmed taking mescaline. BBC, 2021. Available at https://www.bbc.co.uk/programmes/w3ct1z6v [verified 15 February 2023].
81 Guttmann E. Artificial psychoses produced by mescaline. J Ment Sci 1936; 82: 203-221.
| Crossref | Google Scholar |
82 Vamvakopoulou IA, Narine KAD, Campbell I, Dyck JRB, Nutt DJ. Mescaline: The forgotten psychedelic. Neuropharmacology 2023; 222: 109294.
| Crossref | Google Scholar |
83 Osmond H, Smythies J. Schizophrenia: A new approach. J Ment Sci 1952; 98: 309-315.
| Crossref | Google Scholar |
84 Denber HCB, Merlis S. Studies on mescaline I. Action in schizophrenic patients. Psychiatr Q 1955; 29: 421-429.
| Crossref | Google Scholar |
86 Rickli A, Moning OD, Hoener MC, Liechti ME. Receptor interaction profiles of novel psychoactive tryptamines compared with classic hallucinogens. Eur Neuropsychopharmacol 2016; 26: 1327-1337.
| Crossref | Google Scholar |
87 Miller GM. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J Neurochem 2011; 116: 164-176.
| Crossref | Google Scholar |
88 Kolaczynska KE, Luethi D, Trachsel D, Hoener MC, Liechti ME. Receptor interaction profiles of 4-alkoxy-3,5-dimethoxy-phenethylamines (mescaline derivatives) and related amphetamines. Front Pharmacol 2022; 12: 794254.
| Crossref | Google Scholar |
89 Agin-Liebes G, Haas TF, Lancelotta R, Uthaug MV, Ramaekers JG, Davis AK. Naturalistic use of mescaline is associated with self-reported psychiatric improvements and enduring positive life changes. ACS Pharmacol Transl Sci 2021; 4: 543-552.
| Crossref | Google Scholar |
90 Uthaug MV, Davis AK, Haas TF, Davis D, Dolan SB, Lancelotta R, Timmermann C, Ramaekers JG. The epidemiology of mescaline use: Pattern of use, motivations for consumption, and perceived consequences, benefits, and acute and enduring subjective effects. J Psychopharmacol 2022; 36: 309-320.
| Crossref | Google Scholar |
91 Glennon RA, Titeler M, McKenney JD. Evidence for 5-HT2 involvement in the mechanism of action of hallucinogenic agents. Life Sci 1984; 35: 2505-2511.
| Crossref | Google Scholar |
92 Parker MA, Marona-Lewicka D, Lucaites VL, Nelson DL, Nichols DE. A novel (benzodifuranyl)aminoalkane with extremely potent activity at the 5-HT2A receptor. J Med Chem 1998; 41: 5148-5149.
| Crossref | Google Scholar |
93 Braden MR, Parrish JC, Naylor JC, Nichols DE. Molecular interaction of serotonin 5-HT2A receptor residues Phe339(6.51) and Phe340(6.52) with superpotent N-benzyl phenethylamine agonists. Mol Pharmacol 2006; 70: 1956-1964.
| Crossref | Google Scholar |
94 Glennon RA, Dukat M, El-Bermawy M, Law H, De Los Angeles J, Teitler M, King A, Herrick-Davis K. Influence of amine substituents on 5-HT2A versus 5-HT2C binding of phenylalkyl- and indolylalkylamines. J Med Chem 1994; 37: 1929-1935.
| Crossref | Google Scholar |
95 Nichols DE. Structure–activity relationships of serotonin 5-HT2A agonists. WIREs Membr Transp Signal 2012; 1: 559-579.
| Crossref | Google Scholar |
96 Jacob III P, Shulgin AT. Sulfur analogs of psychotomimetic agents. 2. Analogs of (2,5-dimethoxy-4-methylphenyl)- and of (2,5-dimethoxy-4-ethylphenyl)isopropylamine. J Med Chem 1983; 26: 746-752.
| Crossref | Google Scholar |
97 Cunningham MJ, Bock HA, Serrano IC, Bechand B, Vidyadhara DJ, Bonniwell EM, Lankri D, Duggan P, Nazarova AL, Cao AB, Calkins MM, Khirsariya P, Hwu C, Katritch V, Chandra SS, McCorvy JD, Sames D. Pharmacological mechanism of the non-hallucinogenic 5-HT2A agonist ariadne and analogs. ACS Chem Neurosci 2023; 14: 119-135.
| Crossref | Google Scholar |
98 Hansen M, Phonekeo K, Paine JS, Leth-Petersen S, Begtrup M, Bräuner-Osborne H, Kristensen JL. Synthesis and structure–activity relationships of N-benzyl phenethylamines as 5-HT2A/2C agonists. ACS Chem Neurosci 2014; 5: 243-249.
| Crossref | Google Scholar |
99 Hansen M, Jacobsen SE, Plunkett S, Liebscher GE, McCorvy JD, Bräuner-Osborne H, Kristensen JL. Synthesis and pharmacological evaluation of N-benzyl substituted 4-bromo-2,5-dimethoxyphenethylamines as 5-HT2A/2C partial agonists. Biorg Med Chem 2015; 23: 3933-3937.
| Crossref | Google Scholar |
101 Poulie CBM, Jensen AA, Halberstadt AL, Kristensen JL. Dark classics in chemical neuroscience: NBOMes. ACS Chem Neurosci 2020; 11: 3860-3869.
| Crossref | Google Scholar |
102 Isberg V, Paine J, Leth-Petersen S, Kristensen JL, Gloriam DE. Structure-activity relationships of constrained phenylethylamine ligands for the serotonin 5-HT2 receptors. PLoS One 2013; 8: e78515.
| Crossref | Google Scholar |
103 Nichols DE. Hallucinogens. Pharmacol Ther 2004; 101: 131-181.
| Crossref | Google Scholar |
104 Nichols DE. Psychedelics. Pharmacol Rev 2016; 68: 264-355.
| Crossref | Google Scholar |
105 Glennon RA. The 2014 Philip S. Portoghese Medicinal Chemistry Lectureship: The “phenylalkylaminome” with a focus on selected drugs of abuse. J Med Chem 2017; 60: 2605-2628.
| Crossref | Google Scholar |
107 Chewy. ‘It Almost Killed Me: An Experience with 25I-NBOMe (exp95622)’, Erowid. 2012. Available at https://erowid.org/experiences/exp.php?ID=95622 [verified 25 February 2023].
108 Krasowski S. Poisons Standard February 2022. Australian Federal Department of Health and Aged Care; 2022. Available at https://www.legislation.gov.au/Series/F2022L00074 [verified 25 February 2023].
109 Benzenhöfer U, Passie T. Rediscovering MDMA (ecstasy): the role of the American chemist Alexander T. Shulgin. Addiction 2010; 105: 1355-1361.
| Crossref | Google Scholar |
112 Passie T. The early use of MDMA (‘Ecstasy’) in psychotherapy (1977–1985). Drug Sci Policy Law 2018; 4: 205032451876744.
| Crossref | Google Scholar |
113 Australian Institute of Health and Welfare. Illicit Drug Use. Canberra: AIHW; 2022. Available at https://www.aihw.gov.au/reports/illicit-use-of-drugs/illicit-drug-use [verified 19 February 2023].
114 Nichols DE. Differences between the mechanism of action of MDMA, MBDB, and the classic hallucinogens. Identification of a new therapeutic class: Entactogens. J Psychoactive Drugs 1986; 18: 305-313.
| Crossref | Google Scholar |
115 Winter JC. Effects of the phenethylamine derivatives, BL-3912, fenfluramine, and Sch-12679, in rats trained with LSD as a discriminative stimulus. Psychopharmacology 1980; 68: 159-162.
| Crossref | Google Scholar |
116 Freudenmann RW, Spitzer M. The neuropsychopharmacology and toxicology of 3,4-methylenedioxy-N-ethyl-amphetamine (MDEA). CNS Drug Rev 2004; 10: 89-116.
| Crossref | Google Scholar |
119 Johnston TH, Millar Z, Huot P, Wagg K, Thiele S, Salomonczyk D, Yong‐Kee CJ, Gandy MN, McIldowie M, Lewis KD, Gomez‐Ramirez J, Lee J, Fox SH, Martin‐Iverson M, Nash JE, Piggott MJ, Brotchie JM. A novel MDMA analogue, UWA-101, that lacks psychoactivity and cytotoxicity, enhances l-DOPA benefit in parkinsonian primates. FASEB J 2012; 26: 2154-2163.
| Crossref | Google Scholar |
120 Oeri HE. Beyond ecstasy: Alternative entactogens to 3,4-methylenedioxymethamphetamine with potential applications in psychotherapy. J Psychopharmacol 2021; 35: 512-536.
| Crossref | Google Scholar |
121 Marona-Lewicka D, Rhee G-S, Sprague JE, Nichols DE. Reinforcing effects of certain serotonin-releasing amphetamine derivatives. Pharmacol Biochem Behav 1996; 53: 99-105.
| Crossref | Google Scholar |
122 Nichols DE. Entactogens: How the name for a novel class of psychoactive agents originated. Front Psychiatry 2022; 13: 863088.
| Crossref | Google Scholar |
124 Parrott AC. Oxytocin, cortisol and 3,4-methylenedioxymethamphetamine: neurohormonal aspects of recreational ‘ecstasy’. Behav Pharmacol 2016; 27: 649-658.
| Crossref | Google Scholar |
125 Multidisciplinary Association for Psychedelic Studies. MDMA-Assisted Therapy for PTSD. 2023. Available at https://maps.org/mdma/ptsd/ [verified 25 February 2023].
126 Oehen P, Gasser P. Using a MDMA- and LSD-group therapy model in clinical practice in Switzerland and highlighting the treatment of trauma-related disorders. Front Psychiatry 2022; 13: 863552.
| Crossref | Google Scholar |
127 Therapeutic Goods Administration. Notice of final decisions to amend (or not amend) the current Poisons Standard in relation to psilocybine and MDMA. Australian Government Department of Health and Aged Care; 2023. Available at https://www.tga.gov.au/resources/publication/scheduling-decisions-final/notice-final-decision-amend-or-not-amend-current-poisons-standard-june-2022-acms-38-psilocybine-and-mdma [verified 25 February 2023].
128 Molliver ME, Berger UV, Mamounas LA, Molliver DC, O’Hearn E, Wilson MA. Neurotoxicity of MDMA and related compounds: Anatomic studies. Ann N Y Acad Sci 1990; 600: 640-661.
| Crossref | Google Scholar |
129 Hysek CM, Simmler LD, Ineichen M, Grouzmann E, Hoener MC, Brenneisen R, Huwyler J, Liechti ME. The norepinephrine transporter inhibitor reboxetine reduces stimulant effects of MDMA (“ecstasy”) in humans. Clin Pharmacol Ther 2011; 90: 246-255.
| Crossref | Google Scholar |
131 Bertron JL, Seto M, Lindsley CW. Dark classics in chemical neuroscience: Phencyclidine (PCP). ACS Chem Neurosci 2018; 9: 2459-2474.
| Crossref | Google Scholar |
132 Morris H, Wallach J. From PCP to MXE: A comprehensive review of the non-medical use of dissociative drugs. Drug Test Anal 2014; 6: 614-632.
| Crossref | Google Scholar |
133 Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999; 20: 201-225.
| Crossref | Google Scholar |
134 Roth BL, Gibbons S, Arunotayanun W, Huang X-P, Setola V, Treble R, Iversen L. The ketamine analogue methoxetamine and 3- and 4-methoxy analogues of phencyclidine are high affinity and selective ligands for the glutamate NMDA receptor. PLoS One 2013; 8: e59334.
| Crossref | Google Scholar |
135 Stevens CL, Klundt IL, Munk ME, Pillai MD. Amino ketone rearrangements. IV. Thermal rearrangements of α-amino methyl ketones. J Org Chem 1965; 30: 2967-2972.
| Crossref | Google Scholar |
136 Tyler MW, Yourish HB, Ionescu DF, Haggarty SJ. Classics in chemical neuroscience: Ketamine. ACS Chem Neurosci 2017; 8: 1122-1134.
| Crossref | Google Scholar |
137 Muetzelfeldt L, Kamboj SK, Rees H, Taylor J, Morgan CJA, Curran HV. Journey through the K-hole: Phenomenological aspects of ketamine use. Drug Alcohol Depend 2008; 95: 219-229.
| Crossref | Google Scholar |
138 Jansen K. Near death experience and the NMDA receptor. BMJ 1989; 298: 1708.
| Crossref | Google Scholar |
139 Hartvig P, Valtysson J, Lindner K-J, Kristensen J, Karlsten R, Gustafsson LL, Persson J, Svensson JO, Øye I, Antoni G, Westerberg G, Långström B. Central nervous system effects of subdissociative doses of (S)-ketamine are related to plasma and brain concentrations measured with positron emission tomography in healthy volunteers. Clin Pharmacol Ther 1995; 58: 165-173.
| Crossref | Google Scholar |
140 Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, Alkondon M, Yuan P, Pribut HJ, Singh NS, Dossou KSS, Fang Y, Huang XP, Mayo CL, Wainer IW, Albuquerque EX, Thompson SM, Thomas CJ, Zarate Jr CA, Gould TD. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016; 533: 481-486.
| Crossref | Google Scholar |
141 Rolland B, Jardri R, Amad A, Thomas P, Cottencin O, Bordet R. Pharmacology of hallucinations: Several mechanisms for one single symptom? BioMed Res Int 2014; 2014: 307106.
| Crossref | Google Scholar |
142 Dunk CW, Lebel T, Keane PJ. Characterisation of ectomycorrhizal formation by the exotic fungus Amanita muscaria with Nothofagus cunninghamii in Victoria, Australia. Mycorrhiza 2012; 22: 135-147.
| Crossref | Google Scholar |
143 Flament E, Guitton J, Gaulier J-M, Gaillard Y. Human poisoning from poisonous higher fungi: Focus on analytical toxicology and case reports in forensic toxicology. Pharmaceuticals 2020; 13: 454.
| Crossref | Google Scholar |
145 Tsai M‐J, Huang Y‐B, Wu P‐C. A novel clinical pattern of visual hallucination after zolpidem use. J Toxicol Clin Toxic 2003; 41: 869-872.
| Crossref | Google Scholar |
146 Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: A potent naturally occurring nonnitrogenous κ opioid selective agonist. Proc Natl Acad Sci 2002; 99: 11934-11939.
| Crossref | Google Scholar |
148 Timmermann C, Roseman L, Williams L, Erritzoe D, Martial C, Cassol H, Laureys S, Nutt D, Carhart-Harris R. DMT models the near-death experience. Front Psychol 2018; 9: 1424.
| Crossref | Google Scholar |
150 Frolov RV, Ignatova II, Singh S. Inhibition of hERG potassium channels by celecoxib and its mechanism. PLoS One 2011; 6: e26344.
| Crossref | Google Scholar |
151 Straub CJ, Rusali LE, Kremiller KM, Riley AP. What we have gained from ibogaine: α3β4 Nicotinic acetylcholine receptor inhibitors as treatments for substance use disorders. J Med Chem 2023; 66: 107-121.
| Crossref | Google Scholar |