

Why do organic aerosols exist? Understanding aerosol lifetimes using the two-dimensional volatility basis set
N. M. Donahue A G , W. Chuang A , S. A. Epstein A F , J. H. Kroll B , D. R. Worsnop C D , A. L. Robinson A , P. J. Adams A and S. N. Pandis A EA Center for Atmospheric Particle Studies, Carnegie Mellon University, 5000 Forbes Avenue, Pittsburgh, PA 15217, USA.
B Department of Civil and Environmental Engineering and Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA.
C Aerodyne Research, Inc., 45 Manning Road, Billerica, MA 01821, USA.
D University of Helsinki, Department of Physics, FI-00014 Helsinki, Finland.
E Department of Chemical Engineering, University of Patras, Patra, GR-26500, Greece.
F Present address: Department of Chemistry, 1102 Natural Sciences 2, University of California, Irvine, CA 92697, USA.
G Corrsponding author. Email: nmd@andrew.cmu.edu
Environmental Chemistry 10(3) 151-157 https://doi.org/10.1071/EN13022
Submitted: 30 January 2013 Accepted: 26 April 2013 Published: 14 June 2013
Environmental context. Fine particles (aerosols) containing organic compounds are central players in two important environmental issues: aerosol-climate effects and human health effects (including mortality). Although organics constitute half or more of the total fine-particle mass, their chemistry is extremely complex; of critical importance is ongoing oxidation chemistry in both the gas phase and the particle phase. Here we present a method for representing that oxidation chemistry when the actual composition of the organics is not known and show that relatively slow oxidant uptake to particles plays a key role in the very existence of organic aerosols.
Abstract. Organic aerosols play a critical role in atmospheric chemistry, human health and climate. Their behaviour is complex. They consist of thousands of organic molecules in a rich, possibly highly viscous mixture that may or may not be in phase equilibrium with organic vapours. Because the aerosol is a mixture, compounds from all sources interact and thus influence each other. Finally, most ambient organic aerosols are highly oxidised, so the molecules are secondary products formed from primary emissions by oxidation chemistry and possibly non-oxidative association reactions in multiple phases, including gas-phase oxidation, aqueous oxidation, condensed (organic) phase reactions and heterogeneous interactions of all these phases. In spite of this complexity, we can make a strong existential statement about organic aerosol: They exist throughout the troposphere because heterogeneous oxidation by OH radicals is more than an order of magnitude slower than comparable gas-phase oxidation.
Introduction
Organic aerosols are meta-stable intermediates. Most organic emissions are highly reduced,[1,2] and all organics oxidise by complex oxidation pathways in the Earth’s atmosphere.[3–6] Oxidation would convert all reduced carbon in the atmosphere into CO2, given time. Consequently, to first order, the concentration of any given constituent (Ci) will be in a pseudo-steady-state at long enough timescales; given a production rate Pi and a first-order lifetime τi, the pseudo steady-state concentration Ciss will be:

This means that, for a succession of molecules connected by a simple reaction sequence, Mi → Mj → Mk with a shared production rate Pi, the steady-state concentrations of each will be directly proportional to the lifetime. More generally, the total concentration of species at a given generation number in a reaction sequence is related to the (properly weighted) average lifetime of the species in that generation. This simple equation governs organic-aerosol levels, and it shows that the atmospheric lifetime is critical.
To apply Eqn 1 we need to know the lifetimes. Unsaturated organic molecules are typically very short lived, and for the most part the double bonds will be eliminated rapidly.[7] Our main focus here is on later-generation chemistry of organic compounds, where the dominant oxidant is the OH radical. The chemical lifetime of organic vapours is governed by their rate constant for reaction with OH• (kiOH), and the lifetime against oxidation by OH• will be τi = 1/(kiOH COH). For the purposes of this discussion we shall consider COH = ~2 × 106 molecules cm–3, which is a typical daytime average value.[8] If the chemical lifetime is shorter than the deposition timescale, oxidation will in turn dominate the overall atmospheric lifetime. The job of estimating chemical lifetimes thus reduces to estimating OH• oxidation rate constants.
Rate constant estimation faces two challenges. First, the most important rate constants in this exercise involve the reaction of OH• with oxygenated organics, especially those containing multiple functional groups. These are generally large molecules by atmospheric standards, with 5–25 carbon atoms. There are very few kinetic data to draw on.[7] Second, there are thousands, even millions of specific organic molecules involved in chemistry associated with organic aerosols.[1,9] We thus have unmeasured rate constants for unknown molecules.
There are at least two solutions to this problem. One is to take what we do know about the kinetics and mechanisms of organic molecules and train mechanism generators to fill in the rest, connecting precursor molecules to all intermediates and ultimately to CO2.[10] Another is to reduce the complexity by lumping the organics into groupings with similar properties. Here we choose the second approach, employing a discretised two-dimensional space consisting of volatility (saturation concentration, Co in micrograms per cubic metre as the x coordinate and average carbon oxidation state (
Methods
The 2D-VBS facilitates consideration of organic oxidation and phase partitioning without requiring specific knowledge of molecular structures, even carbon numbers. The variables themselves are the fundamental quantities describing phase partitioning and oxidation chemistry. Organic aerosol levels are almost always between 1 and 100 µg m–3, and consequently for organics to reside in the aerosol phase they must have a saturation mass concentration, C* < ~100 µg m–3. Volatility as C* is not the pure-component vapour pressure; at a minimum it includes activity coefficients of organics in the aerosol mixture (Ci* = γi Cio), and more generally it includes any factors governing the equilibrium phase partitioning.[11] The relevant volatility is usually that of any product molecule in the condensed phase (i.e. ammonium oxalate or some other organic salt and not oxalic acid itself).
The equilibrium state says something about organic aerosols; however, the potential for organic aerosols to be out of equilibrium is currently under debate. It is possible that extremely low viscosity (i.e. a glassy state) may inhibit mass transfer within particles.[13–18] However, mass exchange between particles and the gas phase remains a critical topic, and particles do grow and shrink.[19] Considering uptake and condensational growth, most organic molecules that collide with particles do not stick to them. The property governing which compounds stay in the aerosol phase is volatility, and thus the x-axis of the 2D-VBS informs which phase the organic material will be in.
The y-axis of the 2D-VBS concerns oxidation. The oxidation state (
Because we use the 2D-VBS framework, in order to assess organic lifetimes we need to estimate the OH• oxidation rate constants of organics in 2D-VBS bins. Fortunately, we do know something about composition (carbon, hydrogen and oxygen numbers, nC, nH, nO, as well as the more tightly defined O : C and H : C ratios) in the 2D-VBS.[11] There is also strong evidence that the oxidised functional groups consist of approximately equal numbers of =O and –OH groups (the organic compounds appear to be, on average, either hydroxyketones or organic acids or more likely a mixture of both).[30,31] This is enough for us to begin to say something about the gas-phase OH• reactivity in the 2D-VBS. To do this we draw upon and simplify structure–activity relations, starting along the lower edge of the 2D-VBS where the organics are simply hydrocarbons.
We do not know the structure of the typical organics in the 2D-VBS, so we rely on a statistical sampling of kinetic data in the (limited) regime of the 2D-VBS where kinetics are available. The kinetics are distilled by the structure–activity relationship (SAR) of Kwok and Atkinson,[32] which we sample randomly with 35 000 molecules to maintain the proper C : H : O relationships (on average) in the 2D-VBS. However, the basis for the SAR consists almost entirely of small molecules, most with either zero or one oxygenated functional group. To extrapolate this over the full range of the 2D-VBS we smooth the resulting rate-constant surface with a functional form based on three simple assumptions: (1) OH• oxidation rate constants typically increase with increasing carbon number; (2) adding oxygenated functional groups for lightly oxygenated molecules typically increases the OH• reactivity and (3) highly oxygenated molecules ultimately become less reactive due simply to a scarcity of abstractable hydrogens. This final assumption is supported by heterogeneous OH• oxidation experiments on highly oxygenated aerosol surrogates, such as citric acid, which show it to be recalcitrant to oxidation.[33]
The resulting (vapour-phase) rate-constant (kvap, cm3 molecule–1 s–1) expression is given by Eqn 2 and plotted in the 2D-VBS in Fig. 1.
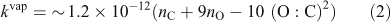

|
The first effect (increasing k with increasing nC) for homologous molecules is very robust.[32] The second two effects counteract each other. We express them with one term dependent on nO alone and a second one dependent on (O : C)2, which is a measure of the H-atom scarcity. Although the specifics of the functional form in Eqn 2 and the actual rate constants must be regarded as quite uncertain, overall Fig. 1 reveals rate constants that vary only modestly throughout the space. The major feature is that kOH increases as Co decreases, but the overall effect is less than a 1 order of magnitude increase in k over a 15 decade decrease in Co. Furthermore, although we include the volatile organic carbon (VOC) region in the 2D-VBS (Co > 3 × 106 µg m–3) for completeness, no realistic simulation would actually use it for a numerical simulation in that region – VOC chemistry is represented by a standard chemical mechanism.
Results and discussion
In Fig. 2a we show the gas-phase chemical lifetime, τ = 1/(kCOH), in days in the 2D-VBS space, assuming COH = ~2 × 106 molecules cm–3. The semi-volatile range is 0.3 ≤ Co ≤ 300 μg m–3, organics with lower Co values will be almost completely in the condensed phase under typical conditions. The critical point is that organic species over the entire range of volatilities representative of aerosols have a gas-phase lifetime of less than 0.2 days. Thus, aerosol species would evolve through upwards of five generations of chemistry in 24 h, if they were in the gas phase. Because the probability of fragmentation and consequent sharp increases in volatility rises rapidly with increasing oxidation state,[25–27] this unrestrained oxidation would sweep the system clean of organic aerosols within a day or two. For example, a C10 backbone, with on average 1.5 oxygen atoms added per generation, will have a simple fragmentation probability (O : C)1/4 of 0, 0.62, 0.74, 0.82, 0.88 and 0.93 for generations 0–5.

|
Organic species, however, do not reside solely in the gas phase. The less volatile ones condense to form organic aerosol, and in the condensed phase they are protected from gas-phase OH• by the diffusion rate of OH• to the particles. For the purposes of this work, the heterogeneous uptake of OH• to particles can be converted into an effective gas-phase rate constant, which depends on particle size.[34,35] Here we assume that OH• will react with unit efficiency with the first organic species it encounters in a particle, and thus the effective rate constant will be independent of composition.[36] For 500-nm diameter particles, the effective rate constant will be keff = 1 × 10–12 cm3 molecule–1 s–1, resulting in an oxidation lifetime of 5.8 days, whereas organics in 200-nm diameter particles would have a lifetime of ~3 days. This will apply regardless of the condensed-phase composition, provided that the pseudo-ideal solution theory relevant to Co holds and that the near-surface particle composition is approximately the same as the bulk organic phase composition. This will be true if the particle viscosity is not so high as to significantly limit condensed-phase mass transfer to the particle surface; the timescale for diffusion within the particles must be faster than the timescale for heterogeneous oxidation (6 days in this example).[36,37] Evidence from relative rate experiments of mixtures made by coating primary organic particles with laboratory secondary organic aerosol (SOA) supports this assumption.[38]
The condensed-phase lifetime of 6 days is thus much longer than the ~0.1-day gas-phase lifetime of the organic molecules in the low-volatility range. However, it is still comparable to the residence time of particles in the atmosphere, so heterogeneous oxidation will play a role even for organics that are more or less homogeneously distributed in the condensed phase. The overall average lifetime of semi-volatile organics will depend on their fraction (ξi) in the condensed phase:
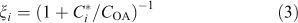
which gives an overall rate constant (kitot) based on the effective heterogeneous rate constant (keff) and the gas-phase rate constant (kivap) from Eqn 2.
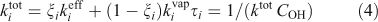
The overall lifetimes including the gas-phase diffusion limitations for heterogeneous oxidation are shown in Fig. 2b for a case with concentration of organic aerosol COA = 1 µg m –3. Based on Eqn 1, the steady-state concentration of species sequestered in particles will thus be ~30 times higher than it would otherwise be if they were oxidised at the rate given by pure gas-phase oxidation shown in Fig. 2a.
In this work we are considering heterogeneous oxidation by OH radical uptake only. Especially at higher relative humidity (and certainly in droplets), aqueous-phase reactions will also contribute to oxidation; it is unclear whether the net effect of these processes will augment or deplete the total organic-aerosol mass, although oxidation of soluble volatile species can clearly contribute to SOA.[39,40] For example, levoglucosan can be oxidised in most particles and droplets with lifetimes in the order of days.[41] However, with a saturation concentration of the order of 7 µg m–3,[42] levoglucosan will reside principally in the gas phase under many circumstances.[12] As Eqn 4 and experimental data show,[43] the majority of oxidation is likely to be by gas-phase OH• reactions. This will apply to other organics even well into the low volatility organic compound region (LVOC) region.
The lifetime contours in Fig. 2b show something profound. The sharp increase in lifetimes with decreasing volatility and thus decreasing gas-phase fractions is the most dramatic feature of the figure (note that the influence of gas-phase oxidation extends to quite low Co). However, just as notable is the broad area of minimum lifetime for semi-volatile organic compounds (SVOC range in green) and intermediate volatility organic compounds (IVOC range in blue). Until recently, many atmospheric models treating SVOCs contained semi-volatile lumped species whose properties (C* and molar yields) were based on empirical fits to smog-chamber data. Often these species were effectively immortal in the model, or at most suffered from deposition to the surface. In fact, these SVOCs have a minimum lifetime and thus will have a minimum steady-state concentration of all molecules in a given reaction sequence. Atmospheric chemistry will thus tend to sweep out the SVOC range with progressive aging downwind of a source region, forming either much lower volatility functionalised products or more volatile fragmentation products and thus producing very low volatility, highly oxidised aerosol in the remote atmosphere. This is exactly what is observed.[29,44]
The lifetime of individual organic molecules is not the same as the lifetime of the aerosol mass itself. So, in order to test our assertion that without condensed-phase sequestration the atmosphere would be rapidly swept clear of organic aerosol, we simulated the evolution of a SOA plume in a simple box model. The model is initiated by SOA formation by ozonolysis of α-pinene, assuming 30 ppb of ozone and 20 µg m–3 of α-pinene (~2 ppbv). The initial fresh SOA formation and subsequent aging chemistry follow the yields and chemistry described previously,[29,45,46] and the model (including slow heterogeneous oxidation) matches observed aging behaviour well.[47] Briefly, OH• reacts with semi-volatile vapours with the rate constants described above. The resulting reaction products are distributed according to two oxidation ‘kernels’, with a ‘functionalisation’ fraction distributed exclusively to lower C* values and a ‘fragmentation’ fraction dispersed widely in volatility space. The probability of fragmentation is assumed to vary as (O : C)1/4, so as organics become highly oxidised they will inevitably fragment towards CO2, which is required by thermodynamics.
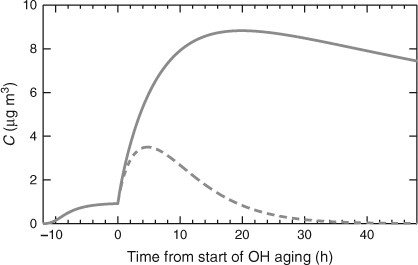
The results of our box-model simulation are shown in Fig. 3. The α-pinene was allowed to react with ozone in the dark for 12 h, forming ~1 µg m–3 of ‘fresh’ SOA with a mass yield of ~5 %. At t = 0 the OH• concentration was suddenly increased to 2 × 106 molecules cm–3. In the base-case simulation the OH• oxidation rate constants are given by Eqn 4. We assume that the mechanism (the product distribution) is not affected by whether the OH• reaction occurs homogeneously in the gas phase or heterogeneously near the particle surface. This is almost certainly an over simplification, but there are insufficient data to describe the chemistry in more detail. The solid curve in Fig. 3 shows the organic aerosol evolution as this aging chemistry progresses. With most of the first-generation ozonolysis products in the vapour phase, the aging causes a dramatic, almost 10-fold, increase in aerosol concentrations over the first half day, but the subsequent aging chemistry is slow because of the slow heterogeneous oxidation. This is broadly consistent with published smog-chamber experiments on SOA aging, although the magnitude of the fractional mass increase is large because of the very low (for a chamber experiment) initial SOA mass loading.[47]
|
In the second, dashed curve in Fig. 3, the heterogeneous aging keff is artificially made equal to the gas-phase OH• oxidation rate constant, so the oxidation proceeds unimpeded by diffusion limitations. In this case the organic aerosol rapidly dissipates, vanishing in ~1 day. This dramatic difference in behaviour shows that the simple steady-state analysis described by Eqn 1 gives a valid picture for bulk organic-aerosol concentrations as well. A typical average age of aerosols in even an urban setting such as Pittsburgh is greater than 2 days,[48] and the ratio in modelled organic aerosol concentrations for 0 ≤ t ≤ 48 h in Fig. 3 is more than a factor of five.
The calculations presented so far have been for ‘typical’ ambient conditions, but in certain times and places the atmosphere can be far more oxidising. One example is the eastern Mediterranean. During the MINOS campaign at the Finokalia research station in Crete in August 2001, ambient OH• levels reached 2 × 107 molecules cm–3.[49] Under these extreme conditions, the gas-phase lifetimes of SVOC within the 2D-VBS are less than 1 h, and even the heterogeneous oxidation timescale is just over 12 h, as shown in Fig. 4 – we show lifetimes for the diurnal maximum OH• levels because under these conditions the oxidation timescales are so fast that the diurnal average is barely meaningful. We thus expect organic aerosols to become completely oxidised very quickly, unless some compounds are able to ‘hide’ in the particles because they are so viscous that they cannot reach the surface to be oxidised.[14,36]

|
Organic-aerosol observations using a unit mass resolution aerosol mass spectrometer (Q-AMS, Aerodyne Inc., Billerica, MA, USA) during the EUCAARI campaigns in May 2008 and February 2009 show that in the late spring organic aerosols arriving at Finokalia were completely oxidised, with no evidence of any primary organic aerosol,[50] whereas Q-AMS observations at the same site in winter 2009 showed significant levels of primary organic aerosol.[44] The highly oxidised aerosols during late spring 2008 were observed even when air-mass back trajectories showed that the sampled air had been in the boundary layer over Athens less than 2 days earlier. The very short lifetimes shown in Fig. 4 confirm this, and the almost complete absence of unoxidised peaks in the mass spectra (even for air recently over Athens) shows that diffusion limitations do not protect extremely low volatility primary compounds within the particles under ambient conditions. Given that the heterogeneous oxidation timescale in Fig. 4 is ~12 h, this suggests that the diffusional timescales for organics within the aerosols sampled during FAME-08 were faster than this. That in turn suggests a diffusion constant D ≥ 10–14 cm 2 s–1, or a dynamic viscosity η ≤ 3 × 105 Pa s,[15] even for quite highly oxidised organic aerosol.
Environmental implications
Oxidation of organic compounds, carried to completion, will form CO2, and consequently oxidation is fundamentally destructive to organic aerosol. However, as Fig. 3 shows, oxidation clearly is a source of organic aerosol as well. Consequently, organic aerosols can only be viewed as meta-stable intermediates.[1] Its concentrations are thus controlled by its lifetime, and as we show the fundamental governor of this lifetime is the delay caused by diffusion of OH radicals to aerosols. Without this, organic aerosol levels would be many times lower than they are, rendering organics virtually irrelevant to aerosol concentrations and properties.
Conversely, although organic compounds can run, they cannot hide. Sequestration into the condensed phase delays oxidation because of diffusion limitations in the gas phase, but heterogeneous oxidation is still important. The near complete oxidation observed in situations where transport occurs within ~1 day from intense sources indicates that the diffusion timescale for organics within particles is (in at least some cases relevant to the atmosphere) less than 1 day.
Acknowledgements
This work was funded by the National Science Foundation, the DOE ASR program and the EPASTAR program.
References
[1] J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. Kessler, M. R. Canagaratna, K. Wilson, K. E. Alteri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb, D.R. Worsnop, Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat. Chem. 2011, 3, 133.| Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovVGhuw%3D%3D&md5=411e8c0cd7c47820db1acdc81ccb0291CAS | 21258386PubMed |
[2] A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons, X. Wang, The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions. Geoscientific Model Development 2012, 5, 1471.
| The model of emissions of gases and aerosols from nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions.Crossref | GoogleScholarGoogle Scholar |
[3] R. Kamens, H. Jeffries, M. Gery, R. Wiener, K. Sexton, G. Howe, The impact of α-pinene on urban smog formation – an outdoor smog chamber study. Atmos. Environ. 1981, 15, 969.
| The impact of α-pinene on urban smog formation – an outdoor smog chamber study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXlsFGmurc%3D&md5=d03a77fd870aab4df560d94cd6886f57CAS |
[4] H. Jeffries, S. Tonnesen, A comparison of 2 photochemical-reaction mechanisms using mass-balance and process analysis. Atmos. Environ. 1994, 28, 2991.
| A comparison of 2 photochemical-reaction mechanisms using mass-balance and process analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXntVOiu7Y%3D&md5=9b642e4b25fdf2006c1e86c3a1559803CAS |
[5] R. M. Kamens, M. Jaoui, Modeling aerosol formation from α-pinene + NOx in the presence of natural sunlight using gas-phase kinetics and gas-particle partitioning theory. Environ. Sci. Technol. 2001, 35, 1394.
| Modeling aerosol formation from α-pinene + NOx in the presence of natural sunlight using gas-phase kinetics and gas-particle partitioning theory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhsFKisb4%3D&md5=3a7d40f431accc009459b782e1046763CAS | 11348073PubMed |
[6] W. Vizuete, H. E. Jeffries, T. W. Tesche, E. P. Olaguer, E. Couzo, Issues with ozone attainment methodology for Houston, TX. J. Air Waste Manag. Assoc. 2011, 61, 238.
| Issues with ozone attainment methodology for Houston, TX.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktFantrg%3D&md5=975db8a82d1421bec2e974bb0f1f5b3eCAS | 21416750PubMed |
[7] R. Atkinson, Gas phase tropospheric chemistry of organic compounds. J. Phys. Chem. Ref. Data 1997, 26, 215.
| Gas phase tropospheric chemistry of organic compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXivVKjt7c%3D&md5=d817872e808dd158fd76aaf166e469f1CAS |
[8] R. Prinn, J. Huang, R. Weiss, D. Cunnold, P. Fraser, P. Simmonds, A. McCulloch, C. Harth, S. Reimann, P. Salameh, S. O’Doherty, R. Wang, L. Porter, B. Miller, P. Krummel, Evidence for variability of atmospheric hydroxyl radicals over the past quarter century. Geophys. Res. Lett. 2005, 32, L07809.
| Evidence for variability of atmospheric hydroxyl radicals over the past quarter century.Crossref | GoogleScholarGoogle Scholar |
[9] A. H. Goldstein, I. E. Galbally, Known and unexplored organic constituents in the Earth’s atmosphere. Environ. Sci. Technol. 2007, 41, 1514.
| Known and unexplored organic constituents in the Earth’s atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXit1Cnuro%3D&md5=bb559320fbd5146133774d732702004cCAS | 17396635PubMed |
[10] B. Aumont, S. Szopa, S. Madronich, Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach. Atmos. Chem. Phys. 2005, 5, 2497.
| Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Kgs7rK&md5=b7731775640661953cf3155ee0d7b99cCAS |
[11] N. M. Donahue, S. A. Epstein, S. N. Pandis, A. L. Robinson, A 2-dimensional volatility basis set: 1. Organic mixing thermodynamics. Atmos. Chem. Phys. 2011, 11, 3303.
| A 2-dimensional volatility basis set: 1. Organic mixing thermodynamics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFWnt7k%3D&md5=88c8540f7bf39a565336ea6d632aeb42CAS |
[12] N. M. Donahue, J. H. Kroll, A. L. Robinson, S. N. Pandis, A 2-dimensional volatility basis set: 2. Diagnostics of laboratory and ambient organic aerosol. Atmos. Chem. Phys. 2012, 12, 615.
| A 2-dimensional volatility basis set: 2. Diagnostics of laboratory and ambient organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XltFShtLg%3D&md5=2d5e8832d506c2d707350bf0b7ecee3dCAS |
[13] B. Zobrist, C. Marcolli, D. A. Pedernera, T. Koop, Do atmospheric aerosols form glasses? Atmos. Chem. Phys. 2008, 8, 5221.
| Do atmospheric aerosols form glasses?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlCntbfL&md5=ead15b08ae8e1655ac09b7723699b395CAS |
[14] A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirila, J. Leskinen, J. M. Makela, J. K. Holopainen, U. Poeschl, M. Kulmala, D. R. Worsnop, A. Laaksonen, An amorphous solid state of biogenic secondary organic aerosol particles. Nature 2010, 467, 824.
| An amorphous solid state of biogenic secondary organic aerosol particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1yhsLnN&md5=4b3f0c1639337cc4b61d104f9f5ca929CAS | 20944744PubMed |
[15] T. Koop, J. Bookhold, M. Shiraiwa, U. Poeschl, Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere. Phys. Chem. Chem. Phys. 2011, 13, 19238.
| Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlKjurjI&md5=ed7c11497d7a64b5803a4e4119f5759eCAS | 21993380PubMed |
[16] T. D. Vaden, D. Imre, J. Ber’anek, M. Shrivastava, A. Zelenyuk, Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol. Proc. Natl. Acad. Sci. USA 2011, 108, 2190.
| Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitFKqt7o%3D&md5=cdf7fa52da4f9ca7663b16cc5a4a6ae4CAS | 21262848PubMed |
[17] M. Shiraiwa, M. Ammann, T. Koop, U. Poeschl, Gas uptake and chemical aging of semisolid organic aerosol particles. Proc. Natl. Acad. Sci. USA 2011, 108, 11 003.
| Gas uptake and chemical aging of semisolid organic aerosol particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptV2ht7o%3D&md5=5b9bc22339f34cfe9308d8d6582cc9c1CAS |
[18] V. Perraud, E. A. Bruns, M. J. Ezell, S. N. Johnson, Y. Yu, M. L. Alexander, A. Zelenyuk, D. Imre, W. L. Chang, D. Dabdub, J. F. Pankow, B. J. Finlayson-Pitts, Nonequilibrium atmospheric secondary organic aerosol formation and growth. Proc. Natl. Acad.Sci. USA 2012, 109, 2836.
| Nonequilibrium atmospheric secondary organic aerosol formation and growth.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjsFyisrk%3D&md5=234ff351a64c52676e6d05c792314385CAS | 22308444PubMed |
[19] A. P. Grieshop, N. M. Donahue, A. L. Robinson, Is the gas-particle partitioning in α-pinene secondary organic aerosol reversible? Geophys. Res. Lett. 2007, 34, L14810.
| Is the gas-particle partitioning in α-pinene secondary organic aerosol reversible?Crossref | GoogleScholarGoogle Scholar |
[20] J. A. Logan, M. J. Prather, S. C. Wofsy, M. B. McElroy, Tropospheric chemistry: a global perspective. J. Geophys. Res. – Atmos. 1981, 86, 7210.
| Tropospheric chemistry: a global perspective.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXlslCmtro%3D&md5=48aefba3f2338dfba46569596be17494CAS |
[21] Y. Rudich, Laboratory perspectives on the chemical transformations of organic matter in atmospheric particles. Chem. Rev. 2003, 103, 5097.
| Laboratory perspectives on the chemical transformations of organic matter in atmospheric particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXmsl2gu7Y%3D&md5=cc6af945c99341c075e638a2af3f5fb3CAS | 14664645PubMed |
[22] Y. Rudich, N. M. Donahue, T. F. Mentel, Aging of organic aerosol: bridging the gap between laboratory and field studies. Annu. Rev. Phys. Chem. 2007, 58, 321.
| Aging of organic aerosol: bridging the gap between laboratory and field studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlslSitrY%3D&md5=803b60d7d15db857f9e72799004bbea6CAS | 17090227PubMed |
[23] M. Kalberer, D. Paulsen, M. Sax, M. Steinbacher, J. Dommen, A. S. H. Prévôt, R. Fisseha, E. Weingartner, V. Frankevic, R. Zenobi, U. Baltensperger, Identification of polymers as major components of atmospheric organic aerosols. Science 2004, 303, 1659.
| Identification of polymers as major components of atmospheric organic aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhvFCnsbc%3D&md5=22b3ffa7e5313fa78091bc3619175ae8CAS | 15016998PubMed |
[24] M. Tolocka, M. Jang, J. Ginter, F. Cox, R. Kamens, M. Johnston, Formation of oligomers in secondary organic aerosol. Environ. Sci. Technol. 2004, 38, 1428.
| Formation of oligomers in secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXntVaksw%3D%3D&md5=db598c55b8b61241fd9ee7fb51d488cbCAS | 15046344PubMed |
[25] J. H. Kroll, J. D. Smith, D. L. Che, S. H. Kessler, D. R. Worsnop, K. R. Wilson, Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol. Phys. Chem. Chem. Phys. 2009, 11, 8005.
| Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOrsLzF&md5=64f9a1b3081e1fd55ce59a27a87effa3CAS | 19727507PubMed |
[26] H. J. Chacon-Madrid, A. A. Presto, N. M. Donahue, Functionalization vs fragmentation: n-aldehyde oxidation mechanisms and secondary organic aerosol formation. Phys. Chem. Chem. Phys. 2010, 12, 13 975.
| Functionalization vs fragmentation: n-aldehyde oxidation mechanisms and secondary organic aerosol formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSqu7bF&md5=8fc5d098d481daace399f1401971e402CAS |
[27] H. J. Chacon-Madrid, N. M. Donahue, Fragmentation v. functionalization: chemical aging and organic aerosol formation. Atmos. Chem. Phys. 2011, 11, 10 553.
| Fragmentation v. functionalization: chemical aging and organic aerosol formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtlSjtrw%3D&md5=168c7634ed4990fa54271d3d7f46cefaCAS |
[28] A. T. Lambe, T. B. Onasch, D. R. Croasdale, J. P. Wright, A. T. Martin, J. P. Franklin, P. Massoli, J. H. Kroll, M. R. Canagaratna, W. H. Brune, D. R. Worsnop, P. Davidovits, Transitions from functionalization to fragmentation reactions of laboratory secondary organic aerosol (SOA) generated from the OH oxidation of alkane precursors. Environ. Sci. Technol. 2012, 46, 5430.
| Transitions from functionalization to fragmentation reactions of laboratory secondary organic aerosol (SOA) generated from the OH oxidation of alkane precursors.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmtVGktLY%3D&md5=99547f8e5dfb54b9a22b366d952fb49bCAS | 22534114PubMed |
[29] J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prévôt, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. D. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, C. E. Kolb, U. Baltensperger, D. R. Worsnop, Evolution of organic aerosols in the atmosphere: a new framework connecting measurements to models. Science 2009, 326, 1525.
| Evolution of organic aerosols in the atmosphere: a new framework connecting measurements to models.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFensbjE&md5=70447e216a6708480c842ef4b6cac6a3CAS | 20007897PubMed |
[30] C. L. Heald, J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. DeCarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer, P. Artaxo, A simplified description of the evolution of organic aerosol composition in the atmosphere. Geophys. Res. Lett. 2010, 37, L08803.
| A simplified description of the evolution of organic aerosol composition in the atmosphere.Crossref | GoogleScholarGoogle Scholar |
[31] N. L. Ng, M. R. Canagaratna, J. L. Jimenez, P. S. Chhabra, J. H. Seinfeld, D. R. Worsnop, Changes in organic aerosol composition with aging inferred from aerosol mass spectra. Atmos. Chem. Phys. 2011, 11, 6465.
| Changes in organic aerosol composition with aging inferred from aerosol mass spectra.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1yksL3E&md5=8dc244cca7fb9787946e0ef053eb32c3CAS |
[32] E. S. C. Kwok, R. Atkinson, Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: an update. Atmos. Environ. 1995, 29, 1685.
| Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: an update.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmvFCms70%3D&md5=fb28d7e29756c5f9a16e407c80314a3fCAS |
[33] S. H. Kessler, T. Nah, K. E. Daumit, J. D. Smith, S. R. Leone, C. E. Kolb, D. R. Worsnop, K. R. Wilson, J. H. Kroll, OH initiated heterogeneous aging of highly oxidized organic aerosol. J. Phys. Chem. A 2012, 116, 6358.
| OH initiated heterogeneous aging of highly oxidized organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XltVyksb4%3D&md5=bc8ec89d34eac131fe72f769e5fb51b8CAS | 22483038PubMed |
[34] A. T. Lambe, M. A. Miracolo, C. J. Hennigan, A. L. Robinson, N. M. Donahue, Effective rate constants and uptake coefficients for the reactions of organic molecular markers (n-alkanes, hopanes and steranes) in motor oil and diesel primary organic aerosols with OH radicals. Environ. Sci. Technol. 2009, 43, 8794.
| Effective rate constants and uptake coefficients for the reactions of organic molecular markers (n-alkanes, hopanes and steranes) in motor oil and diesel primary organic aerosols with OH radicals.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlWisrfN&md5=44c40614427ea80a4cf532588376c850CAS | 19943648PubMed |
[35] N. M. Donahue, A. L. Robinson, E. R. Trump, I. Riipinen, J. H. Kroll, Volatility and aging of atmospheric organic aerosols. Top. Curr. Chem. in press.
| Volatility and aging of atmospheric organic aerosols.Crossref | GoogleScholarGoogle Scholar |
[36] N. M. Donahue, A. L. Robinson, K. E. Huff Hartz, A. M. Sage, E. A. Weitkamp, Competitive oxidation in atmospheric aerosols: the case for relative kinetics. Geophys. Res. Lett. 2005, 32, L16805.
| Competitive oxidation in atmospheric aerosols: the case for relative kinetics.Crossref | GoogleScholarGoogle Scholar |
[37] A. L. Robinson, N. M. Donahue, W. F. Rogge, Photochemical oxidation and changes in molecular composition of organic aerosol in the regional context. J. Geophys. Res. – Atmos. 2006, 111, D03302.
| Photochemical oxidation and changes in molecular composition of organic aerosol in the regional context.Crossref | GoogleScholarGoogle Scholar |
[38] E. A. Weitkamp, A. T. Lambe, N. M. Donahue, A. L. Robinson, Laboratory measurements of the heterogeneous oxidation of condensed-phase organic molecular makers for motor vehicle exhaust. Environ. Sci. Technol. 2008, 42, 7950.
| Laboratory measurements of the heterogeneous oxidation of condensed-phase organic molecular makers for motor vehicle exhaust.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFeqs7vF&md5=0ce62727c9468dfbe79c306c4cfa6a87CAS | 19031886PubMed |
[39] B. Ervens, B. J. Turpin, R. J. Weber, Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies. Atmos. Chem. Phys. 2011, 11, 11 069.
| Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisFOisLw%3D&md5=181a6701e42d0b51b62842e66c2a2585CAS |
[40] Y. Tan, Y. B. Lim, K. E. Altieri, S. P. Seitzinger, B. J. Turpin, Mechanisms leading to oligomers and SOA through aqueous photooxidation: insights from OH radical oxidation of acetic acid and methylglyoxal. Atmos. Chem. Phys. 2012, 12, 801.
| Mechanisms leading to oligomers and SOA through aqueous photooxidation: insights from OH radical oxidation of acetic acid and methylglyoxal.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XltFShtbY%3D&md5=746e6cf5980a9f614fe57e12fecafa8bCAS |
[41] D. Hoffmann, A. Tilgner, Y. Iinuma, H. Herrmann, Atmospheric stability of levoglucosan: a detailed laboratory and modelling study. Environ. Sci. Technol. 2010, 44, 694.
| Atmospheric stability of levoglucosan: a detailed laboratory and modelling study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGkurfP&md5=15c7898bef2c0c0b0a000ef6ab740a58CAS | 20000815PubMed |
[42] A. A. May, R. Saleh, C. J. Hennign, N. M. Donahue, A. L. Robinson, Volatility of organic molecular markers used for source apportionment analysis: measurements and atmospheric implications. Environ. Sci. Technol. 2012, 46, 12 435.
| Volatility of organic molecular markers used for source apportionment analysis: measurements and atmospheric implications.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVWhtLbF&md5=eb391e7ce7f8b608c8972e0e3e625001CAS |
[43] C. J. Hennigan, A. P. Sullivan, J. Jeffrey, L. Collett, A. L. Robinson, Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals. Geophys. Res. Lett. 2010, 37, L09806.
| Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals.Crossref | GoogleScholarGoogle Scholar |
[44] L. Hildebrandt, E. Kostenidou, V. A. Lanz, A. S. H. Prévôt, U. Baltensperger, N. Mihalopoulos, A. Laaksonen, N. M. Donahue, S. N. Pandis, Sources and atmospheric processing of organic aerosol in the Mediterranean: insights from aerosol mass spectrometer factor analysis. Atmos. Chem. Phys. 2011, 11, 12 499.
| Sources and atmospheric processing of organic aerosol in the Mediterranean: insights from aerosol mass spectrometer factor analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVKltrY%3D&md5=5e5580ab5d065126d7cea01e8b20f001CAS |
[45] A. A. Presto, N. M. Donahue, Investigation of α-pinene + ozone secondary organic aerosol formation at low total aerosol mass. Environ. Sci. Technol. 2006, 40, 3536.
| Investigation of α-pinene + ozone secondary organic aerosol formation at low total aerosol mass.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjvVKjsLs%3D&md5=6d2a4bc12491c7ba5328ecdbec1135faCAS | 16786691PubMed |
[46] B. N. Murphy, N. M. Donahue, C. Fountoukis, M. Dall’Osto, C. O’Dowd, A. Kiendler-Scharr, S. N. Pandis, Functionalization and fragmentation during ambient organic aerosol aging: application of the 2-D volatility basis set to field studies. Atmos. Chem. Phys. 2012, 12, 10 797.
| Functionalization and fragmentation during ambient organic aerosol aging: application of the 2-D volatility basis set to field studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFOht7w%3D&md5=e2d5737a7dc8316b688535a9ea63591bCAS |
[47] N. M. Donahue, K. M. Henry, T. F. Mentel, A. K. Scharr, C. Spindler, B. Bohn, T. Brauers, H. P. Dorn, H. Fuchs, R. Tillmann, A. Wahner, H. Saathoff, K. H. Naumann, O. Möhler, T. Leisner, L. Müller, M.-C. Reinnig, T. Hoffmann, K. Salow, M. Hallquist, M. Frosch, M. Bilde, T. Tritscher, P. Barmet, A. P. Praplan, P. F. DeCarlo, J. Dommen, A. S. H. Prévôt, U. Baltensperger, Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions. Proc. Natl. Acad. Sci. USA 2012, 109, 13 503.
| Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVaqurjI&md5=ba600c91d2bd730a3f73dea7f2449ab6CAS |
[48] K. M. Wagstrom, S. N. Pandis, Determination of the age distribution of primary and secondary aerosol species using a chemical transport model. J. Geophys. Res. – Atmos. 2009, 114, D14303.
| Determination of the age distribution of primary and secondary aerosol species using a chemical transport model.Crossref | GoogleScholarGoogle Scholar |
[49] H. Berresheim, C. Plass-Dulmer, T. Elste, N. Mihalopoulos, F. Rohrer, OH in the coastal boundary layer of Crete during MINOS: measurements and relationship with ozone photolysis. Atmos. Chem. Phys. 2003, 3, 639.
| OH in the coastal boundary layer of Crete during MINOS: measurements and relationship with ozone photolysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXpsFClsbY%3D&md5=c05a1edeb04a290549a6d63761d32c3bCAS |
[50] L. Hildebrandt, E. Kostenidou, B. H. Lee, N. Mihalopoulos, D. R. Worsnop, N. M. Donahue, S. N. Pandis, Formation of low-volatility oxygenated organic aerosol in the atmosphere: insights from the Finokalia Aerosol Measurement Experiments. Geophys. Res. Lett. 2010, 37, L23801.
| Formation of low-volatility oxygenated organic aerosol in the atmosphere: insights from the Finokalia Aerosol Measurement Experiments.Crossref | GoogleScholarGoogle Scholar |