

Coupling between dimethylsulfide emissions and the ocean–atmosphere exchange of ammonia
M. T. Johnson A B and T. G. Bell AA School of Environmental Sciences, University of East Anglia, Norwich, NR4 7TJ, UK.
B Corresponding author. Email: martin.johnson@uea.ac.uk
Environmental Chemistry 5(4) 259-267 https://doi.org/10.1071/EN08030
Submitted: 22 May 2008 Accepted: 15 July 2008 Published: 19 August 2008
Environmental context. Dimethylsulfide (DMS) is recognised as a potentially significant climate-forcing gas, owing to its role in particle and cloud formation in the marine atmosphere, where it is the dominant source of acidity. Ammonia, the dominant naturally occurring base in the atmosphere, plays an important role in neutralising particles formed from DMS oxidation products and may even enhance the formation rate of new particles. A biogeochemical coupling has previously been proposed between DMS and ammonia fluxes from the ocean to the atmosphere, in the form of coproduction of the two gases in seawater. We revise this suggestion by introducing the concept of ‘co-emission’ of the gases, where DMS emission controls the rate of emission of ammonia from the ocean by acidifying the atmosphere.
Abstract. A strong correlation between aerosol ammonium and non-sea salt sulfate is commonly observed in the remote marine boundary layer. It has been suggested that this relationship implies a biogeochemical linkage between the nitrogen (N) and sulfur (S) cycles at the cellular biochemical level in phytoplankton in the ocean, or a linkage in the atmosphere (see P. S. Liss and J. N. Galloway, Interactions of C, N, P and S biogeochemical cycles and global change (Springer, 1993), and P. K. Quinn et al. in J. Geophys. Res. – Atmos. 1990, 95). We argue that an oceanic linkage is unlikely and draw on mechanistic and observational evidence to make the argument that the atmospheric connection is based on simple physical chemistry. Drawing on an established analogous concept in terrestrial trace gas biogeochemistry, we propose that any emission of dimethylsulfide (DMS) from the ocean will indirectly influence the flux of NH3 from the ocean, through the neutralisation of acidic DMS oxidation products and consequent lowering of the partial pressure of NH3 in the atmosphere. We present a simple numerical model to investigate this hypothesised phenomenon, using a parameterisation of the rate and thermodynamics of gas-to-particle conversion of NHx and explicitly modelled ocean–atmosphere NH3 exchange. The model indicates that emission of acidic sulfur to the atmosphere (e.g. as a product of DMS oxidation) may enhance the marine emission of NH3. It also suggests that the ratio of ammonium to non-sea salt sulfate in the aerosol phase is strongly dependent on seawater pH, temperature and wind speed – factors that control the ocean–atmosphere ammonia flux. Therefore, it is not necessary to invoke a stoichiometric link between production rates of DMS and ammonia in the ocean to explain a given ammonium to non-sea salt sulfate ratio in the aerosol. We speculate that this mechanism, which can provide a continuous resupply of ammonia to the atmosphere, may be involved in a series of biogeochemical-climate feedbacks.
Introduction
Dimethylsulfide (DMS)
DMS is a biogenic trace gas whose flux is consistently from ocean to atmosphere. Its emission is of particular interest owing to the potential climate-forcing effect of its oxidation products, particularly H2SO4, the dominant source of strong acidity in the atmosphere and key species in new particle formation and growth. This is the basis of the ‘CLAW’ hypothesis,[ 1 ] which suggests a biological climate control (i.e. negative feedback) mechanism, although this is still open to debate (see ref. [2] and ‘opinion’ articles immediately following in the same issue). What is known with certainty is that there is a marine source of DMS[ 3 – 6 ] and that a substantial proportion of fine-mode aerosol in the marine boundary layer (MBL) is composed of non-sea salt sulfate (nss-SO4 2–), which in remote regions is derived from the oxidation of DMS and is the predominant source of aerosol acidity.[ 7 ]
The ocean–atmosphere flux of DMS is unidirectional because (i) DMS is a relatively insoluble gas (Henry’s law constant, KH ≈ 0.5 M atm–1 [ 8 ]), and (ii) it is relatively short-lived in the atmosphere (lifetime ≈ 1 day[ 9 ]) with relatively rapid removal via oxidation by various pathways.[ 10 , 11 ] These factors combine to maintain a strong concentration gradient (and thus flux) across the ocean–atmosphere interface.
Once in the atmosphere, DMS will oxidise on a timescale of hours by reaction with NO3 radical (at night) and BrO radical,[ 11 ] or days with oxidation by OH radical.[ 10 ] The end points of these oxidation reactions are MSA (methanesulfonic acid, HOSO2CH3) and sulfuric acid (H2SO4), the partitioning between these species being dependent on temperature and the relative concentrations of available oxidants.[ 11 ] These processes are effectively irreversible in Earth’s oxidising atmosphere.
A proportion of the H2SO4 will be taken up by coarse mode sea salt aerosol,[ 12 ] this proportion being strongly dependent on wind speed and the enrichment of CaCO3 alkalinity in the coarse mode aerosol.[ 13 ] The remaining H2SO4 can condense spontaneously to form sulfate particles (classically considered to occur via binary nucleation with water molecules) and will also condense onto or dissolve into existing fine mode particles in the atmosphere.[ 14 , 15 ] Whereas the propensity for H2SO4 to enter the particulate phase is governed by equilibrium thermodynamics (higher temperatures favouring the gas phase), it is generally thought that it will not re-enter the gas phase under normal atmospheric conditions owing to its extremely high solubility (in aqueous solutions) and ‘stickiness’ to dry surfaces.[ 16 ] Thus the gas-to-particle conversion of sulfuric acid can also be considered as an effectively irreversible process.
Ammonia (NH3)
NH3 and its protonated equivalent (ammonium, NH4 +), together referred to henceforth as NHx, are ubiquitous in the ocean and in the atmosphere. In the latter, NH3 is the dominant gaseous base and neutraliser of strong acidity. NH3 is emitted to the atmosphere in large quantities by anthropogenic activities, both industrial and agricultural,[ 17 , 18 ] as well as by natural biogenic production in the terrestrial and marine environments. Anthropogenic emissions have led to perturbations in the global NHx cycle many times greater than the pre-industrial fluxes.[ 19 , 20 ] However, NHx is relatively short-lived in the atmosphere (of the order of days), so the majority of NHx from strong terrestrial sources tends to be deposited from the atmosphere before reaching remote marine environments.[ 17 , 21 , 22 ]
In contrast to DMS, all of the processes linking dissolved ammonium in the marine surface layer (NH4 + (sw)) with particulate ammonium in the atmosphere (NH4 + (p)) are completely reversible. NH4 + (sw) exists in thermodynamic equilibrium with NH3(sw) (dissolved, non-solvated NH3), the protonation reaction occurring near-instantaneously to maintain equilibrium.[ 22 ] Under typical seawater conditions, ~1–10% of the total NHx(sw) exists as NH3(sw).[ 23 ]
Ammonia is considerably more soluble than DMS (KH ≈ 60 M atm–1 [ 8 ]). Its solubility means that for typical surface seawater and MBL concentrations, the ocean and atmosphere are generally close to equilibrium with respect to NH3 [ 21 , 24 – 27 ] and thus its ocean–atmosphere exchange can be considered a bidirectional process that is highly sensitive to temperature, pH and concentrations of NH3 in both surface ocean and MBL.[ 24 ]
Once in the atmosphere, NH3(g) will react readily with acidic gases and particles to enter the particulate phase as NH4 + (p). The direction of NH3(g) flux between the gas and aerosol phases is determined by the difference in concentration between NH3(g) and pNH3(g) (the partial pressure of NH3 over the aerosol phase). In aqueous aerosol, this process is reversible,[ 22 ] as it is with dry ammonium salts of nitrate and chloride.[ 21 , 28 ] The lower partial pressure of NH3 over the much less volatile ammonium sulfate ((NH4)2SO4) and ammonium bisulfate (NH4HSO4) means that any NH4 + (p) in dry salts formed with nss-SO4 2– are likely to be irreversibly reacted. However, these salts are highly deliquescent (i.e. water-attracting), so are unlikely to remain in the solid phase in the relatively humid MBL.[ 21 ]
Quinn et al.[ 22 ] considered all components of the ‘multiphase’ ammonia system (NH4 + (sw) ↔ NH3(sw) ↔ NH3(g) ↔ NH4 + (p)) in the remote marine environment. They found that the characteristic time for equilibration between the surface ocean and NH3(g) in the atmosphere is of the order of a few days to a week (dependent on wind speed), whereas the equilibration between gas and aerosol (or cloud) waters occurs on a timescale of hours. Oxidation of NH3(g) by OH radical is rather slow, and considered insignificant relative to other removal processes (wet and dry deposition, mostly from the particulate phase).[ 17 , 22 ] There is generally a disequilibrium observed between the atmosphere and ocean with respect to ammonia,[ 22 , 24 , 27 , 29 , 30 ] supporting the suggestion that air–sea equilibration is a relatively slow process in the system. Therefore, the major control on the concentration of NH3(g) in the remote MBL (neglecting advection from other sources) must be the equilibrium between gas and particle phases; i.e. on timescales of hours, NH3(g) will tend to reach equilibrium with pNH3(g).
Proposed biogeochemical couplings
Quinn et al.[ 29 ] observed average NH4 + : nss-SO4 2– molar ratios of 1.5 ± 0.44 in air of remote marine source (and substantially different and more variable ratios in air of terrestrial and volcanic sources). In addition, relatively consistent NH4 + : nss-SO4 2– molar ratios (typically between 1 and 2) have been observed in marine-source aerosols sampled in the remote MBL of the Atlantic (T. G. Bell, A. R. Baker and T. D. Jickells, unpubl. data). An exception to this trend in clean marine air is the data of Savoie et al.,[ 31 ] who observed substantially lower ratios (annual minimum of 0.6) at Mawson, Antarctica. In addition, their data suggest strong coupling of the seasonal trends in nss-SO4 2– (p) and NHx(p) concentrations over a 5-year period at several Antarctic and Southern Ocean sites.
Quinn et al.[ 29 ] propose that the relative constancy of the NH4 + : nss-SO4 2– ratio in clean marine air may indicate a biogeochemical linkage between the processes that produce DMS and NH4 + in the surface ocean, although they do not observe any relationship between the concentrations of these two species in seawater. Links at the biochemical level have been proposed, for example the possible coproduction of the osmolytes DMSP (dimethylsulfoniopropionate, the precursor to DMS) and glycine betaine (or 2-trimethylammonioacetate, the nitrogenous analogue to DMSP) (see ref. [32], after ref. [33]).
Liss and Galloway[ 32 ] propose that the linkage is more likely to be in the atmosphere and driven by the scavenging of NH3 by acidic sulfate particles, and that the resulting NH4 + : nss-SO4 2– ratio would be dependent on the relative amounts of NH3 and H2SO4 available from various sources (marine emissions, transport from continental regions, etc.). There is substantial evidence for an atmospheric coupling through the titration of aerosol acid sulfate with ammonia. A
Following the hypothesis of Quinn et al.,[ 29 ] Liss and Galloway[ 32 ] invoke ‘coproduction’; i.e. they assert that a given NH4 + : nss-SO4 2– ratio in the remote marine environment (away from continental sources) must represent the ratio of the production of ammonia and DMS in the surface ocean. We argue that this is unlikely, because the source regions of the NH4 + and nss-SO4 2– found in a given aerosol particle will be spatially separated owing to the timescale of DMS oxidation. Thus the likelihood of uniform coupling between the N and S cycles in the marine environment over such scales of space and time is unlikely. Second, only a tiny proportion of the NH4 + produced by biological activity in the ocean will be emitted to the atmosphere; relatively constant surface seawater concentrations are a result of the tight coupling between rapid uptake and regeneration by the mixed plankton community.[ 34 – 36 ] In other words, sea-to-air flux is not the dominant loss of NHx from seawater and the same appears to be true for DMS.[ 37 ] Finally, only a proportion of the DMS emitted from the ocean will be oxidised to nss-SO4 2–.[ 10 ]
However, in a system where all of the atmospheric N and S is derived from the surface ocean, it must be the case that a given NH4 + : nss-SO4 2– molar ratio in the aerosol will have resulted from emission of a stoichiometrically equivalent ratio of ammonia and DMS-derived sulfate. Therefore, there must be a process by which there is atmospheric control on the rate of emission of one or both of the gases in question and we invoke the concept of ‘co-emission’, a recognised process in the terrestrial environment, to explain this.
Coupling of DMS emission and NH3 flux
We hypothesise that away from significant terrestrial influence, any un-neutralised sulfate acidity in the atmosphere will scavenge NH3 towards a ‘titration end-point’. This depletion of gas-phase NH3 will favour the ocean-to-atmosphere flux of NH3, which, in the case of the remote marine atmosphere where nss-SO4 2– is predominantly derived from DMS emissions, can be considered a DMS-driven ‘co-emission’ of ammonia. B Ammonia, however, cannot have the same driving effect on DMS emission owing to the irreversible nature of the processes transforming DMS(sw) to nss-SO4 2– (p).
This hypothesised neutralisation mechanism for atmospheric acidity is consistent with the observed constancy in NH4 + : nss-SO4 2– ratios in the marine environment. Whenever atmospheric acidity increases, pNH3(g) over the aerosol decreases, leading to enhanced uptake of NH3(g) onto the aerosol and consequent increased flux of NH3 from the ocean to compensate the change.
As ocean–atmosphere exchange is the slowest process in the multiphase ammonia system in the marine environment, we predict that factors affecting the flux of ammonia from the ocean will be key controls on the NH4 + : nss-SO4 2– ratio. We have identified in Johnson et al.[ 24 ] that temperature is a fundamental constraint on the magnitude and direction of ocean–atmosphere ammonia exchange such that, at a global scale, it outweighs the effects of biological activity. We predict that temperature is therefore likely to be a key control on the NH4 + : nss-SO4 2– ratio. However, on a regional scale, where temperature is relatively invariable, biological activity and associated concentration and pH changes are likely to be important determinants of the NH4 + : nss-SO4 2– ratio.
Analogous processes in the terrestrial environment
An analogous process is well established in studies of terrestrial trace gas biogeochemistry – that of ‘co-deposition’ of NH3 and SO2 to wet and dry surfaces.[ 38 , 39 ] The concept was first suggested by Brimblecombe,[ 40 ] where NH3 was invoked as a neutralising agent facilitating the dissolution of SO2 onto the ‘dew’ on the surface of leaves. Subsequent laboratory studies observed enhanced efficiency of deposition of both NH3 and SO2 in the presence of both gases,[ 38 ] particularly under high relative humidities. The mechanism invoked to explain this co-deposition is the ‘suppression of the pH limitation to solubility for either gas in the presence of the other’.
In data presented by Sutton et al.,[ 38 ] we see a process more directly analogous to that which we propose for the marine environment (Fig. 1). The process, which they term ‘co-emission’, is driven by SO2 release due to drying on the leaf surfaces in a wheat field. Prior to drying, the atmosphere and wet leaf surfaces were close to equilibrium. Following drying and subsequent emission of SO2, NH3 was depleted by gas-to-particle conversion to below a ‘compensation point’ i.e. pNH3(g) over the substomatal cavities of leaf surfaces.[ 41 ] This led to an emission of NH3 from the surface as a direct result of the sulfur emissions. This is a process that occurs on a small timescale relative to our hypothesised process, but evidence for longer-term connections between atmospheric ammonium and sulfur in the terrestrial realm are demonstrated by Fowler et al.,[ 42 ] who find strong relationships between deposited ammonium and SO2 surface equilibrium concentrations over a 2-year period.
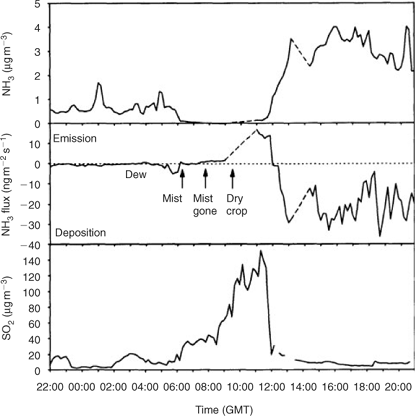
|
Applicability to less remote regions
We focus in the present manuscript on the remote marine environment, where the system of interest is relatively close to equilibrium and thus our hypothesised process should be most easy to identify. However, the implications of the proposed mechanism also apply to more complex (e.g. terrestrially influenced) systems. For instance, wherever there is a net flux of sulfuric acidity from the land surface over the ocean (the norm in many regions of the world[ 17 , 43 ]), gas-phase ammonia concentrations are likely to rapidly fall under control of the aerosol phase (i.e. tend towards pNH3(g)) as the air moves away from terrestrial source regions, owing to the relative slowness of air–sea exchange and oxidation by OH. Low aerosol pH will result in low pNH3(g) and thus enhance the flux of NH3 from the ocean, or inhibit influx, depending on ambient conditions and concentrations. Zhuang and Huebert[ 44 ] present data from a Lagrangian experiment, which show an increasing NH4 + : nss-SO4 2– ratio as sulfate-rich air is advected over the ocean, with evidence of rapid conversion of ocean-emitted NH3 to the particulate phase, providing strong circumstantial evidence of sulfate-driven ammonia emission in continental air masses.
Observed coupling in Antarctic data
The data of Savoie et al.[ 31 ] show strong evidence of a coupling between the emissions of NH3 and DMS, with strong synchronicity between nss-SO4 2– (p) and NH4 + (p) (Fig. 2a). This synchronicity cannot be explained by the seasonal cycles of NHx(sw) observed in the Southern Ocean, or the Antarctic coastal seas (Fig. 2c, d). The Joint Global Ocean Flux Study (JGOFS) Southern Ocean dataset (http://usjgofs.whoi.edu/jg/dir/jgofs/southern/, accessed April 2008) and the Rothera Antarctic Time Series data[ 45 ] show NHx(sw) at its most depleted during the months where the data of Savoie et al.[ 31 ] demonstrates the greatest aerosol ammonium (December, January). This is consistent with our current understanding of the seasonal cycle of NHx(sw) in temperate and high-latitude oceans where the nutrient-limited months of high summer tend to efficiently re-use remineralised N, keeping concentrations low.[ 21 , 34 ] The peaks in NH4 + (p) also occur out of phase with surface temperatures, which reach their annual maximum in March.
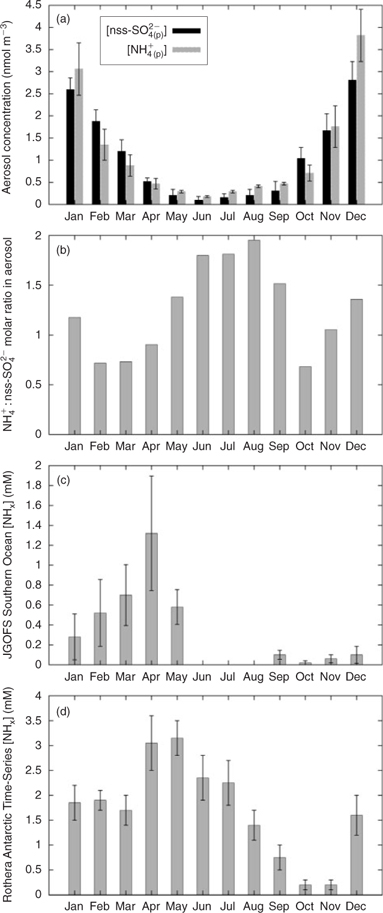
|
We therefore argue that the only likely explanation for the synchronicity between NH4 + (p) and nss-SO4 2– (p) in Antarctic air is our hypothesised process of coupled DMS and NH3 fluxes from the ocean. In doing so we make some assumptions: (i) windspeed and ocean pH are relatively small controls over the ocean–atmosphere NH3 flux over commonly observed ranges[ 21 , 22 ] and therefore the seasonal changes of these parameters in the Antarctic are unlikely to be responsible for the observed changes in NH4 + (p); and (ii) the data of Savoie et al.[ 31 ] are (as indicated by the authors) under strong influence of seawater emissions, i.e. not dominated by advection or local point sources such as penguin colonies, which are substantial sources of ammonia[ 46 ] and can affect local NH4 + : nss-SO4 2– ratios substantially.[ 47 ]
A simple model of the system
Below we present the details of a simple zero-dimensional finite difference box model of the ocean–atmosphere NHx–SO4 2– system, which is intended to test the hypotheses: (i) that stoichiometric production of DMS and NH4 + is not required in seawater to give a constant NH4 + : nss-SO4 2– ratio in aerosol; and (ii) that acidic DMS oxidation products added to the atmosphere can drive NH3 emission from the ocean by depletion of NH3(g). Only the nitrogen side of the system is explicitly modelled, i.e. we do not attempt to investigate the effects of the relative timescales of DMS and NH3 release and subsequent incorporation into aerosol, as this is not practicably possible in a model with no horizontal extent. In brief, the model is driven by input of SO4 2– directly into the aerosol phase, and the response of the NHx system and the NH4 + : nss-SO4 2– ratio are investigated.
Model description
Input parameters, variables and boundary conditions are listed in Table 1 and full details of the model can be found in the Accessory publication. Ocean–atmosphere exchange of NH3 is calculated using the thin film model (concentration difference) approach[ 48 ] using a scheme identical to that presented by Johnson et al.[ 24 ] Equilibration time across the air–sea interface according to this scheme is on the order of 100 h under typical conditions.[ 21 ]

|
This is much slower than the characteristic time of the other modelled process, the interconversion between NH3(g) and NH4 +, which occurs on a timescale of between 0.3 and 7 h.[ 22 ] We employ an equilibrium scheme where the rate of flux between gas and particle phases is proportional to the difference between the concentration of NH3(g) and pNH3(g) (Eqn A6, Accessory publication). We use our own parameterisation of pNH3(g), based on output of the Aerosol Inorganics Model (AIM, http://www.aim.env.uea.ac.uk/aim/aim.html, accessed September 2007)[ 49 ] (Eqn A7 and associated text, Accessory publication).
The NH4 + : nss-SO4 2– ratio in the model is found to be very insensitive to the rate of equilibration between gas and aerosol phases except at very low rates (equilibration times of tens to hundreds of hours), where it approaches the characteristic time of the ocean–atmosphere flux. We adopt a time constant for this process that results in 95% equilibration over ~1 h, in keeping with the observations and models of Harrison and Kitto[ 50 ] and Quinn et al.[ 22 ]
Initial conditions and default parameter values are selected to represent typical ‘average’ conditions in the global remote ocean and MBL, particularly the Southern Ocean. The seawater ammonium concentration is taken as a high-latitude average concentration of 150 nM after Johnson[ 21 ]; the sulfate input is considered as a per-unit area flux and is taken as the mean value of the Southern Ocean DMS flux observed by Shon et al.[ 51 ] (1.3 μmol m–2 day–1), multiplied by their mean observed SO2 conversion factor of 0.6, giving a sulfate input of 0.5 nmol m–2 min–1. The aerosol loss term is selected to maintain a constant sulfate loading, although the NH4 + : nss-SO4 2– ratio is entirely insensitive to this term as ammonium and sulfate are removed from the model atmosphere in the same ratio as they exist in the aerosol. The model is found to be very insensitive to initial values of NH3(g), NH4 + (p) and nss-SO4 2– (p) concentrations over a wide range of values, so arbitrary values of 1, 2 and 3 nmol m–3 respectively are adopted as defaults, based on previous observations.[ 19 , 22 , 28 ] Sensitivity of the model to other factors is considered below.
Model results
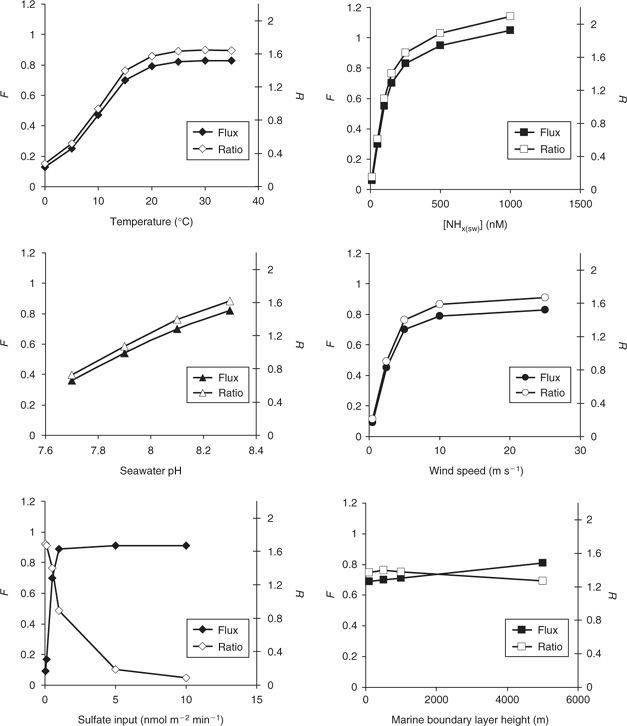
The model is run from starting conditions (concentrations of NH3(g), NH4 + (p), and SO4 2– (p)) until a dynamic steady-state (constant values of all fluxes and concentrations) is reached. Generally, such a state is reached within 48 h of model time, and in all cases within 7 days. The results of the model over a range of values of input variables and boundary conditions are presented in Fig. 3. Note that the NH4 + : nss-SO4 2– ratio rarely exceeds a value of just over 2. This is due to the rapid increase in pNH3(g) when the NH4 + : nss-SO4 2– ratio approaches or surpasses neutralisation, inhibiting further gas-to-particle flux.
|
Changes in [NHx(sw)], temperature (t), wind speed and pH all lead to a directly proportional response in NH4 + : nss-SO4 2– ratio and sea–air NH3 flux. This is because all of these parameters directly affect the rate of sea–air NH3 flux and thus the NH4 + : nss-SO4 2– ratio, in agreement with our hypothesised effect, sea–air flux being the rate-limiting step in the system.
The effect of sulfate input rate on the NH4 + : nss-SO4 2– ratio and sea–air NH3 flux is different. As would be expected, increasing the sulfate input to the system causes the NH4 + : nss-SO4 2– ratio to decrease, as the rate-limiting sea–air NH3 flux struggles to compensate. However, the sea–air NH3 flux increases (up to a maximum value), because the increase in aerosol acidity is leading to uptake of NH3(g) and consequent enhancement of sea–air NH3 flux, demonstrating our hypothesised DMS-driven co-emission.
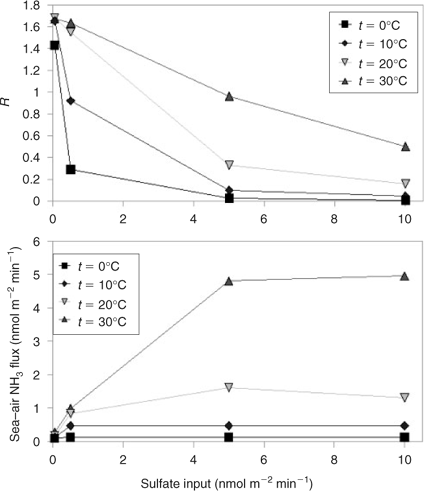
To further investigate this interaction between sulfate input and sea–air NH3 flux in the model, we compare the effect of sulfate input and temperature (as a proxy for all of the parameters that enhance sea–air ammonia flux) on the NH4 + : nss-SO4 2– ratio (Fig. 4). The maximum value of air–sea flux is reached in each case when the sulfate loading is sufficient to consume effectively all of the available NH3(g) at each timestep. This demonstrates that although DMS-driven co-emission of DMS and ammonia may occur over the range of conditions encountered in the remote marine environment, low sulfate emission rate and/or a combination of factors favouring enhanced sea–air NH3 flux are required to ensure substantial neutralisation of the aerosol through emission of ammonia.
|
Discussion
Our results indicate that in the remote marine environment, any DMS-derived sulfate can enhance the rate of sea–air NH3 flux, as hypothesised. Furthermore, it appears that the physical factors controlling ocean–atmosphere ammonia exchange are of fundamental importance to the NH4 + : nss-SO4 2– molar ratio in the marine atmosphere, i.e. biological production alone cannot account for high ratios in the marine environment. Although DMS emission is clearly a significant control on the NH4 + : nss-SO4 2– ratio in remote marine environments through increasing the concentration of nss-SO4 2–, this is somewhat moderated, particularly at higher temperatures and wind speeds, by the enhanced flux of NH3 from the ocean. Thus, physical and chemical forcings (temperature, wind speed and pH) have a greater than expected control on the NH4 + : nss-SO4 2– ratio and consequently any climate–biogeochemical feedbacks in the Earth-system involving the reduced N and S cycles. For instance, the continuous resupply of NH3 to the remore MBL through co-emission may be important in enhancing new particle formation through ternary nucleation[ 52 , 53 ] or other mechanisms.[ 15 , 54 ] This is not currently considered in models of particle formation over the ocean. C In this role, our proposed mechanism provides a possible positive reinforcement of the CLAW hypothesis. Thus long-term changes in ocean pH, temperature or wind fields may have significant effects on the radiative budget in Earth’s past or future climate.
These same factors (t, pH and wind) and consequent changes in atmospheric acidity through changing neutralisation by NH3 may have important effects on aerosol iron processing and consequent availability of Fe to plankton in iron-limited regions of the World’s oceans. Whether these potential feedbacks are positive or negative at a given point in Earth’s history (or future) in part depends on the relative changes in magnitude of t and pH with changing atmospheric CO2 (pCO2) as they tend to act against each other in their effect on NH3 flux (i.e. an increase in pCO2 will tend to decrease ocean pH, thus inhibiting marine NH3 emission, while increased temperature will favour NH3 emission).
Conclusions
We have proposed a mechanism by which observed NH4 + : nss-SO4 2– ratios can be explained without the need to invoke the stoichiometric coproduction of the precursor gases NH3 and DMS in seawater. Our simple model has demonstrated that in the remote MBL, where the predominant sources of NH4 + (p) and nss-SO4 2– (p) are marine and where the system is relatively close to equilibrium with respect to NHx, the hypothesised mechanism appears to exert a significant control on the NH4 + : nss-SO4 2– ratio. However, the model only solves for a dynamic steady-state and therefore accounts for neither fine-scale variation in marine emissions nor the advection of air masses enriched in either NHx or SO4 2– from the terrestrial realm. For large tracts of the contemporary ocean, these are likely to alter the signal that might be expected from our proposed process.
However, in pre-industrial times, when anthropogenic and natural terrestrial emissions were minor contributors to the reduced N and S global biogeochemical cycles, our hypothesised coupling mechanism is more likely to be applicable over most of the open ocean. Therefore, we argue that the suggestion to assume that marine NH3 emissions scale with DMS emissions globally[ 17 ] is probably reasonable for pre-industrial times but is not likely to be applicable in the modern era owing to large anthropogenic perturbations of the S and reduced N cycles.
To further test the real-world significance of co-emission of DMS and ammonia from the ocean, observational evidence must be combined with analysis using detailed box models of DMS oxidation and the next generation of ocean-coupled atmospheric chemical transport models, which will explicitly model ammonia and DMS fluxes and their interactions.

|
Acknowledgements
We are indebted to Peter Liss, Tim Jickells, Roland von Glasow, Tim Lenton and Simon Clegg for useful and insightful discussions, help and advice and to the three reviewers of this manuscript, who provided positive and extremely constructive criticism.
[1]
R. J. Charlson ,
J. E. Lovelock ,
M. O. Andreae ,
S. G. Warren ,
Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate.
Nature 1987
, 326, 655.
| Crossref | GoogleScholarGoogle Scholar |
[Verified 30 July 2008]
[9]
S. Kloster ,
J. Feichter ,
E. M. Reimer ,
K. D. Six ,
P. Stier ,
P. Wetzel ,
DMS cycle in the marine ocean–atmosphere system – a global model study.
Biogeosciences 2006
, 3, 29.

[10]
I. Barnes ,
J. Hjorth ,
N. Mihalopoulos ,
Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere.
Chem. Rev. 2006
, 106, 940.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[11]
R. von Glasow ,
P. J. Crutzen ,
Model study of multiphase DMS oxidation with a focus on halogens.
Atmos. Chem. Phys. 2004
, 4, 589.
[12]
Y. J. Yoon ,
P. Brimblecombe ,
Modelling the contribution of sea salt and dimethyl sulfide-derived aerosol to marine CCN.
Atmos. Chem. Phys. 2002
, 2, 17.
[13]
H. Sievering ,
J. M. Cainey ,
M. Harvey ,
J. McGregor ,
S. Nichol ,
P. K. Quinn ,
Aerosol non-sea-salt sulfate in the remote marine boundary layer under clear-sky and normal cloudiness conditions: ocean-derived biogenic alkalinity enhances sea-salt sulfate production by ozone oxidation.
J. Geophys. Res. 2004
, 109, D19317.
| Crossref | GoogleScholarGoogle Scholar |
[14]
N. S. Holmes ,
A review of particle formation events and growth in the atmosphere in the various environments and discussion of mechanistic implications.
Atmos. Environ. 2007
, 41, 2183.
| Crossref | GoogleScholarGoogle Scholar |
[15]
M. Kulmala ,
How particles nucleate and grow.
Science 2003
, 302, 1000.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[16]
[17]
F. J. Dentener ,
P. J. Crutzen ,
A three-dimensional model of the global ammonia cycle.
J. Atmos. Chem. 1994
, 19, 331.
| Crossref | GoogleScholarGoogle Scholar |
[18]
F. Dentener ,
J. Drevet ,
J. F. Lamarque ,
I. Bey ,
B. Eickhout ,
A. M. Fiore ,
D. Hauglustaine ,
L. W. Horowitz ,
M. Krol ,
U. C. Kulshrestha ,
M. Lawrence ,
C. Galy-Lacaux ,
S. Rast ,
D. Shindell ,
D. Stevenson ,
T. Van Noije ,
C. Atherton ,
N. Bell ,
D. Bergman ,
T. Butler ,
J. Cofala ,
B. Collins ,
R. Doherty ,
K. Ellingsen ,
J. Galloway ,
M. Gauss ,
V. Montanaro ,
J. F. Muller ,
G. Pitari ,
J. Rodriguez ,
M. Sanderson ,
F. Solmon ,
S. Strahan ,
M. Schultz ,
K. Sudo ,
S. Szopa ,
O. Wild ,
Nitrogen and sulfur deposition on regional and global scales: a multimodel evaluation.
Global Biogeochem. Cy. 2006
, 20, GB4003.
| Crossref | GoogleScholarGoogle Scholar |
[19]
N. Gruber ,
J. N. Galloway ,
An Earth-system perspective of the global nitrogen cycle.
Nature 2008
, 451, 293.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[20]
E. A. Holland ,
F. J. Dentener ,
B. H. Braswell ,
J. M. Sulzman ,
Contemporary and pre-industrial global reactive nitrogen budgets.
Biogeochemistry 1999
, 46, 7.
[21]
[22]
P. K. Quinn ,
W. E. Asher ,
R. J. Charlson ,
Equilibria of the marine multiphase ammonia system.
J. Atmos. Chem. 1992
, 14, 11.
| Crossref | GoogleScholarGoogle Scholar |
[23]
T. G. Bell ,
M. T. Johnson ,
T. D. Jickells ,
P. S. Liss ,
Ammonia/ammonium dissociation coefficient in seawater: a significant numerical correction.
Environ. Chem. 2007
, 4, 183.
| Crossref | GoogleScholarGoogle Scholar |
[24]
M. T. Johnson ,
P. S. Liss ,
T. G. Bell ,
T. J. Lesworth ,
A. R. Baker ,
A. J. Hind ,
T. D. Jickells ,
K. F. Biswas ,
E. M. S. Woodward ,
S. W. Gibb ,
Field observations of the ocean–atmosphere exchange of ammonia: fundamental importance of temperature as revealed by a comparison of high and low latitudes.
Global Biogeochem. Cy. 2008
, 22, GB1019.
| Crossref | GoogleScholarGoogle Scholar |
[25]
P. K. Quinn ,
R. J. Charlson ,
T. S. Bates ,
Simultaneous observations of ammonia in the atmosphere and ocean.
Nature 1988
, 335, 336.
| Crossref | GoogleScholarGoogle Scholar |
[26]
L. L. Sørensen ,
O. Hertel ,
C. A. Skjoth ,
M. Lund ,
B. Pedersen ,
Fluxes of ammonia in the coastal marine boundary layer.
Atmos. Environ. 2003
, 37, 167.
| Crossref | GoogleScholarGoogle Scholar |
[27]
S. W. Gibb ,
R. F. C. Mantoura ,
P. S. Liss ,
Ocean–atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea.
Global Biogeochem. Cy. 1999
, 13, 161.
| Crossref | GoogleScholarGoogle Scholar |
[28]
C. Perrino ,
M. Gherardi ,
Optimization of the coating layer for the measurement of ammonia by diffusion denuders.
Atmos. Environ. 1999
, 33, 4579.
| Crossref | GoogleScholarGoogle Scholar |
[29]
P. K. Quinn ,
T. S. Bates ,
J. E. Johnson ,
D. S. Covert ,
R. J. Charlson ,
Interactions between the sulfur and reduced nitrogen cycles over the central Pacific Ocean.
J. Geophys. Res. – Atmos. 1990
, 95, 16405.
| Crossref | GoogleScholarGoogle Scholar |
[30]
W. A. H. Asman ,
R. M. Harrison ,
C. J. Ottley ,
Estimation of the net air–sea flux of ammonia over the southern bight of the North Sea.
Atmos. Environ. 1994
, 28, 3647.
| Crossref | GoogleScholarGoogle Scholar |
[31]
D. I. Savoie ,
J. M. Prospero ,
R. J. Larsen ,
F. Huang ,
M. A. Izaguirre ,
T. Huang ,
T. H. Snowdon ,
L. Custals ,
C. G. Sanderson ,
Nitrogen and sulfur species in Antarctic aerosols at Mawson, Palmer Station, and Marsh (King George Island).
J. Atmos. Chem. 1993
, 17, 95.
| Crossref | GoogleScholarGoogle Scholar |
[32]
[33]
S. M. Turner ,
G. Malin ,
P. S. Liss ,
D. S. Harbour ,
P. M. Holligan ,
The seasonal variation of dimethyl sulfide and dimethylsulfoniopropionate concentrations in nearshore waters.
Limnol. Oceanogr. 1988
, 33, 364.
[34]
M. Johnson ,
R. Sanders ,
V. Avgoustidi ,
M. Lucas ,
L. Brown ,
D. Hansell ,
M. Moore ,
S. Gibb ,
P. Liss ,
T. Jickells ,
Ammonium accumulation during a silicate-limited diatom bloom indicates the potential for ammonia emission events.
Mar. Chem. 2007
, 106, 63.
| Crossref | GoogleScholarGoogle Scholar |
[35]
A. Bode ,
C. G. Castro ,
M. D. Doval ,
M. Varela ,
New and regenerated production and ammonium regeneration in the western Bransfield Strait region (Antarctica) during phytoplankton bloom conditions in summer.
Deep Sea Res. II 2002
, 49, 787.
| Crossref | GoogleScholarGoogle Scholar |
[36]
P. M. Glibert ,
J. J. McCarthy ,
Uptake and assimilation of ammonium and nitrate by phytoplankton: indices of nutritional status for natural assemblages.
J. Plankton Res. 1984
, 6, 677.
| Crossref | GoogleScholarGoogle Scholar |
[37]
R. Simo ,
Production of atmospheric sulfur by oceanic plankton: biogeochemical, ecological and evolutionary links.
Trends Ecol. Evol. 2001
, 16, 287.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[38]
M. A. Sutton ,
W. A. H. Asman ,
J. K. Schjorring ,
Dry deposition of reduced nitrogen.
Tellus B Chem. Phys. Meterol. 1994
, 46, 255.
| Crossref | GoogleScholarGoogle Scholar |
[39]
N. Van Breemen ,
P. A. Burrough ,
E. J. Velthorst ,
H. F. Vandobben ,
T. Dewit ,
T. B. Ridder ,
H. F. R. Reijnders ,
Soil acidification from atmospheric ammonium sulfate in forest canopy throughfall.
Nature 1982
, 299, 548.
| Crossref | GoogleScholarGoogle Scholar |
[40]
P. Brimblecombe ,
Dew as a sink for sulfur dioxide.
Tellus 1978
, 30, 151.
[41]
G. D. Farquhar ,
P. M. Firth ,
R. Wetselaar ,
B. Weir ,
On the gaseous exchange of ammonia between leaves and the environment: determination of the ammonia compensation point.
Plant Physiol. 1980
, 66, 710.
| PubMed |
[42]
D. Fowler ,
M. A. Sutton ,
C. Flechard ,
J. N. Cape ,
R. L. Storeton-West ,
M. Coyle ,
R. I. Smith ,
The control of SO2 dry deposition onto natural surfaces by NH3 and its effects on regional deposition.
Water Air Soil Pollut. Focus 2001
, 1, 39.
| Crossref | GoogleScholarGoogle Scholar |
[43]
S. C. Doney ,
N. Mahowald ,
I. Lima ,
R. A. Feely ,
F. T. Mackenzie ,
J. F. Lamarque ,
P. J. Rasch ,
Impact of anthropogenic atmospheric nitrogen and sulfur deposition on ocean acidification and the inorganic carbon system.
Proc. Natl. Acad. Sci. USA 2007
, 104, 14580.
| Crossref | GoogleScholarGoogle Scholar |
[44]
L. Zhuang ,
B. J. Huebert ,
Lagrangian analysis of the total ammonia budget during Atlantic Stratocumulus Transition Experiment/Marine Aerosol and Gas Exchange.
J. Geophys. Res. 1996
, 101, 4341.
| Crossref | GoogleScholarGoogle Scholar |
[45]
[46]
T. D. Blackall ,
L. J. Wilson ,
M. R. Theobald ,
C. Milford ,
E. Nemitz ,
J. Bull ,
P. J. Bacon ,
K. C. Hamer ,
S. Wanless ,
M. A. Sutton ,
Ammonia emissions from seabird colonies.
Geophys. Res. Lett. 2007
, 34, L10801.
| Crossref | GoogleScholarGoogle Scholar |
[47]
M. Legrand ,
F. Ducroz ,
D. Wagenbach ,
R. Mulvaney ,
J. Hall ,
Ammonium in coastal Antarctic aerosol and snow: role of polar ocean and penguin emissions.
J. Geophys. Res. – Atmos. 1998
, 103, 11043.
| Crossref | GoogleScholarGoogle Scholar |
[48]
P. S. Liss ,
P. G. Slater ,
Flux of gases across the air–sea interface.
Nature 1974
, 247, 181.
| Crossref | GoogleScholarGoogle Scholar |
[49]
S. L. Clegg ,
P. Brimblecombe ,
A. S. Wexler ,
Thermodynamic model of the system H+–NH4
+–SO4
2––NO3
––H2O at tropospheric temperatures.
J. Phys. Chem. A 1998
, 102, 2137.
| Crossref | GoogleScholarGoogle Scholar |
[50]
R. M. Harrison ,
A. M. N. Kitto ,
Estimation of the rate constant for the reaction of acid sulfate aerosol with NH3 gas from atmospheric measurements.
J. Atmos. Chem. 1992
, 15, 133.
| Crossref | GoogleScholarGoogle Scholar |
[51]
Z. H. Shon ,
D. Davis ,
G. Chen ,
G. Grodzinsky ,
A. Bandy ,
D. Thornton ,
S. Sandholm ,
J. Bradshaw ,
R. Stickel ,
W. Chameides ,
G. Kok ,
L. Russell ,
L. Mauldin ,
D. Tanner ,
F. Eisele ,
Evaluation of the DMS flux and its conversion to SO2 over the Southern Ocean.
Atmos. Environ. 2001
, 35, 159.
| Crossref | GoogleScholarGoogle Scholar |
[52]
D. J. Coffman ,
D. A. Hegg ,
A preliminary study of the effect of ammonia on particle nucleation in the marine boundary layer.
J. Geophys. Res. – Atmos. 1995
, 100, 7147.
| Crossref | GoogleScholarGoogle Scholar |
[53]
P. Korhonen ,
M. Kulmala ,
A. Laaksonen ,
Y. Viisanen ,
R. McGraw ,
J. H. Seinfeld ,
Ternary nucleation of H2SO4, NH3, and H2O in the atmosphere.
J. Geophys. Res. – Atmos. 1999
, 104, 26349.
| Crossref | GoogleScholarGoogle Scholar |
[54]
T. Anttila ,
H. Vehkamaki ,
I. Napari ,
M. Kulmala ,
Effect of ammonium bisulphate formation on atmospheric water–sulphuric acid–ammonia nucleation.
Boreal Environ. Res. 2005
, 10, 511.
[55]
W. D. Scott ,
F. C. R. Cattell ,
Vapor pressure of ammonium sulfates.
Atmos. Environ. 1979
, 13, 307.
| Crossref | GoogleScholarGoogle Scholar |
[56]
D. V. Spracklen ,
K. S. Carslaw ,
M. Kulmala ,
V.-M. Kerminen ,
G. W. Mann ,
S.-L. Sihto ,
The contribution of boundary layer nucleation events to total particle concentrations on regional and global scales.
Atmos. Chem. Phys. 2006
, 6, 5631.
A Harrison and Kitto[
50
] found kinetic control of aerosol sulfate neutralisation by NH3 during a ‘connected flow’ study over S.E. England. They observed that the pseudo-first order rate constant (with respect to NH3) for the reaction decreases with increasing neutralisation (Eqn 1).
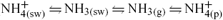 where K has units of s–1. Although these observations were made under a very different biogeochemical regime to that of the remote marine atmosphere, they strongly indicate a decrease in reaction rate towards aerosol neutralisation.
Quinn et al.[
22
] predict exponentially increasing pNH3(g) over aerosol tending towards neutralisation in their thermo-dynamic model of the atmospheric NHx system. The authors do not present the details of their model, but we have closely reproduced their findings using the Aerosol Inorganics Model (AIM) described in Clegg et al.[
49
] and in a related model, PITZ93, which is more reliable at near-neutral pH (S. Clegg, pers. comm.). Furthermore, the strong pH dependence of pNH3(g) over ammonium sulfate aerosols has been observed in laboratory studies.[
55
]
where K has units of s–1. Although these observations were made under a very different biogeochemical regime to that of the remote marine atmosphere, they strongly indicate a decrease in reaction rate towards aerosol neutralisation.
Quinn et al.[
22
] predict exponentially increasing pNH3(g) over aerosol tending towards neutralisation in their thermo-dynamic model of the atmospheric NHx system. The authors do not present the details of their model, but we have closely reproduced their findings using the Aerosol Inorganics Model (AIM) described in Clegg et al.[
49
] and in a related model, PITZ93, which is more reliable at near-neutral pH (S. Clegg, pers. comm.). Furthermore, the strong pH dependence of pNH3(g) over ammonium sulfate aerosols has been observed in laboratory studies.[
55
]
B In regions where seawater temperatures are low and ambient ammonia fluxes are likely to be from atmosphere to ocean (owing to advection from source regions), the coupling of the fluxes may in fact be via an inhibition of NH3 flux into the ocean, rather than enhanced emission of NH3 from the sea surface.
C Recent modelling studies have suggested that new particle formation is rare or non-existent in the MBL, owing to high temperatures inhibiting particle formation.[
56
] These modelling studies consider only binary homogeneous nucleation between sulfuric acid and water (probably not the only process in new particle formation in the marine atmosphere[
15
]) and thus may not be entirely correct. Either way, it has no bearing on our hypothesised process: the co-emitted DMS and ammonia are already spatially and temporally separated owing to the oxidation time for DMS and an extension to this separation while new particles sink back into the MBL is of little consequence at large scales of space and time.