

Thermolysis of RAFT-Synthesized Poly(Methyl Methacrylate)
Bill Chong A , Graeme Moad A B , Ezio Rizzardo A , Melissa Skidmore A and San H. Thang AA CSIRO Molecular and Health Technologies, Bag 10, Clayton South, VIC 3169, Australia.
B Corresponding author. Email: graeme.moad@csiro.au
Australian Journal of Chemistry 59(10) 755-762 https://doi.org/10.1071/CH06229
Submitted: 30 June 2006 Accepted: 2 October 2006 Published: 30 October 2006
Abstract
Thermolysis provides a simple and efficient way of eliminating thiocarbonylthio groups from RAFT-synthesized polymers. The course of thermolysis of poly(methyl methacrylate) (PMMA) prepared with dithiobenzoate and trithiocarbonate RAFT agents was followed by thermogravimetric analysis (TGA), 1H NMR spectroscopy, and gel permeation chromatography (GPC). The weight loss profile observed depends strongly on the RAFT agent used during polymer synthesis. PMMA with a methyl trithiocarbonate end group undergoes loss of that end group at ~180°C, at least in part, by a mechanism believed to involve homolysis of the C–CS2SCH3 bond and subsequent depropagation. In contrast, PMMA with a dithiobenzoate end appears more stable. Only the end group is lost at ~180°C and the dominant mechanism is proposed to be a concerted elimination process analogous to that involved in the Chugaev reaction.
Introduction
Radical polymerization with thiocarbonylthio RAFT (reversible addition–fragmentation chain transfer) agents is arguably one of the most versatile processes for living free-radical polymerization in displaying superior flexibility with respect to monomers and reaction conditions.[1–3] A key feature of RAFT polymerization is that the thiocarbonylthio group(s), present in the initial RAFT agent, is(are) retained in the polymeric product(s). The retention of these groups is responsible for the polymers’ living character. However, the presence of the thiocarbonylthio groups also means that the polymers are usually coloured. The polymers may also, in some cases, be odorous or release an odor over time due to decomposition of the thiocarbonylthio groups and the evolution of volatile sulfur compounds. The presence of such colour and odor can be disadvantageous in some applications. Even though many of these issues are mitigated or overcome by appropriate selection of the initial RAFT agent, these issues have nonetheless provided an incentive for developing effective methods for treatment of RAFT-made polymers to cleave and remove the thiocarbonylthio groups in a post-polymerization process. Cleavage of polymer chains at the thiocarbonylthio groups can also be an aid in providing information on the mechanism of RAFT polymerization.[4,5]
The chemistry of the thiocarbonylthio group is well known from small molecule chemistry[6,7] and much of this knowledge has been shown to be applicable to transforming the thiocarbonylthio groups in RAFT-synthesized polymers.[2] Thiocarbonylthio groups undergo reaction with nucleophiles and ionic reducing agents (e.g. amines,[8–15] hydroxide,[16,17] borohydride[18,19]) to provide thiols. They also react with various oxidizing agents (including NaOCl, H2O2, ButOOH, peracids, ozone)[2,20–22] and are sensitive to UV irradiation.[23,24] However these processes, while they result in loss of the thiocarbonylthio group, leave reactive end group functionality and thus are not suitable in many circumstances. Thermolysis[5,8,25,26] and various radical-induced reactions (reduction,[5,8,27–29] termination[5,30]) provide solutions to this dilemma and can provide desulfurization by complete end-group elimination/transfer.
It was recently reported that butyl trithiocarbonate groups were readily cleaved from RAFT-synthesized polystyrene[5,8,25] and poly(butyl acrylate) (PBA)[25,26] by heating at ~180°C under nitrogen. The dominant mechanism of end group loss was found to depend on the particular polymer, involving concerted elimination in the case of polystyrene (Scheme 1) and initial C–S bond homolysis followed by backbiting and β-scission in the case of PBA (Scheme 2). The thermolysis of xanthate-terminated polymers has also been reported.[26,31] For the case of both S-polystyrene and S-poly(t-butyl acrylate) O-isobutyl xanthate, the reported mechanism involves selective elimination to provide 2-butene and a polymer with a thiol end group.[31] In contrast, for the case of O-ethyl S-poly(vinyl acetate) xanthate,[26] the observed products are consistent with involvement of the poly(vinyl acetate) (PVAc) propagating radical and suggest a mechanism involving initial C–S bond homolysis analogous to that shown in Scheme 2. The thermolysis of RAFT agents has also been studied; cumyl dithiobenzoate was reported to reversibly eliminate dithiobenzoic acid when heated at 120°C while phenylethyl and benzyl dithiobenzoate are comparatively stable under these conditions.[32]
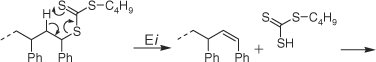
|
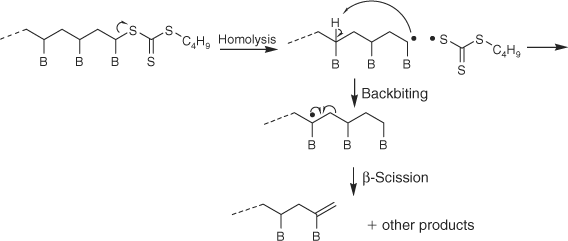
|
Thermogravimetric analysis (TGA) of poly(methyl methacrylate) (PMMA) with dithiobenzoate end groups has been reported by Lima et al.[13] (telechelic PMMA
In this paper, we further explore the applicability of thermolysis as a method of end-group removal to a series of PMMA synthesized by high-conversion RAFT polymerization with either methyl trithiocarbonate or dithiobenzoate RAFT agents and report on the dependence of the thermolysis mechanism and product distribution on the RAFT agent-derived end groups.
Experimental
General
Solvents used for column chromatography and polymerization were of AR grade and were distilled. MMA (Aldrich) was filtered through neutral alumina (70–230 mesh), fractionally distilled under reduced pressure, and redistilled under reduced pressure immediately before use. The RAFT agents, 2-cyanoprop-2-yl methyl trithiocarbonate 1[34] and 2-cyanoprop-2-yl dithiobenzoate 3[35,36] were synthesized as previously described.
Nuclear magnetic resonance (NMR) spectra were obtained with a Bruker AC200 or a Bruker AV400 spectrometer. Chemical shifts are reported in ppm from external tetramethylsilane. Gel permeation chromatography (GPC) was performed on a Waters 515 HPLC pump and Waters 717 Plus Autosampler equipped with Waters 2414 refractive index detector and three Mixed C and one mixed E PLgel column (each 7.5 mm × 300 mm) from Polymer Laboratories. Tetrahydrofuran (flow rate 1.0 mL min–1) was used as eluent at 22 ± 2°C. The columns were calibrated with narrow polydispersity polystyrene standards (Polymer Laboratories). A third-order polynomial was used to fit the log10M versus time calibration curve which was almost linear across the molecular weight range 2 × 102–2 × 106 g mol–1. Thermogravimetric analysis (TGA) was carried out with a Mettler TGA/SDTA521 thermobalance equipped with a sample robot. Experiments were performed under nitrogen (50 mL min–1).
Poly(Methyl Methacrylate)
The following procedure is typical. A stock solution was prepared comprising MMA (11.25 mL), benzene (3.75 mL), and azobis(1-isobutyronitrile) (0.0155 g). Aliquots of this solution (2.5 mL) were transferred to a vial containing the RAFT agent 3 (0.0075 mg). The contents of the vial were transferred to an ampoule that was degassed through three freeze–evacuate–thaw cycles, sealed under nitrogen and heated in a constant temperature bath at 60°C for 16 h. The ampoule was then cooled, opened, diluted with benzene, and precipitated into methanol.
The monomer conversions (Table 1) were calculated by determining the residual monomer in the 1H NMR spectrum of small samples of the polymerization mixtures. The molecular weights shown in Table 1 are for the precipitated samples. Some loss of low molecular weight material on precipitation probably accounts for the discrepancy between found and calculated molecular weights.

|
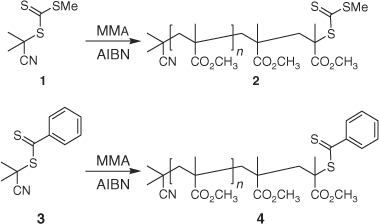
|
Thermolysis Procedure
Dynamic Thermolysis
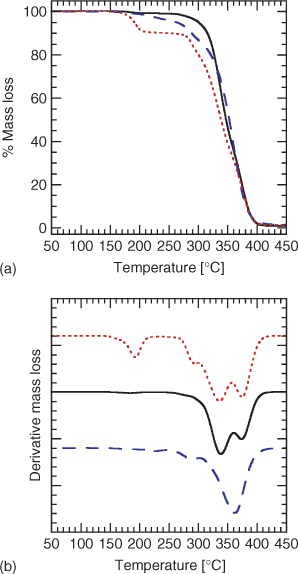
Samples of PMMA (~5 mg) were placed in alumina crucibles and transferred to the carousel of the sample robot coupled to the thermogravimetric balance. The samples were heated at 5°C min–1 under nitrogen from 50 to 600°C. Example thermograms are shown in Figs 1 and 2.
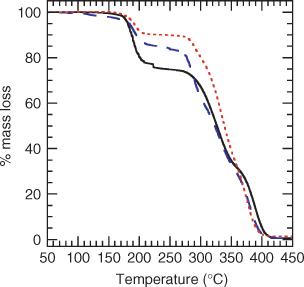
|
|
Isothermal Thermolysis
Small scale preparative thermolysis was also carried out using the thermogravimetric balance. Samples were heated from 50°C to the requisite temperature (140, 160, 180, 200, 220, or 240°C) at 5°C min–1 then maintained at that temperature for 20 min. The entire process was carried out under a flow of nitrogen.
The following procedure is typical. A sample of pale yellow PMMA 2a (18 mg,

|
Results
Thermolysis of Poly(Methyl Methacrylate) with Methyl Trithiocarbonate End Groups
The TGA thermogram of PMMA 2 with a methyl trithiocarbonate end group displays two mass loss regions (Fig. 1), the first between 170 and 220°C and another beginning at 280°C that results in complete degradation of the polymer. The mass loss between 170 and 220°C is substantially greater than that expected on the basis of loss of the end group alone. For PMMA 2a (
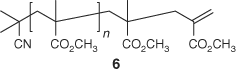
Isothermal thermolysis at 180°C of the pale yellow trithiocarbonate-terminated PMMA 2 (Scheme 1), provides a colourless product. The 1H NMR spectrum of 2a (Fig. 3) shows disappearance of the broad singlet for the trithiocarbonate methyl at δH 2.65 and the appearance of signals attributable to macromonomer chain end 6 (Scheme 4) at δH 6.2, 5.5, and 2.5. Signals for the olefinic hydrogens of entrapped MMA also appear at δH 6.1 and 5.6. The end group concentrations for 2a and its thermolysis products obtained at various temperatures were estimated by integrating the signals associated with the end groups versus that for the PMMA methoxy and are summarized in Table 2. Because of the small end group concentrations, the error in this data is large (~ ±20%) nonetheless it provides an indication of how the product distribution depends on thermolysis temperature. The concentration of macromonomer ends in the thermolysis product appears to be significantly less than the concentration of trithiocarbonate ends in the precursor 2a.
|

|
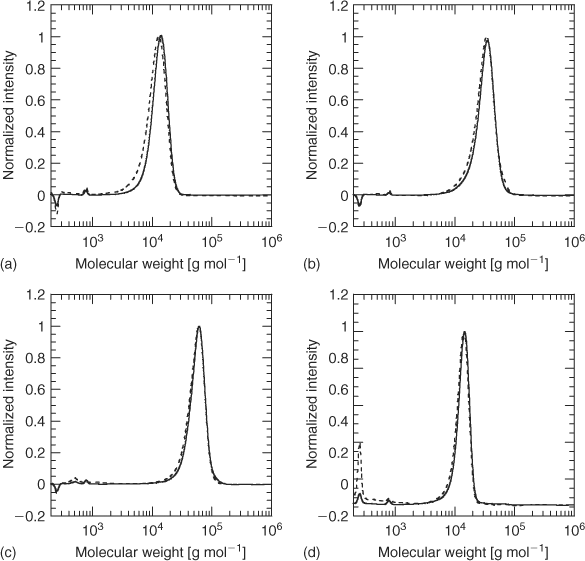
GPC analysis of the thermolysis product from 2 at 240°C shows a small yet significant lowering of the number average molecular weight (Figs 4a–4c). This is most noticeable for the thermolysis of the lowest molecular weight sample (2a;
|
Thermolysis of Poly(Methyl Methacrylate) with Dithiobenzoate End Groups
The PMMA 4 synthesized with a dithiobenzoate RAFT agent displays thermolysis behaviour that is quite different to that of the PMMA with a methyl trithiocarbonate end (2). The thermogram (not shown) for the lower molecular weight sample 4a (
The PMMA 4a was heated isothermally at 140, 160, 180, 200, 220, and 240°C for 20 min. The samples heated at 140 and 160 remained pink. The samples heated at 180 and 200°C became colourless. Those heated at higher temperatures showed some yellow discolouration.
The 1H NMR spectrum of the unheated PMMA sample 4a shows signals at δH 7.8 and 8.0 (ratio ~3:1) which are believed to correspond to the ortho aromatic hydrogens for the diastereomeric dithiobenzoate end groups (Fig. 5). The sample of 4a heated at 140°C shows that the smaller of these has disappeared and that signals at δH 6.3, 5.5, and 2.5, associated with the olefinic hydrogens and the methylene adjacent to the double bond of the PMMA 6, have appeared. The difference in stability of the two diastereomers is likely to be steric in origin and related to the ease of adoption of the transition state for concerted elimination.[39] The 1H NMR spectrum of 4a heated at 180°C shows the complete disappearance of the ortho hydrogen signals of the dithiobenzoate group and the appearance of peaks corresponding to macromonomer 6 and methyl methacrylate. The precise fate of the dithiobenzoate group is unknown and signals in the aromatic region remain. The 1H NMR spectrum of the thermolysis product obtained at 200 or 220°C is similar to that obtained at 180°C. Signals due to entrapped MMA monomer are also seen in the thermolysis products. The GPC, even for the lowest molecular weight sample 4a at 240°C, suggests little change in molecular weight (Fig. 4d).

|
Discussion
Analogous dithiobenzoate- and trithiocarbonate-terminated PMMA show quite different thermolysis behavior. PMMA is known to degrade by unzipping or depropagation which can be initiated at weak links or by random chain scission.[40–48] Any structures that are less stable than the backbone or side chain bonds and which give rise to propagating radicals may constitute weak links. PMMA formed by conventional radical polymerization in the absence of transfer agents is known to contain weak links formed as a consequence of termination by combination or disproportionation.[40] The appearance of thermograms of PMMA can depend strongly on such factors as sample size and heating rate[42] but typically show two or three stages of degradation. The thermogram (Fig. 2) of PMMA 5 synthesized using bulk polymerization with AIBN initiator illustrates the three stages of degradation. The first stage with onset under our conditions at ~160°C, is attributed to degradation initiated by scission of those chains containing head-to-head linkages formed by termination by combination. The second stage of degradation, with onset ~240°C is associated with those chains possessing unsaturated end groups (analogous to 6) formed by disproportionation undergoing radical-induced decomposition. A final stage of degradation is attributed to chains undergoing degradation initiated by random chain scission.
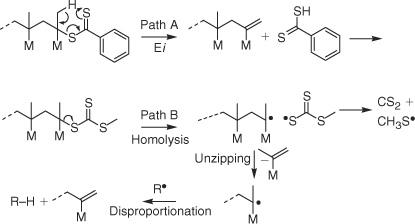
Two possible thermolysis mechanisms for RAFT-synthesized PMMA are shown in Scheme 5. It is proposed that the dithiobenzoate terminated PMMA degrades by Path A which involves concerted elimination of the end group possibly by a process analogous to that involved in the Chugaev reaction[39,49] to form PMMA with an unsaturated chain end. The observation of MMA in the 1H NMR (Fig. 5) may result from unzipping of propagating radicals formed by degradation of small amount of PMMA with unstable head to head linkages formed during polymer synthesis as a consequence of termination by combination.[41] The extent of termination by radical–radical reaction and the combination disproportionation ratio in RAFT polymerization is expected to be similar to that for a conventional radical polymerization carried out under similar reaction conditions.
|
PMMA with a trithiocarbonate chain end is proposed to primarily degrade by Path B which involves homolysis of the C–S bond joining PMMA chain to the trithiocarbonate end to generate propagating radicals which then unzip or depropagate to form MMA until terminated by transfer or radical–radical reaction.
There is no direct correlation between the mass loss through unzipping (region of 150–240°C) and the concentration of trithiocarbonate functionality. However, it is clear that the higher the concentration of trithiocarbonate groups, the greater number of radicals produced through homolysis of the C–S bond, the greater the percentage mass lost through unzipping, which is consistent with the proposed mechanism.
We are unable to reconcile our data for PMMA with dithiobenzoate ends with recent work of Xu et al.[33] These authors reported that PMMA with dithiobenzoate ends degraded at substantially lower temperature than found in this work or in other recent studies.[13,15] They used a higher heating rate, 20°C min–1 versus 5°C min–1. We briefly explored the effect of heating rate and sample size and found as expected that the onset temperature decomposition increased for higher temperatures for higher heating rate and for larger sample sizes. Xu et al.[33] suggest in their paper that their lower degradation temperature with respect to other literature reports[13,15] may be an effect of molecular weight. We have performed thermogravimetric analysis of a PMMA of similar molecular weight (
Notwithstanding these issues we concur with Xu et al.[33] on the finding that thermolysis is a suitable method for cleanly removing dithiobenzoate chain ends of PMMA prepared by RAFT polymerization albeit that a somewhat higher temperature is required.
Conclusions
Thermolysis can be used to cleanly remove dithiobenzoate chain ends from PMMA resulting in a macromonomer product with molecular weight and polydispersity similar to that of the precursor polymer. Thermolysis is less suitable for removal of the trithiocarbonate end groups from PMMA since end group loss is accompanied by some polymer degradation and lowering of molecular weight.
The mechanism of the thiocarbonylthio end group cleavage from RAFT-synthesized polymers during thermolysis depends on both the polymer and the type of RAFT agent used in its synthesis. End group loss during thermolysis of PMMA, PBA[25,26] synthesized with alkyl trithiocarbonate RAFT agents, and poly(vinyl acetate)[26] synthesized with an O-ethyl xanthate RAFT agent follows a mechanism involving initial C–S bond homolysis (path B). Elimination of the end groups of PMMA synthesized with dithiobenzoate RAFT agent and polystyrene synthesized with trithiocarbonate RAFT agent[5,8,25] follows a concerted mechanism (path A).
Acknowledgments
We are grateful to Dr Wendy Tian for performing some of the thermogravimetric analyses and to DuPont Performance Coatings for their support of this work.
[1]
G. Moad,
E. Rizzardo,
S. H. Thang,
Aust. J. Chem. 2005, 58, 379.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
