

Reconciling measurement and modelling studies of the sources and fate of perfluorinated carboxylates
Ian T. Cousins A B , Deguo Kong A and Robin Vestergren AA Department of Applied Environmental Science (ITM), Stockholm University, SE-106 91 Stockholm, Sweden.
B Corresponding author. Email: ian.cousins@itm.su.se

Ian Cousins trained as an environmental chemist at Lancaster University (Ph.D.) and is now an Associate Professor at the Department of Applied Environmental Science (ITM) at Stockholm University in Sweden. His research program comprises a combination of experimental and modelling approaches to investigate the sources, transport, fate and exposure of organic pollutants, including perfluorinated alkyl substances (PFAS). The two coauthors of this critical review article are Ph.D. students in Ian's research group. |

Deguo Kong is a Ph.D. student at ITM. His main research interest is investigating the sources, environmental dynamics and fate of persistent organic pollutants (POPs) in the Arctic region. The tools used in his research are fugacity-based multimedia models which can be combined with field data to provide insights into the environmental distribution and levels of various POPs, including long-chain perfluorinated alkyl acids (PFAAs). |

Robin Vestergren is a Ph.D. student at ITM. His research focuses on elucidating the pathways of human exposure to long-chain perfluorinated alkyl acids (PFAAs) employing a combining measurement and modelling techniques. He is currently developing analytical methods to measure PFAAs at low concentrations in a variety of food matrices. |
Environmental Chemistry 8(4) 339-354 https://doi.org/10.1071/EN10144
Submitted: 31 December 2010 Accepted: 16 February 2011 Published: 19 August 2011
Journal Compilation © CSIRO Publishing 2011 Open Access CC BY-NC-ND
Environmental context. Five years ago a well-cited review of the sources and fate of perfluorinated carboxylates was published. The findings of that review are revisited here in light of recently published measurement and modelling studies of the sources and fate of these compounds, and an attempt is made to reconcile the many seemingly disparate findings reported. This review also aims to provide a road map for future research on the sources and fate of perfluorinated carboxylates and related compounds.
Abstract. This study critically evaluates the recently published measurement and modelling studies of the sources and fate of perfluorinated carboxylates (PFCAs). It is concluded that modelling studies provide support to the ‘direct hypothesis’ for PFOA and PFNA (i.e. the global dominance of direct sources (mainly from fluoropolymer manufacturing)). Empirical evidence for the importance of direct sources of PFOA and PFNA is provided by PFNA : PFOA ratios and isomer profiles of PFOA in ocean water. However, homologue patterns of long-chain PFCAs in biota from remote regions suggest that indirect sources (mainly from precursor degradation) are proportionally more important for PFCAs with more than 10 carbons. Temporal data in biotic and abiotic media are reviewed and an increasing trend to 2000 is observed for all PFCAs, with discrepancies in time trends reported after that period. Some studies on temporal patterns report a levelling off or decline in the latter part of the 2000s for PFOA and PFNA, whereas others show a continual increase throughout the study period. Differences in temporal patterns result from the fact that some environments respond faster to emission changes than others and may thus be useful to elucidate the importance of direct and indirect sources to different regions.
Introduction
The long-chain perfluorinated carboxylates (PFCAs) (CnF2n+1COOH, n > 7) are global contaminants of concern due to their widespread presence in wildlife[1] and humans,[2] persistence,[3] bioaccumulative potential[4] and toxicity.[5] In order to understand how wildlife and humans are exposed, it is important to quantify source emissions. Global historical emission estimates for PFCAs were first made 5 years ago by Prevedouros et al.[6] The study divides emissions of PFCAs into two source categories; releases from direct and indirect sources. Following the definition of Prevedouros et al.,[6] direct emission sources are those that come from the manufacture, intentional use and disposal of PFCAs throughout their product life-cycle. For example, emissions from the manufacture, use and disposal of PFCAs are direct sources. Indirect sources are emissions of a given PFCA that is present as an impurity in a product, or formed by degradation (in the environment, wildlife or humans) of a precursor substance (e.g. PFCA formed from the atmospheric degradation of perfluorooctane sulfonamido ethanol or from the biotransformation of a fluorotelomer alcohol). The estimates of Prevedouros et al.[6] suggested the predominance of direct emission sources for PFCAs,[6] particularly from manufacturing of fluoropolymers. Prior to this study, the degradation of volatile precursor compounds such as fluorotelomer alcohols had been postulated as the major source of PFCAs in the environment[7] and is still considered by many researchers to have a large contribution to global sources of PFCAs. The work of Prevedouros et al.[6] clearly showed that when considering the amount of chemical that is presently in the environment, one must consider the historical emissions (starting from c. 1950) and not just what is being released today. It was hypothesised that most of the PFCAs released historically reside in the world’s oceans where they will only be slowly ‘removed’ by vertical mixing into the deep oceans. It should be noted that the conclusions in Prevedouros et al.[6] were biased by the fact that total PFCA releases have been dominated by perfluorooctanoic acid (PFOA), which was used as production aid in the manufacture of polytetrafluoroethylene (PTFE). When considering emissions of individual homologues, the relative proportions of direct and indirect sources may differ from those of the total PFCA emissions.
Concerns about rising concentrations of PFOS (perfluorooctane sulfonate) and PFOA in human blood and consequent risks prompted one large US manufacturer (3M Co.) to announce the phase-out of production of perfluorooctane sulfonyl fluoride (POSF)-based products, including PFOA, in May 2000.[8] Since then PFOS has been listed in Annex B of the Stockholm Convention (i.e. its uses are restricted), although it is noted that there are numerous exceptions for the use of PFOS in continued industrial applications. Recently, many major PFCA, fluoropolymer and fluorotelomer manufacturers committed to reduce industrial emissions and presence in sales products of PFOA, potential precursors and higher homologue chemicals by 95% by the year-end 2010 as well as to work towards elimination of these chemicals by 2015.[9] There has been a trend in the fluorochemical industry to replace longer chain homologues with compounds containing shorter perfluorinated chains (e.g. the 3M Co. replaced PFOS with perfluorobutane sulfonate (PFBS)). The reasoning is that although these shorter chain compounds are equally persistent, they are much less bioaccumulative.[4,10]
This study aims to re-evaluate the findings of the previous critical review of Prevedouros et al.[6] in light of recently published measurement and modelling studies of the sources and fate of PFCAs. To this end we carefully reconcile recently published modelling and measurement studies with the hypotheses previously presented in Prevedouros et al.[6] This is done in several ways, namely: (a) by considering whether there is a mass balance between the amounts of PFCAs and their precursors estimated to have been released globally and the inventories in the environment; (b) by determining if the spatial distribution of PFCAs in the environment is consistent with manufacturing sites being the major sources of PFCAs; (c) by comparing homologue patterns of PFCAs observed in the environment, wildlife and humans with amounts of homologues estimated to be released; (d) by comparing isomer patterns in production sources with isomer patterns observed in the environment; and (e) by determining if time trends in the environment, wildlife and humans are consistent with time trends that we would expect from our understanding of sources, transport and fate. Throughout the review, we highlight areas where additional research is needed.
Abbreviations are often used for the perfluoroalkyl substances discussed in this review. A list of abbreviations is included in Table A1 of the Accessory publication (see http://www.publish.csiro.au/?act=view_file&file_id=EN10144_AC.pdf).
Review of emission estimates for PFCAs
Total global historic PFCA emissions estimates were reported (1951–2004) in Prevedouros et al.[6] and in Armitage et al.[11] projected PFOA emission estimates were made (2005–2050) based on reduction commitments made by industry. We will not repeat details of these calculations here and instead refer readers to these earlier publications. The global historical (1951–2004) industry-wide emissions of total PFCAs from 14 known sources comprising direct (manufacture, use, consumer products) and indirect (either PFCA impurities or precursors) sources were estimated to be 3200–7300 t.
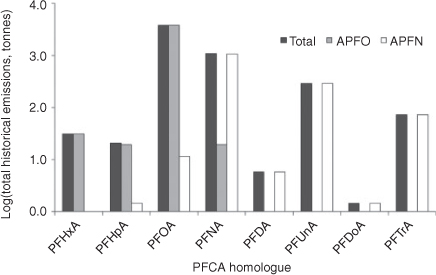
Prevedouros et al.[6] estimated that direct sources account for ~97% of total historical emissions of PFCAs and of these manufacture of ammonium perfluorooctanoate (APFO) and ammonium perfluorononanoate (APFN) and their use in fluoropolymer (FP) manufacturing accounts for ~93% of total historical emissions. Armitage et al.[12] provided an estimate of the historical PFCA homologue composition for manufacture and use of APFO and APFN in FP manufacturing. In Fig. 1 we provide the estimated historical emissions (1951–2010) of PFCA homologues from APFO manufacture and use, APFN manufacture and use, and from APFO plus APFN manufacture and use. The original data for generating these figures were taken from Armitage et al.[12] and we provide the full dataset in the Accessory publication. The estimated homologue profiles for APFO and APFN used to estimate the emissions are based on analysis of PFCA homologues in the commercial products (Fluorad FC-143 and Surflon S-111) used in FP manufacturing.[6] We use the information contained in Fig. 1 later in the paper to determine if homologue patterns in sources match up to those observed in the abiotic environment and in biota and humans after correction for bioaccumulation potential. If direct sources are as dominant as estimated by Prevedouros et al.,[6] then the homologue patterns of APFO and APFN or the mixed direct source profile should be reflected in the environmental samples.
|
Prevedouros et al.[6] assumed that the majority of PFCA sources, manufacture and use emissions occurred and will continue to occur in the northern hemisphere. Will et al.[13] provided a comprehensive analysis of the location and production capacity of 33 FP manufacturing facilities throughout the world for the year 2005. All of the plants listed were in the northern hemisphere; 10 were located in the US, seven in China, six in Japan and most of the rest were in Western European countries. The overall continental distribution of fluoropolymer manufacturing capacity was respectively 44, 27 and 29% for North America, Europe and Russia and Asia. It is noteworthy that the proportion of manufacturing in Asia had increased compared with a previous marketing report by Ring et al.[14] Although no known manufacture of PFCAs occurred in the southern hemisphere, industrial and consumer use of products from both direct and indirect PFCA sources has occurred and continues in South America and Australia[15,16] and presumably in other locations. We later determine if this spatial distribution of major emission sources is consistent with observations.
Mass balance modelling
In the same paper that the source estimates of PFCAs were published,[6] some simple calculations were presented to determine if global emissions balanced with global inventories of PFOA in environmental media. It was determined that the oceans contained most of historically released PFOA and that emissions balanced reasonably well with estimated inventories in the oceans. The publication of the source emissions for PFCAs allowed modellers to undertake more sophisticated fate and mass balance calculations using computer models. Since the emission data were published, several modelling papers have followed. These modelling studies either simulate the fate of the PFCAs from direct release only[11,12,17,18] or simultaneously model the fate of precursor chemicals and PFCAs (from either direct release or precursor degradation).[19–22] Below we review these modelling studies and explain what each has contributed.
The first published modelling study reported a global-scale model simulation of the fate and transport of PFOA emitted from direct sources conducted using a latitudinally resolved model (GloboPOP).[11] The justification of modelling only direct releases was that they were considered to be by far the most important source category and could account for the majority of PFCAs present in the environment according to Prevedouros et al.[6] PFOA emission estimates between 1950 and 2004 were taken from Prevedouros et al.[6] and in addition, projected PFOA emission estimates were made (2004–2050) based on reduction commitments made by industry. It was assumed that the fate and transport of this substance could be represented by the anion (PFO) because the majority of the compound (>99%) was expected to be in the ionised form in the environmental media included in the model (e.g. fresh and ocean water, soils, sediments). The main purpose of the initial study was to evaluate emission estimates and model performance through comparisons between modelled and observed concentrations in surface ocean waters, the compartment representing the most significant global reservoir in terms of contaminant mass. There was reasonable agreement between modelled and observed concentrations in the oceans, which supported the plausibility of the historical emission estimates, the dominance of direct emissions and importance of the oceans as the principal recipient storage medium as well as vector to the Arctic. However, several limitations were noted in this initial study, namely precursor compounds were not included in the simulations, substantial overestimation of observed concentrations in the central Pacific Ocean suggested the potential value of adopting a more spatially resolved model, and only the anion (PFO) was explicitly modelled, whereas in reality, both the neutral and anionic forms will be present in the environment. The first limitation was addressed by Wallington et al.,[19] Wania,[20] Schenker et al.[21] and Yarwood et al.,[22] who all included precursor compounds in model simulations. The studies of Wallington et al.[19] and Yarwood et al.[22] used spatially and temporally resolved atmospheric models to simulate the transport and degradation pathways of FTOHs in the atmosphere alone. Wallington et al.[19] used the IMPACT model including a detailed treatment of FTOH atmospheric degradation mechanism to estimate degradation of FTOHs via reaction intermediates to form PFCAs, and modelled atmospheric transport of PFCAs and FTOHs. Wallington et al.[19] used a simple estimate of global emissions of FTOHs (1000 t year–1) and made no attempt to compare their predicted concentrations with atmospheric monitoring data. They report a calculated 8 : 2 FTOH air concentration value of ‘0.5–5 × 105 molecules cm–3 in remote ocean and Arctic locations in the northern hemisphere’, which converts to 38–385 pg m–3. This is clearly much higher than the measured air concentrations in this area (5.8–26 pg m–3) reported by Shoeib et al.[23] and suggests that the emission estimate in the model was at least a factor of five too high. Yarwood et al.[22] used a highly spatially and temporally resolved model of the North American continent incorporating a detailed treatment of FTOH atmospheric degradation mechanisms to simulate FTOH degradation via intermediates to PFCAs and model atmospheric transport of PFCAs and FTOHs. The higher spatial and temporal resolution provided an enhanced ability to distinguish spatial regions with high and low concentrations of atmospheric nitrogen oxides (NOx). This is important as PFCA formation via the gas-phase oxidation of FTOH is known to be sensitive to local NOx concentrations.[7] A bottom-up emission inventory for PFCAs and FTOHs was developed from sales and product composition data. Predicted FTOH air concentrations were in good agreement with available monitoring data, providing confidence in North American emission estimates, which were also consistent with emissions of FTOHs included in Prevedouros et al.[6]
In order to incorporate degradation of precursors to PFCAs in multimedia models such as GloboPOP and CliMoChem, a simplified treatment of atmospheric degradation and transport was used. Degradation was calculated using a first-order degradation rate of the parent precursor species and overall degradation yield for transformation from the parent precursor to end product (i.e. PFCA). Wania[20] redid the multimedia modelling simulations of Armitage et al.[11] using GloboPOP, but also included emission, transport and degradation of FTOHs to PFCAs in the mass balance. Model estimated FTOH concentrations agreed well with measurements and model estimated atmospheric deposition fluxes of PFO to the Arctic Ocean were orders of magnitude lower than those entering via ocean transport. Wania[20] also showed that PFCAs have a relatively high Arctic contamination potential (as measured by ACP10), ~10 times higher than the ACP10 related to transport and degradation of FTOHs. In another global modelling study using CliMoChem, Schenker et al.[21] simulated and compared the importance of two classes of precursors (perfluorooctyl sulfonamidoethanols (FOSEs) and FTOHs) for PFO deposition to the Arctic. The model predicted that modelled deposition fluxes of PFO to the Arctic originating from FOSEs and FTOHs were of a similar magnitude prior to 2002 before the major manufacturer phased out POSF-based production. Modelled air concentrations of FOSEs were shown to decrease rapidly after phase-out, whereas modelled FTOH concentrations did not decline between 2001 and 2006. Interestingly, the modelled air concentrations for FOSEs did not agree with field measurements, indicating that the sources of FOSEs to the atmosphere are not entirely understood. This finding will be further discussed in the section on temporal patterns.
Further limitations of the initial modelling study of Armitage et al.[11] were addressed in a later study[12] that investigated the global-scale fate and transport of PFOA by conducting a series of model simulations using a multispecies global-scale model (a modified version of BETR-Global) with both latitudinal and longitudinal resolution. Atmospheric transport has previously been discounted as a likely LRT mechanism for long-chain PFCAs, in part because they were assumed to exist almost exclusively as anions in the environment based on measurements and calculations of pKa, primarily performed for PFOA, ranging from 0 to 2.8.[24–28] However, the pKa of PFOA was more recently estimated to be ~3.8 at infinite dilution using potentiometric titration.[29] Although the pKa of PFOA at environmentally relevant concentrations remains controversial,[30–33] it is now recognised that the role of the neutral acid cannot be completely neglected in environmental fate models. As the neutral acid has an appreciable vapour pressure, it is subject to surface-air exchange processes and will also be present in the gas-phase of the atmosphere. It is known that PFOA is emitted directly to the atmosphere at fluoropolymer manufacturing sites.[34] The model simulations in Armitage et al.[12] suggested that the atmospheric transport of directly emitted PFOA can deliver this substance via atmospheric deposition to terrestrial environments remote from sources. We revisit this hypothesis in the next section when we consider the spatial distribution of PFCAs in the environment.
Consistent with previous studies, Armitage et al.[12] found that ocean transport was the dominant pathway for delivering PFOA associated with direct sources to the Arctic marine environment, regardless of model assumptions. Although the spatial resolution and description of ocean transport were greatly improved in the modified BETR-Global model relative to earlier studies, it was still highly simplified relative to more detailed ocean transport models. It was therefore encouraging that model simulations undertaken with a highly spatially resolved ocean transport model,[18] using similar physical-chemical input values as in the BETR-Global study, provided estimated concentrations and fluxes to the Arctic Ocean that were in very good agreement with the earlier modelling results.
The multispecies modelling results for PFOA from Armitage et al.[12] raised the possibility that direct sources of other PFCAs contribute to the contamination of both terrestrial and marine environments through atmospheric LRT of neutral species. A follow up study was therefore undertaken[17] to model the multispecies fate of PFOA to PFTrA emitted from direct sources. Emission estimates for PFCA homologues were extracted from information provided in Prevedouros et al.[6] Model simulations for PFOA, PFNA, and PFUnA indicated that mass fluxes to the Arctic marine environment associated with oceanic transport were in excess of mass fluxes from atmospheric transport of precursor substances and subsequent degradation to PFCAs. Although modelled concentrations of PFOA and PFNA were consistent with what one observes in surface ocean waters and in biota, it was not possible to conduct mass balances for other compounds owing to lack of ocean water data for homologues greater than PFNA. It was also not possible to reconcile modelled concentration ratios of PFDA to PFUnA with wildlife biomonitoring data. We revisit these findings in more detail in a later section of this paper, which discusses homologue patterns in monitoring data.
Spatial distribution of PFCAs in the environment
Two main hypotheses have been put forward regarding the dominant sources of PFCAs to the global environment. One hypothesis, the ‘indirect hypothesis’, is that PFCAs in the environmental largely originate from the abiotic or biotic transformation of precursor compounds. In contrast, the ‘direct hypothesis’ postulates that the presence of PFCAs in the environment is largely due to direct emissions (those resulting from manufacture and use of PFCAs).[6] Supporters of the direct hypothesis have noted that even directly released PFCAs can be transported via the atmosphere or via oceanic currents.[6,11,12,17] Although proving to be useful concepts for driving the research of PFCAs in the environment, the division into two diametrically divergent hypotheses is overly simplistic. More realistically the relative importance of each source type will depend on the environment and PFCA homologue being studied. Here we consider in turn the relative importance of direct and indirect sources to: (a) different hemispheres; (b) the oceans; (c) rivers; (d) inland areas; and (e) the atmosphere.
All known manufacturing of PFCAs occurs in the northern hemisphere. However, products containing PFCAs have been used in the southern hemisphere so we can expect PFCAs to be present in the environment, although at lower concentrations compared with the northern hemisphere. If direct sources are the dominant source category we should expect a large gradient in observed concentrations in ocean water on either side of the equator. Indeed, measurements of PFCAs in the oceans in the northern and southern hemispheres[35,36] have revealed that PFCAs can only be detected in oceans north of the equator. However, due to the lower population of the southern hemisphere, concentrations of volatile precursors in the remote environments originating from consumer products should also be lower in the southern hemisphere. Again our understanding is consistent with measurements. Air concentrations of volatile precursors (fluorotelomer alcohols (FTOHs) as well as N-alkylated fluorooctane sulfonamides and sulfonamidoethanols) are on average a factor of ten higher in the remote atmosphere of the northern hemisphere (maximum concentration 8 : 2-FTOH, 190 pg m–3) compared with the southern hemisphere (maximum concentration 8 : 2 FTOH, 14 pg m–3).[37] Despite the lower environmental concentrations present in the southern hemisphere, PFCAs have been detected in the serum of Australians at similar levels to humans in industrialised countries in the northern hemisphere, indicating that either localised environmental or consumer exposure from product use are similar in industrial societies in both hemispheres.[15,16]
Within the northern hemisphere, as direct emissions to surface waters have occurred for many decades, it can be expected that a large proportion of water soluble PFCAs have been transported to the oceans. Modelling studies[11] support this hypothesis and suggest that the majority of global historical emissions of PFCAs, which are dominated by direct sources, reside in the oceans. Armitage et al.[12] undertook a spatially resolved global modelling exercise and showed that ocean coastal regions in proximity to point sources on US East Coast, North Sea and seas surrounding Japan have higher estimated ocean water concentrations of PFOA and PFNA than open sea areas. This simulated pattern of ocean contamination is confirmed by ocean monitoring data.[38,39] Ocean transport is a slow process with mixing occurring over decades. Modelling studies predict that if emissions of PFCAs are effectively eliminated the concentrations in different regions will not converge until the end of the 21st century.[11]
McLachlan et al.[40] measured PFOA in major European rivers and determined that most rivers were not affected by direct manufacturing sources. In the one case where a fluoropolymer manufacturing plant was located on the River Po, it was clear that this was the major source of PFOA. Indeed McLachlan et al.[40] estimated that the River Po discharge accounted for two-thirds of the riverine outflow of PFOA in Europe. Rivers to which there are no known direct manufacturing discharges have been shown to contain PFCAs in the range 0.26 to 60.0 ng L–1 (sum PFCAs).[40] Although the study by McLachlan et al.[40] provides support for the direct hypothesis, it also illustrates that there are widely distributed non-point sources of PFOA that contribute to the contamination of European rivers. PFCAs can enter rivers from a combination of wastewater input from municipal and industrial sources as well as urban runoff from precipitation.[41–44] A recent Japanese study[45] concluded that loadings of PFOA from runoff to rivers were greater than or equal to the loadings from wastewater treatment plants. PFOA present in street dust from urban sources may contribute an additional loading to runoff. Murakami et al.[43] showed that PFOA concentrations were significantly higher in surface runoff than in rainfall, suggesting that PFOA in urban runoff is partially derived from dust deposited on impervious surfaces. Clara et al.[46] analysed PFCAs in wastewaters from municipal and industrial facilities. PFCAs were found in nearly equal concentrations in municipal and industrial wastewaters (14–459 and 3–664 ng L–1 respectively (∑(PFHxA to PFDoA)). The origin of PFCAs in municipal wastewaters is likely from residual PFOA in fluorinated polymeric products and degradation of residual precursors still used in household products such as stain repellents and waterproof and greaseproof paper coatings, and in the cases where it is not separated, from urban runoff. Maybe surprisingly, effluents from textile and paper industries did not result in significantly elevated concentrations of PFCAs compared with municipal wastewater.[46] Even rivers and lakes that do not have wastewater inputs have been found to be contaminated with PFCAs from precipitation and runoff.[47–50] For example, concentrations of the sum of PFCAs have been reported in precipitation in Northern Germany at 0.7 to 41.9 ng L–1 by Dreyer et al.,[49] and Kwok et al.[47] reported that the average total PFCAs concentrations ranged from 1.4 to 15.2 ng L–1.
Remote inland environments (including inland lakes[51,52] and the High Arctic[53]) only receive inputs from the atmosphere. Above we mentioned that PFCAs are present in precipitation and thus these inland areas are not free from contamination. However, the concentrations are an order of magnitude lower than in areas with wastewater input.[40] The ultimate source of PFCAs in the atmosphere is uncertain. There still remains the possibility that atmospheric PFCAs can be derived by either direct release from manufacturing facilities and subsequent atmospheric transport or from precursor release, transport and degradation. Recently, marine aerosol transport has also been proposed,[54,55] although this has been estimated to be of minor importance on a global scale.[12] Armitage et al.[12,17] have shown that atmospheric transport of PFCAs released from manufacturing sources could be a previously underestimated transport pathway. The relative importance of either indirect or direct atmospheric transport pathways for PFCAs is difficult to estimate using models because of controversy surrounding the concentration dependence of the acid dissociation constant (pKa).[30] A growing body of theoretical[30–32] and experimental[33,56] evidence contradicts the proposed monomeric pKa of 3.8 for PFOA.[29] It has been shown[32] that long-chain PFCAs form premicellular aggregates (or dimers) at relatively low concentrations, which may cause an increase of their pKa to somewhere between 2 and 3, in agreement with historical measurements.[24–28] A recent study by Kaiser et al.[57] demonstrated a pH dependent water-air transfer of PFOA from acidic process sumps consistent with an aggregated pKa of 2.8. Thus, due to aggregation or dimerisation of long-chain PFCAs increasing the pKa, there may be some direct atmospheric transport of PFCAs and non-negligible amounts of neutral PFOA may partition to the gas phase. To date PFCAs have not been measured in the gas phase in ambient air. Although they may be present there, sampling gas phase PFCAs using traditional high volume sampling methods is not possible because PFCAs sorb strongly to the glass fibre filters.[58] Innovative measurement techniques for elucidating gas–particle partitioning of PFCAs are needed.
It is interesting to note that levels of volatile precursors of PFCAs[7,19,59] and levels of PFCAs in precipitation[47–49] are observed to be highest near to urban areas, suggesting that urban areas are sources of PFCAs to the atmosphere. Previous models that included precursors[21] were able to predict levels of selected precursors in the background atmosphere but underpredicted levels of PFOA observed in atmospheric deposition by an order of magnitude.[47,48] If there is direct atmospheric release and transport of PFCAs from fluoropolymer manufacturing plants, higher levels of PFCAs would be expected in precipitation near to the plants. There are studies showing that fluoropolymer plants are a source of PFCAs to the atmosphere[34]; however, monitoring around these plants has been limited and the importance of this atmospheric source has not been properly elucidated. It is thus currently not possible to determine if model underestimation of precipitation levels in Schenker et al.[21] are due to (a) not considering direct atmospheric sources of PFCAs or (b) not including all relevant precursor compounds contributing to PFCA atmospheric levels. Thus, the sources of PFCAs to either air or precipitation is an area where more research is needed.
Homologue patterns in abiotic and biotic environmental samples
In the following sections, PFCA homologue patterns observed in surface waters (rivers, lakes and marine waters), groundwater, drinking water, wastewater, sediments, precipitation and biota are reviewed and compared with the patterns in our source estimates. Tables and figures summarising these data are included in the Accessory publication (see Tables A3–A5 therein). It is important when doing any comparison that the relative importance of point-source inputs and diffuse inputs at the sampling location is considered. It is probable that homologue patterns in environments affected by point sources (e.g. spill of aqueous film forming foam (AFFF), manufacturing emission, etc.) will be quite different to environments affected by diffuse sources only (e.g. atmospheric deposition resulting from long-range atmospheric deposition). Ocean waters are considered to be the best environmental medium for comparing homologue source profiles of total global emissions, because the oceans are the ultimate reservoir for the majority of PFCAs historically released.
Natural waters
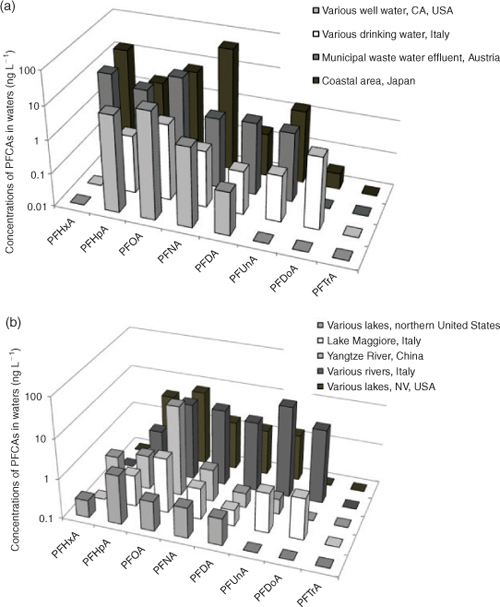
Surface water concentrations of PFCAs have been reported for PFCA homologues in rivers, lakes, drinking waters, marine waters, wastewaters and ground waters.[39] Concentrations reported are typically for PFHxA, PFHpA, PFOA and PFNA, although a few studies have reported concentrations for longer chain length homologues (Fig. 2). Ocean water data display a distinct PFOA > PFNA pattern in the Atlantic Ocean as well as the Western Pacific, with PFOA levels typically being approximately a factor of five higher than PFNA levels.[36,38,60,61] Few data points are available for other PFCA homologues in the open ocean as the concentrations are generally non-detectable with current analytical techniques. Some data have recently become available for PFDA, PFUnA,[35] but the number of samples reported are currently too few to make meaningful comparisons with source homologue profiles. It is hoped that in the near future a larger quantity of reliable ocean water data will become available for the long-chain homologues, which will provide an ideal dataset for testing the reliability of PFCA emission estimates. Coastal waters of Europe, North America and China display higher concentrations compared with the open ocean although with a similar PFOA : PFNA ratio indicating that the homologue pattern of continental water discharge is conserved in the open ocean. Interestingly, water samples from Japanese coastal areas and Tokyo bay display a somewhat different homologue pattern (PFNA > PFOA and PFUnA > PFDA) indicating a regional variability in sources.[62,63] The observed homologue pattern in Japanese coastal waters may reflect the presence local APFN sources in line with the predicted emission estimates of Prevedouros et al.[6] In contrast to the estimated source emission profile, PFHxA and PFHpA have been reported in coastal waters[64] and river water[40] at concentrations comparable to those of PFOA within a factor of 2–5. Sources of PFHxA and PFHpA cannot be quantitatively explained by the current emission source understanding, but may include: (i) impurities from direct releases of PFCAs from fluoropolymer manufacturing and and post-manufacture processing (Fig. 1); (ii) PFCA impurities in perfluorohexane sulfonate or 6 : 2 fluorotelomer sulfonate both used in AFFF; or (iii) atmospheric or microbial degradation of precursors containing a six carbon perfluoroalkyl moiety (e.g. 6 : 2-FTOH). Interestingly, Ahrens et al.[35] observed a strong (r = 0.989) statistically significant (P < 0.01) correlation between concentrations PFHxA and PFOA in ocean water samples from the East Atlantic ocean, indicating that these compounds have a common source.
|
In lake, river and drinking water, PFOA has often been reported at concentrations an order of magnitude higher than levels of PFNA, which is consistent with the overall source pattern estimates (Fig. 1). In the presence of point sources, a PFOA : PFNA ratio of ~100 has been observed[40] possibly indicating a more pronounced APFO input. In contrast to the estimated source emission profile, PFHxA and PFHpA have been reported in fresh and coastal waters at levels comparable to, or much higher than PFOA. PFHxA and PFHpA are typically either not measured or detected or reported to be at similar or even higher concentrations (respective levels of PFHxA and PFHpA as high as 372 and 149 μg L–1 have been reported near an air force base).[39,40] Examination of these studies in more detail revealed that high PFHxA and PFHpA levels were often found at locations associated with obvious point sources of PFCAs (e.g. air force bases where AFFF is used or wastewater treatment plants).
Sediments
Several studies have reported PFCA concentrations in sediments.[64–72] Some studies reported higher concentrations of PFHxA than PFOA and PFDA[67,68,73] indicating point sources of PFHxA similar to observations in surface waters. Only Higgins et al.[69] Ma and Shih[67] and Zushi et al.[65] reported a wide range of long-chain PFCA homologues and these studies show inconsistent homologue patterns. For example, Higgins et al.[69] concludes that PFCAs with an even number of carbons dominate with PFOA > PFHxA > PFDA, whereas Zushi et al.[65] find that PFCAs with odd carbons dominate with PFTrA > PFUnA > PFNA > PFOA > PFDoA > PFDA >PFHpA > PFHxA. The Higgins et al.[69] study from the San Francisco Bay cannot be reconciled with direct source patterns (Fig. 1) even if sediment sorption is taken into account. It has been shown that the PFCA sorption increases by a factor of ~3.5 for the addition of each successive CF2 moiety.[74] The study of Zushi et al.[65] undertaken in the Tokyo Bay is more consistent with the results expected from a sediment affected by direct sources.
Biota
A comprehensive review of PFCA biological monitoring data was presented by Houde et al.[75] although a considerable number of studies have been published since. We present a selection of PFCA homologue patterns for various species in Fig. 3.

|
A trend where odd carbon number homologue concentrations are higher than the next lower even homologue (e.g. PFNA >PFOA; PFUnA > PFDA; PFTrA > PFDoA) would be expected based on the homologue pattern in the sources (Fig. 1). This pattern has been observed in some biota (Fig. 3), but the fact that PFDA and PFUnA, as well as PFDoA and PFTrA, are often found at similar concentrations in biota inhabiting both source and remote regions (Fig. 3) cannot be reconciled with the direct source emission estimates (Fig. 1) assuming a positive trend in bioaccumulation potential with increasing chain length in all species. For biota where PFDA ≥ PFUnA and PFDoA ≥ PFTrA, the alternative hypothesis that atmospheric degradation of FTOHs is an important contributing source of longer chain PFCAs appears more consistent with monitoring data for PFCAs, assuming the FTOH reaction scheme in Ellis et al.[7] is correct. Following our previous discussion, it seems likely that the contribution of direct and indirect sources to biota will vary with location. If this hypothesis is correct then Greenland polar bears who ultimately receive contamination from the North Atlantic are more likely to have the direct source pattern (PFNA > PFOA; PFUnA > PFDA; PFTrA > PFDoA), whereas polar bears from inland areas are more likely to have the indirect source pattern (PFNA > PFOA; PFDA ≥ PFUnA; PFDoA ≥ PFTrA). The data for polar bears (Fig. 3b) do support this hypothesis with polar bears from inland areas (High Arctic, NW Territories and S. Baffin Island) showing the indirect source pattern, whereas those from Greenland show the direct source pattern.
It should be noted, however, that it is difficult to constrain the factor by which each additional -CF2- increases the bioaccumulative potential in wildlife (and hence the relative PFCA amplification at each trophic level) in the absence of empirical studies that are spatially and temporally contiguous. The paucity of reliable data in this regard is especially problematic when interpreting homologue patterns in higher trophic level organisms, particularly air-breathing animals that are exposed primarily through their diet.
Precipitation
Homologous patterns of PFCAs have been reported in precipitation from several northern hemisphere sites.[47,49] Dreyer et al.[49] reported that in >50% of samples, PFOA levels were most abundant and in the remainder concentrations of PFHpA or PFNA were most abundant. PFHxA was reported to be the most abundant in only one sample. In ~90% of samples PFHpA was reported to be more abundant than PFHxA. PFCA homologues with less than five carbons were also observed (e.g. TFA, PFPrA, PFBA and PFPeA).[47,49] Kwok et al.[47] reported that PFPrA was the third most abundant PFCA after PFOA and PFNA in Japanese, French and USA precipitation samples. PFCA homologues with more than nine carbons (PFDA and above) were mostly below limits of detection. Some researchers have also reported data for fluorotelomer acids (FTAs) and fluorotelomer unsaturated acids (FTUAs) in the continental precipitation studies.[47,48] FTAs and FTUAs have also been found in precipitation and in Arctic lakes.[53] The presence of FTAs and FTUAs in precipitation and remote surface waters is evidence of a contribution from a fluorotelomer source, but the relative importance of this source has not been quantified. Simcik and Dorweiler[52] assert that the ratio of PFHp : PFO is a unique marker of the relative contribution of atmospheric versus non-atmospheric sources with higher PFHp : PFO ratios if the source is atmospheric and this is verified in precipitation where remote sites had the highest PFHp : PFO ratios.
Isomer profiles
The major industrial synthesis of PFCAs is accomplished primarily by one of two methods: electrochemical fluorination (ECF) and telomerisation. Products of these two synthetic processes can be distinguished on the basis of their associated structural isomers. ECF products are a mixture of structural isomers predominantly composed of the linear perfluoroalkyl chain (70–80%) with smaller quantities of branched chain isomers. Until 2002, 3M was the major global manufacturer of PFOA by the ECF process and accounted for ~80% of the global market. Structural isomers of PFOA have not been found to be a by-product of the telomerisation synthesis process because the linear geometry of the starting materials is apparently conserved in the product. Isomeric profiles of PFCAs thus may provide evidence of the relative importance of different industrial production sources to the environment, biota and humans.[76]
Research has shown that isomer profiles of PFCAs, including PFOA and PFNA, are predominantly linear with a minor (5%) fraction consisting of branched isomers in Arctic polar bears livers and human blood serum[77,78] and this was initially suggested to support the dominance of telomer-based exposure sources. However, toxicokinetic studies in rats indicate that branched isomers are eliminated substantially faster than linear isomers[79–81] demonstrating that the ratio of branched to linear isomers in serum samples cannot be directly compared with the isomer profile of technical mixtures. Isomeric profiles of PFOA in ocean waters are generally consistent with that of an ECF standard (20–30% branched),[82] with the exception of one sampling site in an industrialised harbour area (Tokyo Bay), where a pure linear source contributed over 50% of the PFOA. Surface waters from Lake Ontario displayed some variability in the branched to linear ratio of PFOA with ~29–73% of the input being attributable to ECF-based sources.[80] In contrast, monitoring data from the North American environment indicated that a linear source of PFOA was the predominant input in rain water (>70%) and remote Arctic lakes (>90%).[80] These first observations of isomer profiles are consistent with the hypothesis that historical ECF-derived emissions are the major source of PFOA to the oceans and water bodies with long residence times.[6,11] However, the predominance of linear PFOA in precipitation and remote lakes indicates that remote environments receiving only atmospheric deposition reflect contemporary, rather than historical emission sources. Analysis of isomers is clearly a promising methodology for source elucidation (fingerprinting) that requires further investigation. However, caution is warranted before over-interpreting isomer patterns; differences in properties of isomers (e.g. solubilities, sorption and eliminated rates) are likely to influence the pattern in the environment and biota relative to the technical mixtures.
Temporal patterns in the environment and wildlife
In previous modelling studies[11,20,21] it has been predicted that direct emissions are largely responsible for the burden of PFCAs in Arctic surface water. Ocean transport models typically predict doubling times in the Arctic Ocean of ~7.5–10 years for PFOA surface water concentrations between 1975 and 2004. Furthermore, despite the estimated downturn in direct PFOA emissions in the early 2000s, Arctic seawater levels were predicted to increase until c. 2030 and then only slowly decline. The reason for this slow decline is that PFCAs are not degradable and will only be slowly removed by vertical mixing into the deeper oceans. It was noted by Wania[20] that irrespective of how the PFCAs are transported to the surface water of the Arctic Ocean (through ocean currents or via atmospheric degradation of precursors), the Arctic Ocean will have a very slow response time to changing emissions due to its long water residence time. The same slow response can be expected for other water bodies that have long water residence times (e.g. the Baltic Sea, which has a water residence time of 30 years). Faster responses to changing emissions can be expected for precursor concentrations in the atmosphere because they are relatively quickly removed by oxidation processes. Thus, environments that are affected mainly by atmospheric precursor sources and have removal mechanisms for PFCAs may show fast responses to changing precursor emissions. Furthermore, relatively fast response times to changing emissions can also be expected for PFCAs in water bodies with short residence times (e.g. rivers and coastal waters) that are in close proximity to the emission sources. Most studies of temporal patterns of long-chain PFCAs are conducted in biota due to the relative ease of detection in biota compared with in surface waters. Temporal patterns in biota are likely to follow the temporal patterns of PFCAs in water if exposure is derived via uptake of PFCAs. If exposure occurs via uptake of precursors and subsequent metabolism of the precursor to PFCAs, then the temporal pattern will be dependent on the temporal pattern of precursors in water. Precursor levels in water will respond more quickly to changing emissions due their potential for sea–air transport.
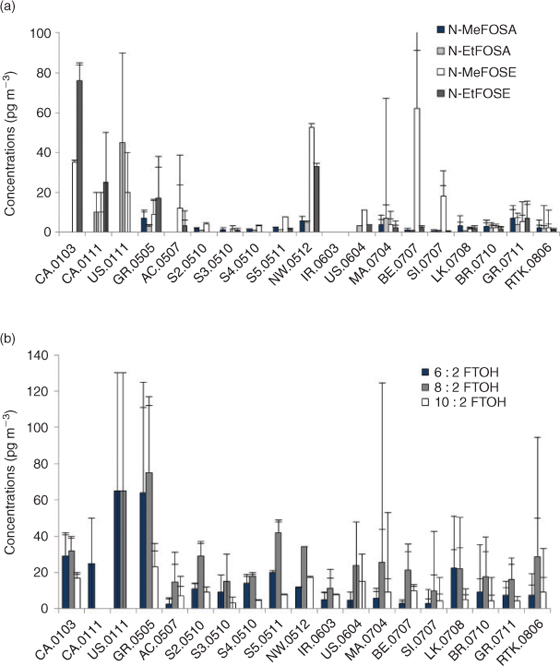
In this section we examine if time trend studies of PFCAs in biota support any of the above hypotheses. Numerous time trend studies have been undertaken globally. For the Arctic environment these have been thoroughly reviewed.[83] In this section, we summarise time trends for numerous Arctic biota and for biota from other regions in Table 1. We also determine if we can draw any conclusions about the temporal patterns of precursors in air based on the numerous reported studies (Fig. 4) and discuss temporal patterns in Arctic deposition fluxes measured in an ice pack.

|
 ’ indicates there is no clear trend. Years are mentioned when there is a break in a time trend (e.g. ↑88↓98↑ means that concentrations increased to 1988 then declined after until 1998 when they began to increase again). A certain amount of scientific judgment was required to compile this table
’ indicates there is no clear trend. Years are mentioned when there is a break in a time trend (e.g. ↑88↓98↑ means that concentrations increased to 1988 then declined after until 1998 when they began to increase again). A certain amount of scientific judgment was required to compile this table
|
Temporal trend studies of PFCAs in biota universally show an increasing trend to 2000, but there are discrepancies in time trends reported after that period. Some studies on temporal patterns in Arctic biota report a levelling off or even in decline in the latter part of the 2000s (lake trout (M. S. Evans, D. Muir, G. Low, et al., unpubl. data, 2006); burbot (G. A. Stern and G. T. Tomy, unpubl. data, 2007); ringed seals from Arviat, Nunavut[84]; beluga whales from Hendrickson Bay (G. Tomy, K. Pleskach, B. Rosenberg and G. Stern, unpubl. data, 2008)). It should be noted that the apparent decrease in these studies is not consistent for all PFCA homologues and the number of data points indicating a decrease are generally few. Therefore, it cannot be ruled out that confounding factors that are independent of chemical transport to the Arctic, contribute to a large interannual variability as seen in temporal trends for PFOS in the Baltic Sea.[85] Several studies show a continued increasing trend in Arctic concentrations throughout the study period (thick-billed murres and northern fulmars[86]; polar bear livers[87–89]; ringed seals from Resolute Bay, Nunavut[84]; and from Greenland (F. Riget et al., unpubl. data); seabirds[90]; beluga from Pangnirtung G. Tomy, et al., unpubl. data, 2008). It is notable that some species show diverging time trends in recent years in different regions (e.g. ringed seals and beluga whales). Continued monitoring of the areas indicating decreasing concentrations is therefore strongly recommended in order to produce more statistically robust time trends. If the discrepancies in time trends are supported by additional data, they may indicate a difference in the relative importance of indirect and direct sources in different locations. Furthermore, previous modelling studies have possibly over-simplified the complexity of ocean transport within the Arctic Ocean. It is known that the European and North American Arctic are affected by distinct ocean waters; the water in the Canadian Archipelago and northern Hudson Bay is of Pacific origin, whereas the water in the European Arctic is mainly influenced by Atlantic Ocean waters.[91,92] It is notable that those studies where decreasing trends have been reported in the Arctic biota are either inland (lake trout, burbot) or from areas of the Arctic Ocean affected by Pacific Ocean waters (ringed seals from Arviat, Nunavut or beluga whales from Hendrickson Bay).
If the transport of PFCAs to certain inland areas of the Arctic where declines in biota concentration have been observed is from atmospheric deposition, then we would expect to see a temporal decline in atmospheric deposition of precursors in air and deposition in recent years. Fig. 4 shows a compilation of measured atmospheric concentrations of major PFCA precursors for different years. In general Fig. 4 shows no apparent decline in FTOHs or POSF-based precursor compounds in recent years, indicative of on-going sources of these compounds. However, there are currently to our knowledge no long-term continuous monitoring activities that can be used to create proper time trends in atmospheric precursor levels and the mixed data set contained in Fig. 4 should not be over-interpreted. Because the data in Fig. 4 originate from different locations and seasons with no concentrations reported before 2001, it cannot be ruled out that any true decrease in POSF precursor concentrations is masked by seasonal and regional variability or an insufficient time span of the data. Nevertheless, we would not expect to observe a strong decline for FTOHs where on-going production sources are documented.[6] However, for POSF-based precursor compounds, a marked decrease after 2000–2002 reflecting reduced emissions might be expected.[21] In fact, ice core data from the high Arctic show that deposition of PFOS decreased by a factor 3–4 after 2002,[93] indicating decreasing concentrations of POSF precursors in the atmosphere. On the other hand, there are certainly on-going sources of POSF-based precursors from: (a) other ongoing manufacture of POSF-based chemical products (e.g. in Asia); (b) from products manufactured historically by 3M that have long lifetimes; and (c) emissions of residual precursors in landfill sites where waste products reside (L. Ahrens, M. Shoeib, S. C. Lee, T. Harner, unpubl. data, 2010).[93,94] Eliminating emissions of POSF-based precursors is expected to take many more years.
For biota from temperate regions there are also contrasting time trends reported. For ringed seals in the German Bight of the North Sea, a slight decrease in PFOA concentrations has been observed between 1999 and 2008.[95] However, no trend in concentrations was observed for PFNA to PFTrA.[95] For loggerhead turtles on the East coast of the US there has been a significant, albeit slow, decline in PFNA concentrations between 2000 and 2008.[96] In stranded whales along the Japanese coast, the concentrations of PFNA to PFDoA increased between 1982 and 2001–02 whereas no clear change in concentration was observed between 2001–02 and 2006.[97] Liver samples of Baikal seals displayed slightly higher concentrations of PFNA and PFDA in 2005 compared with 1992, although the difference was statistically significant only for PFDA.[98] A temporal trend performed on Lake Ontario trout indicated a peak in concentrations for PFNA to PFTA in 1988 followed by an apparent, but not statistically significant, decline until 2004.[99] In coastal environments and lake systems that were affected by direct sources of PFCAs before the phase out of POSF in 2000–2002, emission reductions are expected to cause a faster time response compared with the Arctic.[11] Recently, Zushi et al.[65] reported decreasing levels of PFOS and POSF-based precursors in Tokyo Bay as a consequence of emission reductions. However, concentrations of PFOA continued to increase after the POSF phase out indicating the presence of local telomer-based sources.[65]
In contrast to these studies performed on limnic or marine ecosystems, Swedish Peregrine Falcon eggs collected between 1974 and 2007 in South-Western Sweden displayed an increasing trend for PFNA, PFDA, PFUnA, PFDoA, PFTrA, PFTeA and PFPeDA over the study period.[85] Ahrens et al.[100] examined the temporal patterns in eggs of another terrestrial bird species, the tawny owl, from Central Norway collected over a 24 year period (1986–2009). These authors observed a significant increase in PFDA, PFUnA, PFDoA and PFTriA concentrations with a 4.2–12% increase observed over the study period. These two terrestrial bird studies provide consistent results and are also consistent with the expected lack of decrease in atmospheric deposition of PFCAs, as discussed by Schenker et al.[21]
Time trends of PFCAs in human serum may also be used to formulate hypotheses regarding environmental and consumer product based exposure sources. For PFOA, significant decreasing time trends have been observed in human serum in several industrialised countries after year 2000.[101–105] As noted in the previous section, a rapid decrease, corresponding to ~50% of the estimated the elimination rate, of PFOA in human serum is inconsistent with time trend studies in wildlife, regardless of location. In previous work[106] it has been suggested that the decreasing human serum concentrations of PFOA can be linked to POSF-based consumer that were phased out in 2000–02. Thus the recently observed decline in human serum of PFOA is not believed to reflect an overall decrease in environmental concentrations. In contrast to PFOA, the longer chain PFCAs (PFNA, PFDA and PFUnA) show steady or slightly increasing concentrations after 2000, which is more consistent with trends in wildlife and environmental emissions.
Conclusions
In summary, mass balance modelling, homologue patterns and isomer profiles in ocean water and the majority of temporal trend data are uniformly consistent with the hypothesis that direct sources can account for the majority of PFOA and PFNA present in the global environment. However, similar mass balance calculations for longer chain length PFCAs (PFDA and higher) are complicated by the fact that they are generally not detectable in ocean water samples. Although present in biota, it is not possible to reconcile the homologue pattern of long-chain PFCAs in biota with the estimated pattern in direct sources. Thus, it appears probable that the importance of indirect sources increase for longer chain PFCAs (PFDA and higher).
Fresh waters that are not affected by direct manufacturing sources will be affected by PFCA-laden municipal and industrial wastewaters and from PFCA-laden runoff from precipitation. Inland environments (inland lakes, remote streams, soils, the High Arctic) will be primarily affected by precipitation, but the ultimate origin of the PFCAs in the deposition is uncertain; it could be from direct (manufacturing), indirect (precursor) sources or both. To fully explore the potential contribution of indirect sources to air and deposition, additional precursors of PFCAs need to be included in future mass balance model calculations including fluorotelomer olefins, fluorotelomer iodides, fluorotelomer acrylates and perfluoroalkyl phosphates (PAPs), which are known to degrade and form PFCAs.[7,19,59,107]
Time trend studies for biota show contrasting results. However, in remote (e.g. Arctic) areas, with few exceptions, there is a generally increasing trend of PFCAs in aquatic biota, which is expected according to our source emission and model understanding.
In more populated regions, the variable total concentrations, homologue patterns and isomer profiles indicate that multiple sources (direct and indirect) contribute to PFCA contamination. There have been few temporal studies conducted adjacent to obvious historical sources. Decreasing time trends of PFCAs are observed in biota in some coastal regions where there is likely to be a faster response to recent declines in emissions. There appears to be ongoing sources of POSF-based precursors despite the phase out nearly 10 years ago. The increasing direct and indirect sources from the manufacture and use of perfluorinated chemistry in Asia suggest the likelihood of higher environmental contamination in this region in the future and represent and area where future temporal studies should be conducted.
Acknowledgements
We thank E.I. du Pont de Nemours & Co., Inc. who provided an unrestricted gift to support to Ph.D. studies of Robin Vestergren. The Ph.D. studies of Deguo Kong are funded by ArcRisk, which is a Collaborative Project supported under the Seventh Framework Program of the European Community for research, technological development and demonstration activities (FP7-ENV-2008-1, Grant Agreement Number: 226534). We thank Matthew MacLeod (colleague), Mark H. Russell (E. I. du Pont) and anonymous reviewers for helpful comments on the manuscript.
References
[1] J. P. Giesy, K. Kannan, Global distribution of perfluorooctane sulfonate in wildlife. Environ. Sci. Technol. 2001, 35, 1339.| Global distribution of perfluorooctane sulfonate in wildlife.Crossref | GoogleScholarGoogle Scholar |
[2] K. J. Hansen, L. A. Clemen, M. E. Ellefson, H. O. Johnson, Compound-specific, quantitative characterization of organic: fluorochemicals in biological matrices. Environ. Sci. Technol. 2001, 35, 766.
| Compound-specific, quantitative characterization of organic: fluorochemicals in biological matrices.Crossref | GoogleScholarGoogle Scholar |
[3] B. E. Smart, Characteristics of C–F systems, in Organofluorine Chemistry – Principles and Commercial Applications (Eds R. E. Banks, B. E. Smart, J. C. Tatlow) 1994, pp. 57–88 (Plenum Press: New York).
[4] J. M. Conder, R. A. Hoke, W. De Wolf, M. H. Russell, R. C. Buck, Are PFCAs bioaccumulative? A critical review and comparison with regulatory lipophilic compounds. Environ. Sci. Technol. 2008, 42, 995.
| Are PFCAs bioaccumulative? A critical review and comparison with regulatory lipophilic compounds.Crossref | GoogleScholarGoogle Scholar |
[5] C. Lau, K. Anitole, C. Hodes, D. Lai, A. Pfahles-Hutchens, J. Seed, Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol. Sci. 2007, 99, 366.
| Perfluoroalkyl acids: a review of monitoring and toxicological findings.Crossref | GoogleScholarGoogle Scholar |
[6] K. Prevedouros, I. T. Cousins, R. C. Buck, S. H. Korzeniowski, Sources, fate and transport of perfluorocarboxylates. Environ. Sci. Technol. 2006, 40, 32.
| Sources, fate and transport of perfluorocarboxylates.Crossref | GoogleScholarGoogle Scholar |
[7] D. A. Ellis, J. W. Martin, A. O. De Silva, S. A. Mabury, M. D. Hurley, M. P. S. Andersen, T. J. Wallington, Degradation of fluorotelomer alcohols: a likely atmospheric source of perfluorinated carboxylic acids. Environ. Sci. Technol. 2004, 38, 3316.
| Degradation of fluorotelomer alcohols: a likely atmospheric source of perfluorinated carboxylic acids.Crossref | GoogleScholarGoogle Scholar |
[8] 3M Company, Phase-Out Plan for POSF-Based Products. US EPA Administrative Record AR226–0600 2000 2000. Available at http://www.fluoridealert.org/pesticides/pfos.fr.final.docket.0009.pdf [Verified 16 March 2011].
[9] S. K. Ritter, Fluorochemicals go short. Chem. Eng. News 2010, 88, 12..
[10] J. W. Martin, S. A. Mabury, K. R. Solomon, D. C. G. Muir, Dietary accumulation of perfluorinated acids in juvenile rainbow trout (Oncorhynchus mykiss). Environ. Toxicol. Chem. 2003, 22, 189.
| Dietary accumulation of perfluorinated acids in juvenile rainbow trout (Oncorhynchus mykiss).Crossref | GoogleScholarGoogle Scholar |
[11] J. Armitage, I. T. Cousins, R. C. Buck, K. Prevedouros, M. H. Russell, M. MacLeod, S. H. Korzeniowski, Modeling global-scale fate and transport of perfluorooctanoate emitted from direct sources. Environ. Sci. Technol. 2006, 40, 6969.
| Modeling global-scale fate and transport of perfluorooctanoate emitted from direct sources.Crossref | GoogleScholarGoogle Scholar |
[12] J. M. Armitage, M. MacLeod, I. T. Cousins, Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources. Environ. Sci. Technol. 2009, 43, 5830.
| Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources.Crossref | GoogleScholarGoogle Scholar |
[13] R. Will, T. Kälin, A. Kishi, Fluoropolymers CEH Marketing Research Report 2005 2005 (SRI International: Menlo Park, CA).
[14] K. L. Ring, T. Kalin, A. Kishi, Fluoropolymers CEH Marketing Research Report 2002 2002 (SRI International: Menlo Park, CA).
[15] A. Kärrman, J. F. Mueller, B. Van Bavel, F. Harden, L. M. L. Toms, G. Lindström, Levels of 12 perfluorinated chemicals in pooled Australian serum, collected 2002–2003, in relation to age, gender and region. Environ. Sci. Technol. 2006, 40, 3742.
| Levels of 12 perfluorinated chemicals in pooled Australian serum, collected 2002–2003, in relation to age, gender and region.Crossref | GoogleScholarGoogle Scholar |
[16] L.-M. L. Toms, A. M. Calafat, K. Kato, J. Thompson, F. Harden, P. Hobson, A. Sjödin, J. F. Mueller, Polyfluoroalkyl chemicals in pooled blood serum from infants, children, and adults in Australia. Environ. Sci. Technol. 2009, 43, 4194.
| Polyfluoroalkyl chemicals in pooled blood serum from infants, children, and adults in Australia.Crossref | GoogleScholarGoogle Scholar |
[17] J. M. Armitage, M. MacLeod, I. T. Cousins, Modeling the global fate and transport of perfluorooctanoic acid (PFOA) and perfluorooctanoate (PFO) emitted from direct sources using a multispecies mass balance model. Environ. Sci. Technol. 2009, 43, 6438.
| Modeling the global fate and transport of perfluorooctanoic acid (PFOA) and perfluorooctanoate (PFO) emitted from direct sources using a multispecies mass balance model.Crossref | GoogleScholarGoogle Scholar |
[18] I. Stemmler, G. Lammel, Pathways of PFOA to the Arctic: variabilities and contributions of oceanic currents and atmospheric transport and chemistry sources. Atmos. Chem. Phys. Discuss. 2010, 10, 11577.
| Pathways of PFOA to the Arctic: variabilities and contributions of oceanic currents and atmospheric transport and chemistry sources.Crossref | GoogleScholarGoogle Scholar |
[19] T. J. Wallington, M. D. Hurley, J. Xia, D. J. Wuebbles, S. Sillman, A. Ito, J. E. Penner, D. A. Ellis, J. Martin, S. A. Mabury, O. J. Nielsen, M. P. S. Andersen, Formation of C7F15COOH (PFOA) and other perfluorocarboxylic acids during the atmospheric oxidation of 8:2 fluorotelomer alcohol. Environ. Sci. Technol. 2006, 40, 924.
| Formation of C7F15COOH (PFOA) and other perfluorocarboxylic acids during the atmospheric oxidation of 8:2 fluorotelomer alcohol.Crossref | GoogleScholarGoogle Scholar |
[20] F. Wania, Potential of degradable organic chemicals for absolute and relative enrichment in the arctic. Environ. Sci. Technol. 2006, 40, 569.
| Potential of degradable organic chemicals for absolute and relative enrichment in the arctic.Crossref | GoogleScholarGoogle Scholar |
[21] U. Schenker, M. Scheringer, M. MacLeod, J. W. Martin, I. T. Cousins, K. Hungerbühler, Contribution of volatile precursor substances to the flux of perfluorooctanoate to the Arctic. Environ. Sci. Technol. 2008, 42, 3710.
| Contribution of volatile precursor substances to the flux of perfluorooctanoate to the Arctic.Crossref | GoogleScholarGoogle Scholar |
[22] G. Yarwood, S. Kemball-Cook, M. Keinath, R. L. Waterland, S. H. Korzeniowski, R. C. Buck, M. H. Russell, S. T. Washburn, High-resolution atmospheric modeling of fluorotelomer alcohols and perfluorocarboxylic acids in the North American troposphere. Environ. Sci. Technol. 2007, 41, 5756.
| High-resolution atmospheric modeling of fluorotelomer alcohols and perfluorocarboxylic acids in the North American troposphere.Crossref | GoogleScholarGoogle Scholar |
[23] M. Shoeib, T. Harner, P. Vlahos, Perfluorinated chemicals in the Arctic atmosphere. Environ. Sci. Technol. 2006, 40, 7577.
| Perfluorinated chemicals in the Arctic atmosphere.Crossref | GoogleScholarGoogle Scholar |
[24] N. O. Brace, Long chain alkanoic and alkenoic acids with perfluoroalkyl terminal segments. J. Org. Chem. 1962, 27, 4491.
| Long chain alkanoic and alkenoic acids with perfluoroalkyl terminal segments.Crossref | GoogleScholarGoogle Scholar |
[25] M. Ylinen, A. Kojo, H. Hanhijärvi, P. Peura, Disposition of perfluorooctanoic acid in the rat after single and subchronic administration. Bull. Environ. Contam. Toxicol. 1990, 44, 46.
| Disposition of perfluorooctanoic acid in the rat after single and subchronic administration.Crossref | GoogleScholarGoogle Scholar |
[26] Y. Moroi, H. Yano, O. Shibata, T. Yonemitsu, Determination of acidity constants of perfluoroalkanoic acids. Bull. Chem. Soc. Jpn. 2001, 74, 667.
| Determination of acidity constants of perfluoroalkanoic acids.Crossref | GoogleScholarGoogle Scholar |
[27] J. L. López-Fontán, F. Sarmiento, P. C. Schulz, The aggregation of sodium perfluorooctanoate in water. Colloid Polym. Sc. 2005, 283, 862.
| The aggregation of sodium perfluorooctanoate in water.Crossref | GoogleScholarGoogle Scholar |
[28] S. Igarashi, T. Yotsuyanagi, Homogeneous liquid-liquid extraction by pH dependent phase separation with a fluorocarbon ionic surfactant and its application to the preconcentration of porphyrin compounds. Mikrochim. Acta 1992, 106, 37.
| Homogeneous liquid-liquid extraction by pH dependent phase separation with a fluorocarbon ionic surfactant and its application to the preconcentration of porphyrin compounds.Crossref | GoogleScholarGoogle Scholar |
[29] D. C. Burns, D. A. Ellis, H. Li, C. J. McMurdo, E. Webster, Experimental pK(a) determination for perfluorooctanoic acid (PFOA) and the potential impact of pK(a) concentration dependence on laboratory-measured partitioning phenomena and environmental modeling. Environ. Sci. Technol. 2008, 42, 9283.
| Experimental pK(a) determination for perfluorooctanoic acid (PFOA) and the potential impact of pK(a) concentration dependence on laboratory-measured partitioning phenomena and environmental modeling.Crossref | GoogleScholarGoogle Scholar |
[30] K. U. Goss, The pK(a) values of PFOA and other highly fluorinated carboxylic acids. Environ. Sci. Technol. 2008, 42, 456.
| The pK(a) values of PFOA and other highly fluorinated carboxylic acids.Crossref | GoogleScholarGoogle Scholar |
[31] S. Rayne, K. Forest, Theoretical studies on the pK(a) values of perfluoroalkyl carboxylic acids. J. Mol. Struct. THEOCHEM 2010, 949, 60.
| Theoretical studies on the pK(a) values of perfluoroalkyl carboxylic acids.Crossref | GoogleScholarGoogle Scholar |
[32] K. U. Goss, H. P. H. Arp, Comment on ‘Experimental pK(a) determination for perfluorooctanoic acid (PFOA) and the potential impact of pK(a) concentration dependence on laboratory-measured partitioning phenomena and environmental modeling’. Environ. Sci. Technol. 2009, 43, 5150.
| Comment on ‘Experimental pK(a) determination for perfluorooctanoic acid (PFOA) and the potential impact of pK(a) concentration dependence on laboratory-measured partitioning phenomena and environmental modeling’.Crossref | GoogleScholarGoogle Scholar |
[33] J. Cheng, E. Psillakis, M. R. Hoffmann, A. J. Colussi, Acid dissociation versus molecular association of perfluoroalkyl oxoacids: environmental implications. J. Phys. Chem. A 2009, 113, 8152.
| Acid dissociation versus molecular association of perfluoroalkyl oxoacids: environmental implications.Crossref | GoogleScholarGoogle Scholar |
[34] C. A. Barton, L. E. Butler, C. J. Zarzecki, J. Flaherty, M. Kaiser, Characterizing perfluorooctanoate in ambient air near the fence line of a manufacturing facility: comparing modeled and monitored values. J. Air Waste Manage. Assoc. 2006, 56, 48..
[35] L. Ahrens, Z. Y. Xie, R. Ebinghaus, Distribution of perfluoroalkyl compounds in seawater from Northern Europe, Atlantic Ocean, and Southern Ocean. Chemosphere 2010, 78, 1011.
| Distribution of perfluoroalkyl compounds in seawater from Northern Europe, Atlantic Ocean, and Southern Ocean.Crossref | GoogleScholarGoogle Scholar |
[36] L. Ahrens, J. L. Barber, Z. Xie, R. Ebinghaus, Longitudinal and latitudinal distribution of perfluoroalkyl compounds in the surface water of the Atlantic Ocean. Environ. Sci. Technol. 2009, 43, 3122.
| Longitudinal and latitudinal distribution of perfluoroalkyl compounds in the surface water of the Atlantic Ocean.Crossref | GoogleScholarGoogle Scholar |
[37] A. Jahnke, U. Berger, R. Ebinghaus, C. Temme, Latitudinal gradient of airborne polyfluorinated alkyl substances in the marine atmosphere between Germany and South Africa (53°N–33°S). Environ. Sci. Technol. 2007, 41, 3055.
| Latitudinal gradient of airborne polyfluorinated alkyl substances in the marine atmosphere between Germany and South Africa (53°N–33°S).Crossref | GoogleScholarGoogle Scholar |
[38] N. Yamashita, K. Kannan, S. Taniyasu, Y. Horii, G. Petrick, T. Gamo, A global survey of perfluorinated acids in oceans. Mar. Pollut. Bull. 2005, 51, 658.
| A global survey of perfluorinated acids in oceans.Crossref | GoogleScholarGoogle Scholar |
[39] S. Rayne, K. Forest, Perfluoroalkyl sulfonic and carboxylic acids: a critical review of physicochemical properties, levels and patterns in waters and wastewaters, and treatment methods. J. Environ. Sci. Health Part A Tox. Hazard. Subst. Environ. Eng. 2009, 44, 1145.
| Perfluoroalkyl sulfonic and carboxylic acids: a critical review of physicochemical properties, levels and patterns in waters and wastewaters, and treatment methods.Crossref | GoogleScholarGoogle Scholar |
[40] M. S. McLachlan, K. E. Holmström, M. Reth, U. Berger, Riverine discharge of perfluorinated carboxylates from the European continent. Environ. Sci. Technol. 2007, 41, 7260.
| Riverine discharge of perfluorinated carboxylates from the European continent.Crossref | GoogleScholarGoogle Scholar |
[41] R. Bossi, J. Strand, O. Sortkjaer, M. M. Larsen, Perfluoroalkyl compounds in Danish wastewater treatment plants and aquatic environments. Environ. Int. 2008, 34, 443.
| Perfluoroalkyl compounds in Danish wastewater treatment plants and aquatic environments.Crossref | GoogleScholarGoogle Scholar |
[42] J. Busch, L. Ahrens, R. Sturm, R. Ebinghaus, Polyfluoroalkyl compounds in landfill leachates. Environ. Pollut. 2010, 158, 1467.
| Polyfluoroalkyl compounds in landfill leachates.Crossref | GoogleScholarGoogle Scholar |
[43] M. Murakami, H. Shinohara, H. Takada, Evaluation of wastewater and street runoff as sources of perfluorinated surfactants (PFSs). Chemosphere 2009, 74, 487.
| Evaluation of wastewater and street runoff as sources of perfluorinated surfactants (PFSs).Crossref | GoogleScholarGoogle Scholar |
[44] S.-K. Kim, K. Kannan, Perfluorinated acids in air, rain, snow, surface runoff, and lakes: relative importance of pathways to contamination of urban lakes. Environ. Sci. Technol. 2007, 41, 8328.
| Perfluorinated acids in air, rain, snow, surface runoff, and lakes: relative importance of pathways to contamination of urban lakes.Crossref | GoogleScholarGoogle Scholar |
[45] Y. Zushi, T. Takeda, S. Masunaga, Existence of nonpoint source of perfluorinated compounds and their loads in the Tsurumi River basin, Japan. Chemosphere 2008, 71, 1566.
| Existence of nonpoint source of perfluorinated compounds and their loads in the Tsurumi River basin, Japan.Crossref | GoogleScholarGoogle Scholar |
[46] M. Clara, C. Scheffknecht, S. Scharf, S. Weiss, O. Gans, Emissions of perfluorinated alkylated substances (PFAS) from point sources – identification of relevant branches. Water Sci. Technol. 2008, 58, 59.
| Emissions of perfluorinated alkylated substances (PFAS) from point sources – identification of relevant branches.Crossref | GoogleScholarGoogle Scholar |
[47] K. Y. Kwok, S. Taniyasu, L. W. Y. Yeung, M. B. Murphy, P. K. S. Lam, Y. Horii, K. Kannan, G. Petrick, R. K. Sinha, N. Yamashita, Flux of perfluorinated chemicals through wet deposition in Japan, the United States, and several other countries. Environ. Sci. Technol. 2010, 44, 7043.
| Flux of perfluorinated chemicals through wet deposition in Japan, the United States, and several other countries.Crossref | GoogleScholarGoogle Scholar |
[48] B. F. Scott, C. Spencer, S. A. Mabury, D. C. G. Muir, Poly- and perfluorinated carboxylates in North American precipitation. Environ. Sci. Technol. 2006, 40, 7167.
| Poly- and perfluorinated carboxylates in North American precipitation.Crossref | GoogleScholarGoogle Scholar |
[49] A. Dreyer, V. Matthias, I. Weinberg, R. Ebinghaus, Wet deposition of poly- and perfluorinated compounds in northern Germany. Environ. Pollut. 2010, 158, 1221.
| Wet deposition of poly- and perfluorinated compounds in northern Germany.Crossref | GoogleScholarGoogle Scholar |
[50] S. Taniyasu, K. Kannan, L. W. Y. Yeung, K. Y. Kwok, P. K. S. Lam, N. Yamashita, Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2–C4) in precipitation by liquid chromatography-tandem mass spectrometry: comparison to patterns of long-chain perfluorinated acids (C5–C18). Anal. Chim. Acta 2008, 619, 221.
| Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2–C4) in precipitation by liquid chromatography-tandem mass spectrometry: comparison to patterns of long-chain perfluorinated acids (C5–C18).Crossref | GoogleScholarGoogle Scholar |
[51] E. Sinclair, D. Mayack, K. Roblee, N. Yamashita, K. Kannan, Occurrence of perfluoroalkyl surfactants in water, fish, and birds from New York State. Arch. Environ. Contam. Toxicol. 2006, 50, 398.
| Occurrence of perfluoroalkyl surfactants in water, fish, and birds from New York State.Crossref | GoogleScholarGoogle Scholar |
[52] M. F. Simcik, K. J. Dorweiler, Ratio of perfluorochemical concentrations as a tracer of atmospheric deposition to surface waters. Environ. Sci. Technol. 2005, 39, 8678.
| Ratio of perfluorochemical concentrations as a tracer of atmospheric deposition to surface waters.Crossref | GoogleScholarGoogle Scholar |
[53] N. L. Stock, V. I. Furdui, D. C. G. Muir, S. A. Mabury, Perfluoroalkyl contaminants in the Canadian Arctic: evidence of atmospheric transport and local contamination. Environ. Sci. Technol. 2007, 41, 3529.
| Perfluoroalkyl contaminants in the Canadian Arctic: evidence of atmospheric transport and local contamination.Crossref | GoogleScholarGoogle Scholar |
[54] E. Webster, D. A. Ellis, Potential role of sea spray generation in the atmospheric transport of perfluorocarboxylic acids. Environ. Toxicol. Chem. 2010, 29, 1703.
| Potential role of sea spray generation in the atmospheric transport of perfluorocarboxylic acids.Crossref | GoogleScholarGoogle Scholar |
[55] C. J. McMurdo, D. A. Ellis, E. Webster, J. Butler, R. D. Christensen, L. K. Reid, Aerosol enrichment of the surfactant PFO and mediation of the water–air transport of gaseous PFOA. Environ. Sci. Technol. 2008, 42, 3969.
| Aerosol enrichment of the surfactant PFO and mediation of the water–air transport of gaseous PFOA.Crossref | GoogleScholarGoogle Scholar |
[56] S. Kutsuna, H. Hori, Experimental determination of Henry’s law constant of perfluorooctanoic acid (PFOA) at 298 K by means of an inert-gas stripping method with a helical plate. Atmos. Environ. 2008, 42, 8883.
| Experimental determination of Henry’s law constant of perfluorooctanoic acid (PFOA) at 298 K by means of an inert-gas stripping method with a helical plate.Crossref | GoogleScholarGoogle Scholar |
[57] M. A. Kaiser, B. J. Dawson, C. A. Barton, M. A. Botelho, Understanding potential exposure sources of perfluorinated carboxylic acids in the workplace. Ann. Occup. Hyg. 2010, 54, 915.
| Understanding potential exposure sources of perfluorinated carboxylic acids in the workplace.Crossref | GoogleScholarGoogle Scholar |
[58] H. P. H. Arp, K.-U. Goss, Irreversible sorption of trace concentrations of perfluorocarboxylic acids to fiber filters used for air sampling. Atmos. Environ. 2008, 42, 6869.
| Irreversible sorption of trace concentrations of perfluorocarboxylic acids to fiber filters used for air sampling.Crossref | GoogleScholarGoogle Scholar |
[59] J. W. Martin, D. A. Ellis, S. A. Mabury, M. D. Hurley, T. J. Wallington, Atmospheric chemistry of perfluoroalkanesulfonamides: kinetic and product studies of the OH radical and Cl atom initiated oxidation of N-ethyl perfluorobutanesulfonamide. Environ. Sci. Technol. 2006, 40, 864.
| Atmospheric chemistry of perfluoroalkanesulfonamides: kinetic and product studies of the OH radical and Cl atom initiated oxidation of N-ethyl perfluorobutanesulfonamide.Crossref | GoogleScholarGoogle Scholar |
[60] N. Yamashita, K. Kannan, S. Taniyasu, Y. Horii, T. Okazawa, G. Petrick, T. Gamo, Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry. Environ. Sci. Technol. 2004, 38, 5522.
| Analysis of perfluorinated acids at parts-per-quadrillion levels in seawater using liquid chromatography-tandem mass spectrometry.Crossref | GoogleScholarGoogle Scholar |
[61] S. Wei, L. Q. Chen, S. Taniyasu, M. K. So, M. B. Murphy, N. Yamashita, L. W. Y. Yeung, P. K. S. Lam, Distribution of perfluorinated compounds in surface seawaters between Asia and Antarctica. Mar. Pollut. Bull. 2007, 54, 1813.
| Distribution of perfluorinated compounds in surface seawaters between Asia and Antarctica.Crossref | GoogleScholarGoogle Scholar |
[62] S. Taniyasu, K. Kannan, M. K. So, A. Gulkowska, E. Sinclair, T. Okazawa, N. Yamashita, Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota. J. Chromatogr. A 2005, 1093, 89.
| Analysis of fluorotelomer alcohols, fluorotelomer acids, and short- and long-chain perfluorinated acids in water and biota.Crossref | GoogleScholarGoogle Scholar |
[63] Y. Miyake, N. Yamashita, P. Rostkowski, M. K. So, S. Taniyasu, P. K. S. Lam, K. Kannan, Determination of trace levels of total fluorine in water using combustion ion chromatography for fluorine: a mass balance approach to determine individual perfluorinated chemicals in water. J. Chromatogr. A 2007, 1143, 98.
| Determination of trace levels of total fluorine in water using combustion ion chromatography for fluorine: a mass balance approach to determine individual perfluorinated chemicals in water.Crossref | GoogleScholarGoogle Scholar |
[64] L. Ahrens, S. Taniyasu, L. W. Y. Yeung, N. Yamashita, P. K. S. Lam, R. Ebinghaus, Distribution of polyfluoroalkyl compounds in water, suspended particulate matter and sediment from Tokyo Bay, Japan. Chemosphere 2010, 79, 266.
| Distribution of polyfluoroalkyl compounds in water, suspended particulate matter and sediment from Tokyo Bay, Japan.Crossref | GoogleScholarGoogle Scholar |
[65] Y. Zushi, M. Tamada, Y. Kanai, S. Masunaga, Time trends of perfluorinated compounds from the sediment core of Tokyo Bay, Japan (1950s–2004). Environ. Pollut. 2010, 158, 756.
| Time trends of perfluorinated compounds from the sediment core of Tokyo Bay, Japan (1950s–2004).Crossref | GoogleScholarGoogle Scholar |
[66] T. Sakurai, S. Serizawa, T. Isobe, J. Kobayashi, K. Kodama, G. Kume, J. H. Lee, H. Maki, Y. Imaizumi, N. Suzuki, T. Horiguchi, M. Morita, H. Shiraishi, Spatial, phase, and temporal distributions of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in Tokyo Bay, Japan. Environ. Sci. Technol. 2010, 44, 4110.
| Spatial, phase, and temporal distributions of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in Tokyo Bay, Japan.Crossref | GoogleScholarGoogle Scholar |
[67] R. W. Ma, K. M. Shih, Perfluorochemicals in wastewater treatment plants and sediments in Hong Kong. Environ. Pollut. 2010, 158, 1354.
| Perfluorochemicals in wastewater treatment plants and sediments in Hong Kong.Crossref | GoogleScholarGoogle Scholar |
[68] K. S. Kumar, Y. Zushi, S. Masunaga, M. Gilligan, C. Pride, K. S. Sajwan, Perfluorinated organic contaminants in sediment and aquatic wildlife, including sharks, from Georgia, USA. Mar. Pollut. Bull. 2009, 58, 621.
| Perfluorinated organic contaminants in sediment and aquatic wildlife, including sharks, from Georgia, USA.Crossref | GoogleScholarGoogle Scholar |
[69] C. P. Higgins, J. A. Field, C. S. Criddle, R. G. Luthy, Quantitative determination of perfluorochemicals in sediments and domestic sludge. Environ. Sci. Technol. 2005, 39, 3946.
| Quantitative determination of perfluorochemicals in sediments and domestic sludge.Crossref | GoogleScholarGoogle Scholar |
[70] J. Bao, W. Liu, L. Liu, Y. H. Jin, X. R. Ran, Z. X. Zhang, Perfluorinated compounds in urban river sediments from Guangzhou and Shanghai of China. Chemosphere 2010, 80, 123.
| Perfluorinated compounds in urban river sediments from Guangzhou and Shanghai of China.Crossref | GoogleScholarGoogle Scholar |
[71] J. Bao, Y. H. Jin, W. Liu, X. R. Ran, Z. X. Zhang, Perfluorinated compounds in sediments from the Daliao River system of northeast China. Chemosphere 2009, 77, 652.
| Perfluorinated compounds in sediments from the Daliao River system of northeast China.Crossref | GoogleScholarGoogle Scholar |
[72] K. Senthilkumar, E. Ohi, K. Sajwan, T. Takasuga, K. Kannan, Perfluorinated compounds in river water, river sediment, market fish, and wildlife samples from Japan. Bull. Environ. Contam. Toxicol. 2007, 79, 427.
| Perfluorinated compounds in river water, river sediment, market fish, and wildlife samples from Japan.Crossref | GoogleScholarGoogle Scholar |
[73] M. Clara, O. Gans, S. Weiss, D. Sanz-Escribano, S. Scharf, C. Scheffknecht, Perfluorinated alkylated substances in the aquatic environment: an Austrian case study. Water Res. 2009, 43, 4760.
| Perfluorinated alkylated substances in the aquatic environment: an Austrian case study.Crossref | GoogleScholarGoogle Scholar |
[74] C. P. Higgins, R. G. Luthy, Sorption of perfluorinated surfactants on sediments. Environ. Sci. Technol. 2006, 40, 7251.
| Sorption of perfluorinated surfactants on sediments.Crossref | GoogleScholarGoogle Scholar |
[75] M. Houde, J. W. Martin, R. J. Letcher, K. R. Solomon, D. C. G. Muir, Biological monitoring of polyfluoroalkyl substances: a review. Environ. Sci. Technol. 2006, 40, 3463.
| Biological monitoring of polyfluoroalkyl substances: a review.Crossref | GoogleScholarGoogle Scholar |
[76] J. P. Benskin, A. O. De Silva, J. W. Martin, in Reviews of Environmental Contamination and Toxicology 2010, vol. 208, pp. 111–160 (Springer: New York).
[77] A. O. De Silva, S. A. Mabury, Isolating isomers of perfluorocarboxylates in polar bears (Ursus maritimus) from two geographical locations. Environ. Sci. Technol. 2004, 38, 6538.
| Isolating isomers of perfluorocarboxylates in polar bears (Ursus maritimus) from two geographical locations.Crossref | GoogleScholarGoogle Scholar |
[78] A. O. De Silva, S. A. Mabury, Isomer distribution of perfluorocarboxylates in human blood: potential correlation to source. Environ. Sci. Technol. 2006, 40, 2903.
| Isomer distribution of perfluorocarboxylates in human blood: potential correlation to source.Crossref | GoogleScholarGoogle Scholar |
[79] S. E. Loveless, C. Finlay, N. E. Everds, S. R. Frame, P. J. Gillies, J. C. O’Connor, C. R. Powley, G. L. Kennedy, Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (APFO). Toxicology 2006, 220, 203.
| Comparative responses of rats and mice exposed to linear/branched, linear, or branched ammonium perfluorooctanoate (APFO).Crossref | GoogleScholarGoogle Scholar |
[80] A. O. De Silva, D. C. G. Muir, S. A. Mabury, Distribution of perfluorocarboxylate isomers in select samples from the North American environment. Environ. Toxicol. Chem. 2009, 28, 1801.
| Distribution of perfluorocarboxylate isomers in select samples from the North American environment.Crossref | GoogleScholarGoogle Scholar |
[81] J. P. Benskin, A. O. De Silva, L. J. Martin, G. Arsenault, R. McCrindle, N. Riddell, S. A. Mabury, J. W. Martin, Disposition of perfluorinated acid isomers in sprague-dawley rats. Part 1: single dose. Environ. Toxicol. Chem. 2009, 28, 542.
| Disposition of perfluorinated acid isomers in sprague-dawley rats. Part 1: single dose.Crossref | GoogleScholarGoogle Scholar |
[82] J. P. Benskin, A. Holt, J. W. Martin, Isomer-specific biotransformation rates of a perfluorooctane sulfonate (PFOS)-precursor by cytochrome P450 isozymes and human liver microsomes. Environ. Sci. Technol. 2009, 43, 8566.
| Isomer-specific biotransformation rates of a perfluorooctane sulfonate (PFOS)-precursor by cytochrome P450 isozymes and human liver microsomes.Crossref | GoogleScholarGoogle Scholar |
[83] C. M. Butt, U. Berger, R. Bossi, G. T. Tomy, Levels and trends of poly- and perfluorinated compounds in the arctic environment. Sci. Total Environ. 2010, 408, 2936.
| Levels and trends of poly- and perfluorinated compounds in the arctic environment.Crossref | GoogleScholarGoogle Scholar |
[84] C. M. Butt, D. C. G. Muir, I. Stirling, M. Kwan, S. A. Mabury, Rapid response of arctic ringed seals to changes in perfluoroalkyl production. Environ. Sci. Technol. 2007, 41, 42.
| Rapid response of arctic ringed seals to changes in perfluoroalkyl production.Crossref | GoogleScholarGoogle Scholar |
[85] K. E. Holmström, A. K. Johansson, A. Bignert, P. Lindberg, U. Berger, Temporal trends of perfluorinated surfactants in Swedish peregrine falcon eggs (Falco peregrinus), 1974–2007. Environ. Sci. Technol. 2010, 44, 4083.
| Temporal trends of perfluorinated surfactants in Swedish peregrine falcon eggs (Falco peregrinus), 1974–2007.Crossref | GoogleScholarGoogle Scholar |
[86] C. M. Butt, S. A. Mabury, D. C. G. Muir, B. M. Braune, Prevalence of long-chained perfluorinated carboxylates in seabirds from the Canadian arctic between 1975 and 2004. Environ. Sci. Technol. 2007, 41, 3521.
| Prevalence of long-chained perfluorinated carboxylates in seabirds from the Canadian arctic between 1975 and 2004.Crossref | GoogleScholarGoogle Scholar |
[87] M. Smithwick, R. J. Norstrom, S. A. Mabury, K. Solomon, T. J. Evans, I. Stirling, M. K. Taylor, D. C. G. Muir, Temporal trends of perfluoroalkyl contaminants in polar bears (Ursus maritimus) from two locations in the North American Arctic, 1972–2002. Environ. Sci. Technol. 2006, 40, 1139.
| Temporal trends of perfluoroalkyl contaminants in polar bears (Ursus maritimus) from two locations in the North American Arctic, 1972–2002.Crossref | GoogleScholarGoogle Scholar |
[88] R. Dietz, R. Bossi, F. F. Rigét, C. Sonne, E. W. Born, Increasing perfluoroalkyl contaminants in East Greenland polar bears (Ursus maritimus): a new toxic threat to the arctic bears. Environ. Sci. Technol. 2008, 42, 2701.
| Increasing perfluoroalkyl contaminants in East Greenland polar bears (Ursus maritimus): a new toxic threat to the arctic bears.Crossref | GoogleScholarGoogle Scholar |
[89] R. Bossi, F. F. Riget, R. Dietz, C. Sonne, P. Fauser, M. Dam, K. Vorkamp, Preliminary screening of perfluorooctane sulfonate (PFOS) and other fluorochemicals in fish, birds and marine mammals from Greenland and the Faroe Islands. Environ. Pollut. 2005, 136, 323.
| Preliminary screening of perfluorooctane sulfonate (PFOS) and other fluorochemicals in fish, birds and marine mammals from Greenland and the Faroe Islands.Crossref | GoogleScholarGoogle Scholar |
[90] J. Verreault, U. Berger, G. W. Gabrielsen, Trends of perfluorinated alkyl substances in herring gull eggs from two coastal colonies in Northern Norway: 1983–2003. Environ. Sci. Technol. 2007, 41, 6671.
| Trends of perfluorinated alkyl substances in herring gull eggs from two coastal colonies in Northern Norway: 1983–2003.Crossref | GoogleScholarGoogle Scholar |
[91] E. P. Jones, L. G. Anderson, J. H. Swift, Distribution of Atlantic and Pacific waters in the upper Arctic Ocean: implications for circulation. Geophys. Res. Lett. 1998, 25, 765.
| Distribution of Atlantic and Pacific waters in the upper Arctic Ocean: implications for circulation.Crossref | GoogleScholarGoogle Scholar |
[92] E. P. Jones, J. H. Swift, L. G. Anderson, M. Lipizer, G. Civitarese, K. K. Falkner, G. Kattner, F. McLaughlin, Tracing Pacific water in the North Atlantic Ocean. J. Geophys. Res. – Oceans 2003, 108, 3114.
| Tracing Pacific water in the North Atlantic Ocean.Crossref | GoogleScholarGoogle Scholar |
[93] C. J. Young, V. I. Furdui, J. Franklin, R. M. Koerner, D. C. G. Muir, S. A. Mabury, Perfluorinated acids in Arctic snow: new evidence for atmospheric formation. Environ. Sci. Technol. 2007, 41, 3455.
| Perfluorinated acids in Arctic snow: new evidence for atmospheric formation.Crossref | GoogleScholarGoogle Scholar |
[94] I. Weinberg, A. Dreyer, R. Ebinghaus, Landfills as sources of polyfluorinated compounds, polybrominated diphenyl ethers and musk fragrances to ambient air. Atmos. Environ. 2011, 45, 935.
| Landfills as sources of polyfluorinated compounds, polybrominated diphenyl ethers and musk fragrances to ambient air.Crossref | GoogleScholarGoogle Scholar |
[95] L. Ahrens, U. Siebert, R. Ebinghaus, Temporal trends of polyfluoroalkyl compounds in harbor seals (Phoca vitulina) from the German Bight, 1999–2008. Chemosphere 2009, 76, 151.
| Temporal trends of polyfluoroalkyl compounds in harbor seals (Phoca vitulina) from the German Bight, 1999–2008.Crossref | GoogleScholarGoogle Scholar |
[96] S. G. O’Connell, M. Arendt, A. Segars, T. Kimmel, J. Braun-McNeill, L. Avens, B. Schroeder, L. Ngai, J. R. Kucklick, J. M. Keller, Temporal and spatial trends of perfluorinated compounds in juvenile loggerhead sea turtles (Caretta caretta) along the east coast of the United States. Environ. Sci. Technol. 2010, 44, 5202.
| Temporal and spatial trends of perfluorinated compounds in juvenile loggerhead sea turtles (Caretta caretta) along the east coast of the United States.Crossref | GoogleScholarGoogle Scholar |
[97] K. Hart, K. Kannan, T. Isobe, S. Takahashi, T. K. Yamada, N. Miyazaki, S. Tanabe, Time trends and transplacental transfer of perfluorinated compounds in melon-headed whales stranded along the Japanese coast in 1982, 2001/2002, and 2006. Environ. Sci. Technol. 2008, 42, 7132.
| Time trends and transplacental transfer of perfluorinated compounds in melon-headed whales stranded along the Japanese coast in 1982, 2001/2002, and 2006.Crossref | GoogleScholarGoogle Scholar |
[98] H. Ishibashi, H. Iwata, E. Y. Kim, L. Tao, K. Kannan, M. Amano, N. Miyazaki, S. Tanabe, V. B. Batoev, E. A. Petrov, Contamination and effects of perfluorochemicals in baikal seal (Pusa sibirica). 1. Residue level, tissue distribution, and temporal trend. Environ. Sci. Technol. 2008, 42, 2295.
| Contamination and effects of perfluorochemicals in baikal seal (Pusa sibirica). 1. Residue level, tissue distribution, and temporal trend.Crossref | GoogleScholarGoogle Scholar |
[99] V. I. Furdui, P. A. Helm, P. W. Crozier, C. Lucaciu, E. J. Reiner, C. H. Marvin, D. M. Whittle, S. A. Mabury, G. T. Tomy, Temporal trends of perfluoroalkyl compounds with isomer analysis in lake trout from Lake Ontario (1979–2004). Environ. Sci. Technol. 2008, 42, 4739.
| Temporal trends of perfluoroalkyl compounds with isomer analysis in lake trout from Lake Ontario (1979–2004).Crossref | GoogleScholarGoogle Scholar |
[100] L. Ahrens, D. Herzke, S. Huber, J. O. Bustnes, G. Bangjord, R. Ebinghaus, Temporal trends and pattern of polyfluoroalkyl compounds in tawny owl (Strix aluco) eggs from Norway, 1986–2009. Environ. Sci. Technol. 2011, in press. [Published online ahead of 18 January 2011]
[101] A. M. Calafat, L. Y. Wong, Z. Kuklenyik, J. A. Reidy, L. L. Needham, Polyfluoroalkyl chemicals in the US population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ. Health Perspect. 2007, 115, 1596.
| Polyfluoroalkyl chemicals in the US population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000.Crossref | GoogleScholarGoogle Scholar |
[102] G. W. Olsen, D. C. Mair, W. K. Reagen, M. E. Ellefson, D. J. Ehresman, J. L. Butenhoff, L. R. Zobel, Preliminary evidence of a decline in perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations in American Red Cross blood donors. Chemosphere 2007, 68, 105.
| Preliminary evidence of a decline in perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations in American Red Cross blood donors.Crossref | GoogleScholarGoogle Scholar |
[103] L. S. Haug, C. Thomsen, G. Becher, Time trends and the influence of age and gender on serum concentrations of perfluorinated compounds in archived human samples. Environ. Sci. Technol. 2009, 43, 2131.
| Time trends and the influence of age and gender on serum concentrations of perfluorinated compounds in archived human samples.Crossref | GoogleScholarGoogle Scholar |
[104] H. M. Spliethoff, L. Tao, S. M. Shaver, K. M. Aldous, K. A. Pass, K. Kannan, G. A. Eadon, Use of newborn screening program blood spots for exposure assessment: declining levels of perfluorinated compounds in New York State infants. Environ. Sci. Technol. 2008, 42, 5361.
| Use of newborn screening program blood spots for exposure assessment: declining levels of perfluorinated compounds in New York State infants.Crossref | GoogleScholarGoogle Scholar |
[105] G. W. Olsen, D. C. Mair, T. R. Church, M. E. Ellefson, W. K. Reagen, T. M. Boyd, R. M. Herron, Z. Medhdizadehkashi, J. B. Nobilett, J. A. Rios, J. L. Butenhoff, L. R. Zobel, Decline in perfluorooctanesulfonate and other polyfluoroalkyl chemicals in American Red Cross adult blood donors, 2000–2006. Environ. Sci. Technol. 2008, 42, 4989.
| Decline in perfluorooctanesulfonate and other polyfluoroalkyl chemicals in American Red Cross adult blood donors, 2000–2006.Crossref | GoogleScholarGoogle Scholar |
[106] R. Vestergren, I. T. Cousins, Tracking the pathways of human exposure to perfluorocarboxylates. Environ. Sci. Technol. 2009, 43, 5565.
| Tracking the pathways of human exposure to perfluorocarboxylates.Crossref | GoogleScholarGoogle Scholar |
[107] H. Lee, J. D’eon, S. A. Mabury, Biodegradation of polyfluoroalkyl phosphates as a source of perfluorinated acids to the environment. Environ. Sci. Technol. 2010, 44, 3305.
| Biodegradation of polyfluoroalkyl phosphates as a source of perfluorinated acids to the environment.Crossref | GoogleScholarGoogle Scholar |
[108] K. Hart, V. A. Gill, K. Kannan, Temporal trends (1992–2007) of perfluorinated chemicals in northern sea otters (Enhydra lutris kenyoni) from south-central Alaska. Arch. Environ. Contam. Toxicol. 2009, 56, 607.
| Temporal trends (1992–2007) of perfluorinated chemicals in northern sea otters (Enhydra lutris kenyoni) from south-central Alaska.Crossref | GoogleScholarGoogle Scholar |
[109] R. Bossi, F. F. Riget, R. Dietz, Temporal and spatial trends of perfluorinated compounds in ringed seal (Phoca hispida) from Greenland. Environ. Sci. Technol. 2005, 39, 7416.
| Temporal and spatial trends of perfluorinated compounds in ringed seal (Phoca hispida) from Greenland.Crossref | GoogleScholarGoogle Scholar |
[110] R. Loos, J. Wollgast, T. Huber, G. Hanke, Polar herbicides, pharmaceutical products, perfluorooctanesulfonate (PFOS), perfluorooctanoate (PFOA), and nonylphenol and its carboxylates and ethoxylates in surface and tap waters around Lake Maggiore in Northern Italy. Anal. Bioanal. Chem. 2007, 387, 1469.
| Polar herbicides, pharmaceutical products, perfluorooctanesulfonate (PFOS), perfluorooctanoate (PFOA), and nonylphenol and its carboxylates and ethoxylates in surface and tap waters around Lake Maggiore in Northern Italy.Crossref | GoogleScholarGoogle Scholar |
[111] E. Hoehn, M. H. Plumlee, M. Reinhard, Natural attenuation potential of downwelling streams for perfluorochemicals and other emerging contaminants. Water Sci. Technol. 2007, 56, 59.
| Natural attenuation potential of downwelling streams for perfluorochemicals and other emerging contaminants.Crossref | GoogleScholarGoogle Scholar |
[112] C. González-Barreiro, E. Martínez-Carballo, A. Sitka, S. Scharf, O. Gans, Method optimization for determination of selected perfluorinated alkylated substances in water samples. Anal. Bioanal. Chem. 2006, 386, 2123.
| Method optimization for determination of selected perfluorinated alkylated substances in water samples.Crossref | GoogleScholarGoogle Scholar |
[113] M. K. So, Y. Miyake, W. Y. Yeung, Y. M. Ho, S. Taniyasu, P. Rostkowski, N. Yamashita, B. S. Zhou, X. J. Shi, J. X. Wang, J. P. Giesy, H. Yu, P. K. S. Lam, Perfluorinated compounds in the Pearl River and Yangtze River of China. Chemosphere 2007, 68, 2085.
| Perfluorinated compounds in the Pearl River and Yangtze River of China.Crossref | GoogleScholarGoogle Scholar |
[114] J. W. Martin, D. M. Whittle, D. C. G. Muir, S. A. Mabury, Perfluoroalkyl contaminants in a food web from Lake Ontario. Environ. Sci. Technol. 2004, 38, 5379.
| Perfluoroalkyl contaminants in a food web from Lake Ontario.Crossref | GoogleScholarGoogle Scholar |
[115] M. Smithwick, S. A. Mabury, K. R. Solomon, C. Sonne, J. W. Martin, E. W. Born, R. Dietz, A. E. Derocher, R. J. Letcher, T. J. Evans, G. W. Gabrielsen, J. Nagy, I. Stirling, M. K. Taylor, D. C. G. Muir, Circumpolar study of perfluoroalkyl contaminants in polar bears (Ursus maritimus). Environ. Sci. Technol. 2005, 39, 5517.
| Circumpolar study of perfluoroalkyl contaminants in polar bears (Ursus maritimus).Crossref | GoogleScholarGoogle Scholar |
[116] J. W. Martin, M. M. Smithwick, B. M. Braune, P. F. Hoekstra, D. C. G. Muir, S. A. Mabury, Identification of long-chain perfluorinated acids in biota from the Canadian Arctic. Environ. Sci. Technol. 2004, 38, 373.
| Identification of long-chain perfluorinated acids in biota from the Canadian Arctic.Crossref | GoogleScholarGoogle Scholar |
[117] M. Houde, R. S. Wells, P. A. Fair, G. D. Bossart, A. A. Hohn, T. K. Rowles, J. C. Sweeney, K. R. Solomon, D. C. G. Muir, Polyfluoroalkyl compounds in free-ranging bottlenose dolphins (Tursiops truncatus) from the Gulf of Mexico and the Atlantic Ocean. Environ. Sci. Technol. 2005, 39, 6591.
| Polyfluoroalkyl compounds in free-ranging bottlenose dolphins (Tursiops truncatus) from the Gulf of Mexico and the Atlantic Ocean.Crossref | GoogleScholarGoogle Scholar |
[118] J. W. Martin, D. C. G. Muir, C. A. Moody, D. A. Ellis, W. C. Kwan, K. R. Solomon, S. A. Mabury, Collection of airborne fluorinated organics and analysis by gas chromatography/chemical ionization mass spectrometry. Anal. Chem. 2002, 74, 584.
| Collection of airborne fluorinated organics and analysis by gas chromatography/chemical ionization mass spectrometry.Crossref | GoogleScholarGoogle Scholar |
[119] N. L. Stock, F. K. Lau, D. A. Ellis, J. W. Martin, D. C. G. Muir, S. A. Mabury, Polyfluorinated telomer alcohols and sulfonamides in the North American troposphere. Environ. Sci. Technol. 2004, 38, 991.
| Polyfluorinated telomer alcohols and sulfonamides in the North American troposphere.Crossref | GoogleScholarGoogle Scholar |
[120] A. Jahnke, L. Ahrens, R. Ebinghaus, C. Temme, Urban versus remote air concentrations of fluorotelomer alcohols and other polyfluorinated alkyl substances in Germany. Environ. Sci. Technol. 2007, 41, 745.
| Urban versus remote air concentrations of fluorotelomer alcohols and other polyfluorinated alkyl substances in Germany.Crossref | GoogleScholarGoogle Scholar |
[121] J. L. Barber, U. Berger, C. Chaemfa, S. Huber, A. Jahnke, C. Temme, K. C. Jones, Analysis of per- and polyfluorinated alkyl substances in air samples from northwest Europe. J. Environ. Monit. 2007, 9, 530.
| Analysis of per- and polyfluorinated alkyl substances in air samples from northwest Europe.Crossref | GoogleScholarGoogle Scholar |
[122] A. M. Piekarz, T. Primbs, J. A. Field, D. F. Barofsky, S. Simonich, Semivolatile fluorinated organic compounds in Asian and western US air masses. Environ. Sci. Technol. 2007, 41, 8248.
| Semivolatile fluorinated organic compounds in Asian and western US air masses.Crossref | GoogleScholarGoogle Scholar |
[123] A. Dreyer, I. Weinberg, C. Temme, R. Ebinghaus, Polyfluorinated compounds in the atmosphere of the Atlantic and Southern Oceans: evidence for a global distribution. Environ. Sci. Technol. 2009, 43, 6507.
| Polyfluorinated compounds in the atmosphere of the Atlantic and Southern Oceans: evidence for a global distribution.Crossref | GoogleScholarGoogle Scholar |
[124] M. Shoeib, P. Vlahos, T. Hamer, A. Peters, M. Graustein, J. Narayan, Survey of polyfluorinated chemicals (PFCs) in the atmosphere over the Northeast Atlantic Ocean. Atmos. Environ. 2010, 44, 2887.
| Survey of polyfluorinated chemicals (PFCs) in the atmosphere over the Northeast Atlantic Ocean.Crossref | GoogleScholarGoogle Scholar |
[125] A. Dreyer, R. Ebinghaus, Polyfluorinated compounds in ambient air from ship- and land-based measurements in northern Germany. Atmos. Environ. 2009, 43, 1527.
| Polyfluorinated compounds in ambient air from ship- and land-based measurements in northern Germany.Crossref | GoogleScholarGoogle Scholar |