

On the Stability of Water Oxidation Catalysts: Challenges and Prospects
Alex Izgorodin A , Orawan Winther-Jensen A and Douglas R. MacFarlane A BA Australian Centre for Electromaterials Science, Monash University, Clayton, Vic. 3800, Australia.
B Corresponding author. Email: douglas.macfarlane@monash.edu

Dr. Alex Izgorodin completed his Master of Science degree in electronics and microelectronics at the Ivanovo State University of Chemistry and Technology in 2006. In 2010, Alex completed his Ph.D. in physical chemistry at Monash University entitled ‘Towards Hydrogen Production via Water Splitting’ under the supervision of Professor Douglas R. MacFarlane and is currently contributing his expertise to the area of semiconductors and water splitting. |

Dr. Orawan Winther-Jensen is an Australian Postdoctoral Fellow (ARC) at Monash University. Her main research field covers materials and structures for water-splitting devices where breathable gas-diffusion electrodes and conjugated polymer electro-catalysts are the core areas of interest. |

Professor Doug MacFarlane is an Australian Research Council Federation Fellow at Monash University and leads the Monash Ionic Liquids Group. He is also the program leader of the Energy Program in ACES. He was a Ph.D. graduate of Purdue University in 1982 and after post-doctoral work at Victoria University, Wellington, took up a faculty position at Monash. Professor MacFarlane was elected to the Australian Academy of Sciences in 2007 and to the Australian Academy of Technological Sciences and Engineering in 2009. His research interests include the chemistry and properties of ionic liquids and solids and their application in a wide range of technologies from electrochemical (batteries, fuel cells, solar cells, and corrosion prevention), to biotechnology (drug ionic liquids and protein stabilization) and biofuel processing. He is an Adjunct Professor of the University of Alabama, an International Fellow of the Queens University Ionic Liquid Laboratory, Belfast, and a Visiting Professor of the Chinese Academy of Science. |
Australian Journal of Chemistry 65(6) 638-642 https://doi.org/10.1071/CH12024
Submitted: 18 January 2012 Accepted: 24 February 2012 Published: 16 April 2012
Abstract
Future requirements for water splitting technologies need highly efficient water oxidation catalysts that are sufficiently stable for operation over many years. Recent research has achieved significant progress in improving the electro-catalytic activities of these catalysts. However, there has not been a strong research focus on their long-term mechanical and chemical stability, yet this is critical for commercial application. In this paper we discuss some of the chemical and thermodynamic challenges confronting this goal, as well as some of the strategies that are available to overcome them. The challenge becomes even greater in the area of photo-active electromaterials; fortunately some of the same strategies may allow progress in this area also.
Introduction
Efficient water oxidation is of paramount importance for hydrogen production via water splitting. Typical overpotentials of 400 mV or more are often required to drive this process at reasonable rates, which represent an energy loss of 25 % or more. Significant attention in the recent years has been devoted to the development of novel water oxidation catalysts that can be used in electrolyzers, or as a part of photo-electrochemical devices.[1] However, the prerequisites for electrode materials to be used in technological applications include not only excellent electro-catalytic properties, but also excellent long-term mechanical and chemical stability.[2] While high catalytic activity materials are known in this context, as are adequately stable materials, achieving both properties in a single, inexpensive material comprised of earth abundant elements is very challenging. If one then adds this list of target properties to those that stem from the goal of photo-enhanced water oxidation catalysts, i.e. stability under photo-excitation and an absorption spectrum ideally matched to the solar spectrum, the challenge becomes very great indeed. In this paper we highlight some of the recent progress towards long-term stable materials and discuss some of the challenges involved, as well as some of the strategies available for overcoming them.
Stability of Water Oxidation Catalysts
One of the highest catalytic activities towards water oxidation is observed in RuO2.[1b] However, the catalyst is unstable during operation at high overpotentials and rapidly decomposes in basic electrolytes.[3] Stability was improved using the concept of dimensionally stable anodes (DSA) that consist of active component metal oxides RuO2 and/or IrO2 formed by thermal decomposition onto an inert support (e.g. Ti)[4] or stabilized by inert metal oxides (TiO2, SnO2, Ta2O5, ZrO2).[5] This mixture produces a poorer and more expensive electro-catalyst compared with RuO2, but makes it technologically practical. However, the cost of these rare metals makes these anode materials practical only in specialized applications. In the future, the ultimate global availability of these materials would also be an issue.
Nickel operated in hot alkaline electrolytes forms an oxide coating that is moderately active as a water oxidation catalyst and is quite stable, for reasons discussed further below. Despite the relatively high overpotentials required to drive water oxidation on this material, it remains the main technology used in commercial electrolyzers, due to its low cost and stability under alkaline conditions where high currents are possible.[1b] However, it could not, without significant improvement in activity, i.e. the overpotentials required, satisfy the energy efficiency targets required by future water splitting applications.
The most efficient, currently-known electro-catalysts that are based on earth abundant elements are those from the families of manganese[6] and cobalt oxides.[7] High rates of water oxidation were also observed from hydroxides of transition metals cations and their binary mixtures (i.e. Fe(iii), Co(iii), Mn(iii), Ru(iv), etc).[8] However, long-term stability remains an issue with these materials. In recent work, Kanan and Nocera describe an in situ catalyst generation process that tackles the instability issue in the Co oxide family by providing a regeneration capability. However, catalytic performance is significantly lower than the catalysts described above, especially in alkaline electrolytes.[7b,9]
Nature, of course, has evolved an efficient and highly effective catalytic centre based on a manganese cluster in Photosystem ii (PSii) that is capable of water oxidation with high turnover numbers. Nonetheless, it is clear that nature has also evolved mechanisms of regenerating this centre on a fairly regular basis (days to weeks). The whole mechanism of the degradation process is complex, involving the surrounding protein but it is fair to say that any attempt to mimic PSii in a practical technology either needs to include a regeneration mechanism or to substantially understand and improve the instability issue that is apparent in PSii.[10] Recent experience with bio-inspired molecular catalysts based on Mn clusters has shown that these too are relatively unstable in operation over a period of hours to days, and ultimately transform to nano-particulate manganese oxide phases.[11] These phase transformations at higher oxidizing potentials are thermodynamically expected and will be discussed further below.
The longer term goal of research in this field is to achieve water splitting via solar energy input alone, either via a single material (or structure) supporting both reactions, or via separated electrodes carrying out the individual reactions in a photo-electrochemical cell. One or both of the electrodes can be photo-sensitive in this device, and as a stepping stone to the ultimate performance, some external electrical energy (driving voltage) is often applied. In order to obtain high catalytic efficiency in photo-electrochemical devices, the energy structure of the materials has to ultimately be fine-tuned to accommodate both absorption of a large part of the visible spectrum and the ability to split water directly without the need for an external applied potential. Metal sulfides have been studied extensively in this respect.[12] Their small band-gap energies allow absorption of the visible part of the solar spectrum, leading to high overall energy efficiencies.[13] In particular, CdS is one of the well known visible light driven photo-catalysts for water splitting.[14] Prolonged irradiation of these sulfide materials, however, leads to oxidative photo-corrosion as described by Eqn 1. In this case the metal sulfide, rather than water, is oxidized by the photo-generated holes.[14] Similar effects can be observed even in metal oxides upon band gap excitation, as is shown for ZnO2 in Eqn 2.[14]
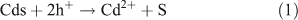
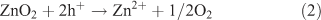
The stability of metal sulfides can be increased by combining them with Pt and RuO2 co-catalysts that assist in transfer of photo-generated electrons and holes away from the photo-active material and also provide catalytic sites.[15] Despite these efforts the catalysts have short lifetimes and photo-corrosion remains a critical issue.[13a]
Other strategies to separate the reactive hole and electron from the photo-active materials have included layered structures based on Si,[16] AlGaAs/Si,[17] InP,[1b] and (Ga1–xZnx)(N1-yOy).[18] The best photo electrochemical cells based on these structures have produced solar water splitting efficiencies of up to 18 %, based on the solar energy to hydrogen conversion. However, similar to metal sulfides, these materials decompose under prolonged operation (>1 day). An attempt to overcome photo-corrosion in these systems was proposed by Khaselev and Turner in a monolithic GaInP2/GaAs photo-electrochemical cell. The use of a p-type material at the electrode-electrolyte interface allowed the surface to be cathodically protected under light illumination and hence reduce the rate of photo corrosion.[19] The reported system was able to produce solar to hydrogen conversion efficiencies as high as 16 %, but these cells deteriorated over 1 day of operation.[20]
The poor applicability of the systems described above to practical applications due to their low overall stability is further exacerbated by the use of rare and expensive materials that are required if high energy efficiencies are to be achieved. An approach to mimic photosynthesis with the use of earth abundant elements and neutral electrolytes reported by Nocera et al. utilizes Si and a cobalt based catalyst that was able to split water with solar to hydrogen efficiency of 2.5 %.[21] A cell based on hematite (α-Fe2O3) developed by Gratzel et al. further develops the concept of the use of inexpensive and earth abundant catalytic materials for water splitting.[22] However, the long-term stability (>1000 h) of these systems has yet to be demonstrated. Despite the fact that these photoactive oxide materials are able to operate under visible light irradiation and are more stable compared with metal sulfides and III–V semiconductor materials, the majority of them require an external potential to be applied due to their less than ideal optoelectronic properties.[23]
Strategies to overcome poor stability by developing systems that can be easily restored in situ, such as Ag/Ag3PO4,[24] are under investigation. Performance of these systems, however, requires further improvement to be sufficient for commercial applications.[25]
Thus, despite recent success in development of stable catalytic materials for water oxidation, stable systems with high solar to hydrogen conversion efficiencies beyond a day of operation have proven elusive, and overcoming these stability issues is considered one of the crucial hurdles in this field.[1b,22,26]
Thermodynamic Challenges and Limits
The fundamental stability of catalysts during oxygen evolution can be estimated from thermodynamic considerations using the Pourbaix diagram as a convenient method of summarizing the available information. These represent equilibrium phase diagrams as a function of potential and pH, indicating the fields of stability of the various known phases. They are readily available for certain conditions of temperature and aqueous solute composition, and can be easily re-calculated for other, more relevant conditions using available software packages (the Geochemist’s Workbench software package was used in this work).
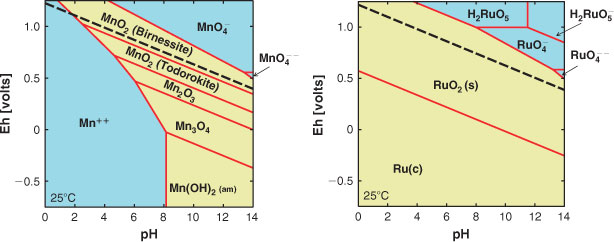
A series of example Pourbaix diagrams relevant to important water oxidation catalytic materials in the Mn and Ru families have been calculated in our work and are shown in Fig. 1. The diagrams allow direct comparison of the equilibrium phase present in aqueous electrolytes at a given potential, with the potential required for water oxidation (dashed black line in Fig. 1). In principle, any species, such as MnO4– that sits well above the water oxidation line should cause water oxidation to take place. However, as is well known, such reactions are frequently sluggish and metastability in that respect is commonplace.
|
Ruthenium oxide is one of the best known catalysts for water oxidation. It can be seen from Fig. 1, however, that in alkaline electrolytes the potential required for oxygen evolution is sufficient to form the soluble RuO42– ion, leading to catalyst dissolution. In neutral and acidic electrolytes, moderate overpotentials (below 200 mV) are sufficient to form other soluble RuOx species including (not shown on the Pourbaix diagram) volatile RuO4.[27] The Pourbaix diagram of manganese shows that the potential required for water oxidation in highly alkaline electrolytes (pH 14) is close to that corresponding to the formation of the soluble MnO42– ion as the equilibrium species, while only small overpotentials (below 300 mV) are required for oxidation to MnO4– in neutral and slightly basic electrolytes.
In the recent research on the Co based catalyst, cyclic voltammetric and electron paramagnetic resonance (EPR) spectroscopic studies were used to determine the nature of the cobalt species present at various pH and electrochemical potentials.[28] The empirically produced data show a high degree of correlation with data derived from thermodynamic free energies as represented by the Pourbaix diagram for Co. Similarly to Ru and Mn this recent research confirms formation of a higher oxidation state of Co (from 3+ to 4+) during catalytic water oxidation (though at higher overpotentials).[27]
As discussed further below, cycling between oxidation states in these materials is a key aspect of their catalytic activity and therefore multiple oxidation states of the metal centre may be seen as an important criterion for high catalytic activity. However, the formation of a transient soluble species during the oxidation/reduction cycle is likely to create a long-term stability problem with respect to loss of material from the electrode. For example, in the manganese oxide system, the transient formation of a high oxidation state, potentially soluble species such as Mn(vii), to even a small fractional extent during each catalytic cycle becomes problematical for several reasons:
-
If the subsequent reduction to lower valent states takes place after dissociation from the solid electrode layer, re-precipitation may not take place on the electrode, leading to formation of inactive precipitates elsewhere in the cell.
-
Reprecipitation onto the surface of the electrode is likely to take place in a less crystallographically ordered way, causing loss of activity and potentially greater probability of dissociation and material loss on a subsequent cycle
On the other hand the Pourbaix diagram for Ni (a recent publication has presented detailed Pourbaix diagrams for Ni as a function of temperature[29]) indicates formation of oxides and hydroxides of nickel in oxidation states between 2 and 4, at potentials required for water oxidation. Importantly, these higher potential phases are solids and therefore Ni electrodes show good anodic stability over a wide range of alkaline pH even at high overpotentials compared with water oxidation potentials. Hence, even though catalytic properties of Ni anodes are significantly worse than the materials described above, their low cost and much better long-term stability are the reasons for the use of Ni in commercial electrolyzers.[2]
The thermodynamic stability of photo-catalysts is a more complex situation. During photo-excitation the electron-hole pairs generated can be considered to represent a large electrochemical potential difference in the semiconductor. In some cases these have sufficient energy to cause decomposition reactions of the type shown in Eqns 1 and 2.[26] These reactions are not typically shown on the Pourbaix diagrams, however given sufficient thermodynamic information, they could be and would represent an upper limit to the phase stability of some of the oxide phases. Placement on the diagram of the valence and conduction band energies, where known, would indicate when, for example, h+ is sufficiently energetic to cause these decomposition reactions to take place.
Challenges in Mechanical Stability
It is understood that in most cases during catalysis of the water oxidation reaction, the active site on the surface of the material undergoes a constant cycling between oxidation states. The cycling involves changes in charge and metal ion coordination.[30] Such drastic changes inevitably affect the binding of the metal ion to the surrounding lattice and hence are a potential source of ultimate mechanical degradation of the catalyst material.
As an example of this effect, a greater stability was obtained in the layered double hydroxide of CoOx, the structure of which allows changes in the oxidation state of the cobalt ion while preserving the metal coordination geometry and structural integrity.[27] The charge increases from CoII → CoIII → CoIV during oxidation and decreases following subsequent oxygen evolution. This process is in turn balanced through the formation of the bridging ligands, or movement of counter ions in and out of the interlayer.[27] The layered double hydroxide structure allows this local change in oxidation state of the metal without substantial strain in the surrounding lattice. Clearly such ‘flexible’ structures are required in long-term stable catalytic materials.
It is known that the method of preparation (e.g. processing temperature) also significantly affects stability of many water oxidation catalysts. Materials with higher crystallinity that are synthesized at high temperature generally show higher stability compared with more amorphous films formed at low temperature. This is presumably a result of a less defected structure which is more able to absorb the lattice strain that occurs during the catalytic cycle.[2]
The microstructure of the catalyst layer also has an impact on the stability with respect to the oxygen gas pressure that can be generated due to the nucleation of small gas bubbles inside the pores, especially at high oxygen production rates. This can also lead to material deterioration during long-term operation.[31]
Strategies for Improved Stability
Nocera and co-workers[7a,32] have introduced an important concept that may assist in designing long-term stability into water electrolysis systems. The approach involves a self-healing mechanism via inclusion in the electrolyte of catalyst components, in this case Co(ii) and phosphate, that have the capacity to form, or reform, the catalytic layer via oxidation and precipitation. This type of self-healing mechanism has tremendous potential across many types of catalytic systems and may be implemented by having a permanent ‘dose’ of the component materials present, or by an occasional dosing of a small amount of material to regenerate the electrode. Operation for more than 100 h has been demonstrated.
Surface modification is an alternate strategy for the stabilization of water oxidation catalysts that has recently been developed in our laboratory. In this approach a surface treatment is applied to a catalyst layer on the electrode, which is designed to modify the surface to create a secondary lattice network. This secondary network potentially stabilizes the surface via two mechanisms: (i) by modifying the thermodynamics of the phases involved, dissolution of high metal oxidation states is avoided, and/or (ii) by providing a means of absorbing the lattice strain during catalytic cycling. For example, we have recently[33] described a surface modification treatment for manganese dioxide electrodes which involves anodizing the catalytic electrode in a phosphate ionic liquid. The process incorporates some phosphate anions into the surface layer of the catalyst (Fig. 2). This was shown to provide a substantial increase in stability of the electrode under both dark water oxidation and photo-assisted water oxidation. Operation for 80 h under intense visible light illumination was demonstrated.

|
Conclusions and Future Prospects
Practical water splitting as a technology, whether purely as an electrolysis process or photo-driven, is currently constrained by the combined impact of one or more of the challenges of energetic efficiency, expensive materials, and limited material stability. The best electro-catalysts are often either precious metal based, or are of limited stability, or both. Some of the stability challenges are thermodynamic at origin and strategies to modify the thermodynamics in a favourable way, such as surface modification processes to create mixed phases, offer a way forward. The development of repair mechanisms also offers a useful approach to dealing with the breakdown process, either as a periodic additive to the electrolyte solution or as a standing component of the electrolyte. In our view it is vital that stability issues be addressed whenever new catalyst systems are designed and tested and we entreat researchers to pursue longer-term tests in their research programs as a matter of course, not only as qualification of the material for practical applications but also to provide a database of stability issues and solutions. In light of the growing demand for efficient and stable water oxidation catalysts, the wider collaborations that are developing between scientists will strongly accelerate research progress towards design of novel catalyst materials that will meet the requirements of the practical applications.
References
[1] (a) M. R. Hoffmann, S. T. Martin, W. Y. Choi, D. W. Bahnemann, Chem. Rev. 1995, 95, 69.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjtF2qur4%3D&md5=a2c095c6cd961275f5015f655349caeaCAS |
(b) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori, N. S. Lewis, Chem. Rev. 2010, 110, 6446.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan, P. Strasser, ChemCatChem 2010, 2, 724.
| Crossref | GoogleScholarGoogle Scholar |
(d) R. M. N. Yerga, M. C. A. Galvan, F. del Valle, J. A. V. de la Mano, J. L. G. Fierro, ChemSusChem 2009, 2, 471.
| Crossref | GoogleScholarGoogle Scholar |
(e) R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince, R. T. Sayre, Science 2011, 332, 805.
| Crossref | GoogleScholarGoogle Scholar |
(f) J. K. Hurst, Science 2010, 328, 315.
| Crossref | GoogleScholarGoogle Scholar |
[2] S. Trasatti, Electrochim. Acta 1984, 29, 1503.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXmvFWqtQ%3D%3D&md5=7f7f65aabdcc13b3627d273bb80491b5CAS |
[3] R. Kotz, S. Stucki, D. Scherson, D. M. Kolb, J. Electroanal. Chem. 1984, 172, 211.
| Crossref | GoogleScholarGoogle Scholar |
[4] H. B. Beer, J. Electroanal. Chem. 1980, 127, 303C.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXltl2rsrc%3D&md5=5d35c83ba7af170c1c62e3e470cd08f1CAS |
[5] S. Trasatti, Electrochim. Acta 2000, 45, 2377.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjvFymur0%3D&md5=4a7941c2f88cba79ce50a58fd59038e0CAS |
[6] (a) F. Jiao, H. Frei, Energ. Environ. Sci. 2010, 3, 1018.
| 1:CAS:528:DC%2BC3cXht1yrsbrK&md5=6a3bb3d58cd5800baca5773d2af8bf55CAS |
(b) M. M. Najafpour, T. Ehrenberg, M. Wiechen, P. Kurz, Angew. Chem. Int. Ed. 2010, 49, 2233.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. M. Robinson, Y. B. Go, M. Greenblatt, G. C. Dismukes, J. Am. Chem. Soc. 2010, 132, 11467.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) A. J. Esswein, Y. Surendranath, S. Y. Reece, D. G. Nocera, Energ. Environ. Sci. 2011, 4, 499.
| 1:CAS:528:DC%2BC3MXivF2msL0%3D&md5=0c6e8ce604f0c9b93b3bc2d2236b01e0CAS |
(b) M. W. Kanan, D. G. Nocera, Science 2008, 321, 1072.
| Crossref | GoogleScholarGoogle Scholar |
(c) Q. S. Yin, J. M. Tan, C. Besson, Y. V. Geletii, D. G. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle, C. L. Hill, Science 2010, 328, 342.
| Crossref | GoogleScholarGoogle Scholar |
(d) O. Metin, E. Kocak, S. Ozkar, React. Kinet., Mech. Catal. 2011, 103, 325.
| Crossref | GoogleScholarGoogle Scholar |
[8] G. L. Elizarova, G. M. Zhidomirov, V. N. Parmon, Catal. Today 2000, 58, 71.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisFyqt7k%3D&md5=5740faa55435e1b99b1ef741b07d70c3CAS |
[9] J. G. McAlpin, Y. Surendranath, M. Dinca, T. A. Stich, S. A. Stoian, W. H. Casey, D. G. Nocera, R. D. Britt, J. Am. Chem. Soc. 2010, 132, 6882.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVeju70%3D&md5=e0d5c4c45a7e130e756a2f625f5c667fCAS |
[10] D. C. I. Yao, D. C. Brune, D. Vavilin, W. F. J. Vermaas, J. Biol. Chem. 2012, 287, 682.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotVGl&md5=058e8748971365d6d386b830771455c4CAS |
[11] R. K. Hocking, R. Brimblecombe, L. Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey, L. Spiccia, Nat. Chem. 2011, 3, 461.
| 1:CAS:528:DC%2BC3MXmtV2htL0%3D&md5=e1a1da8eed8e7449a5936fba7038db72CAS |
[12] M. Badawi, J. F. Paul, S. Cristol, E. Payen, Y. Romero, F. Richard, S. Brunet, D. Lambert, X. Portier, A. Popov, E. Kondratieva, J. M. Goupil, J. El Fallah, J. P. Gilson, L. Mariey, A. Travert, F. Mauge, J. Catal. 2011, 282, 155.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVSns73O&md5=ad4c2bf67c999551aeb1a3cbe8b07b64CAS |
[13] (a) F. E. Osterloh, Chem. Mater. 2008, 20, 35.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVWqsL3E&md5=9ea2398a70b48b0126639f609bbf7158CAS |
(b) S. Ikeda, T. Nakamura, S. M. Lee, T. Yagi, T. Harada, T. Minegishi, M. Matsumura, ChemSusChem 2011, 4, 262.
(c) I. Tsuji, H. Kato, A. Kudo, Angew. Chem. Int. Ed. 2005, 44, 3565.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. Kudo, Y. Miseki, Chem. Soc. Rev. 2009, 38, 253.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFWjtL3P&md5=9be15ab9c4385e4c91950d64e164b96eCAS |
[15] (a) K. Kalyanasundaram, E. Borgarello, D. Duonghong, M. Gratzel, Angew. Chem. Int. Ed. 1981, 20, 987.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. M. T. Khan, R. C. Bhardwaj, C. M. Jadhav, J. Chem. Soc., Chem. Commun. 1985, 1690.
[16] K. Maeda, K. Teramura, K. Domen, J. Catal. 2008, 254, 198.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXisVensrk%3D&md5=1d6974dcbaa79739bec82501f54cfde2CAS |
[17] S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno, H. Tributsch, Int. J. Hydrogen Energy 2001, 26, 653.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXkvVegsLY%3D&md5=1dcbaf7002e48f980b404e04ff100fb9CAS |
[18] (a) K. Maeda, K. Teramura, D. L. Lu, T. Takata, N. Saito, Y. Inoue, K. Domen, Nature 2006, 440, 295.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XitlKgtbc%3D&md5=7cd087a56a8b9223ed860a9e1384d27eCAS |
(b) Y. D. Hou, B. L. Abrams, P. C. K. Vesborg, M. E. Bjorketun, K. Herbst, L. Bech, A. M. Setti, C. D. Damsgaard, T. Pedersen, O. Hansen, J. Rossmeisl, S. Dahl, J. K. Norskov, I. Chorkendorff, Nat. Mater. 2011, 10, 434.
| Crossref | GoogleScholarGoogle Scholar |
[19] O. Khaselev, J. A. Turner, Science 1998, 280, 425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXivVeltro%3D&md5=27e2d14aa02bba8bd74f436628607869CAS |
[20] O. Khaselev, A. Bansal, J. A. Turner, Int. J. Hydrogen Energy 2001, 26, 127.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXosFels74%3D&md5=4d1ae912705e484f600dd4709c909eafCAS |
[21] S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers, D. G. Nocera, Science 2011, 334, 645.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlyqu7vF&md5=e0298161b92e218b9b45c56538dc6eb5CAS |
[22] K. Sivula, F. Le Formal, M. Gratzel, ChemSusChem 2011, 4, 432.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXks1WktLY%3D&md5=2d427f9baccfe01e0c406edebe721136CAS |
[23] C. X. Kronawitter, L. Vayssieres, S. H. Shen, L. J. Guo, D. A. Wheeler, J. Z. Zhang, B. R. Antoun, S. S. Mao, Energ. Environ. Sci. 2011, 4, 3889.
| 1:CAS:528:DC%2BC3MXhsVKitbvK&md5=d55f23c91e04b5cef4104d7f19d02e64CAS |
[24] Z. G. Yi, J. H. Ye, N. Kikugawa, T. Kako, S. X. Ouyang, H. Stuart-Williams, H. Yang, J. Y. Cao, W. J. Luo, Z. S. Li, Y. Liu, R. L. Withers, Nat. Mater. 2010, 9, 559.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnvVOktrc%3D&md5=63796058a8604ba017792d8060edc2efCAS |
[25] K. Sivula, F. Le Formal, M. Gratzel, ChemSusChem 2011, 4, 432.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXks1WktLY%3D&md5=2d427f9baccfe01e0c406edebe721136CAS |
[26] A. J. Nozik, Annu. Rev. Phys. Chem. 1978, 29, 189.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXht1Cnurc%3D&md5=f4909c5e84352ede7628af521bf7fc2dCAS |
[27] J. B. Gerken, J. G. McAlpin, J. Y. C. Chen, M. L. Rigsby, W. H. Casey, R. D. Britt, S. S. Stahl, J. Am. Chem. Soc. 2011, 133, 14431.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVGjtbnN&md5=9618355b89b666e65c02c13ff3f5ba48CAS |
[28] R. Kotz, S. Stucki, D. Scherson, D. M. Kolb, J. Electroanal. Chem. 1984, 172, 211.
| Crossref | GoogleScholarGoogle Scholar |
[29] B. Beverskog, I. Puigdomenech, Corros. Sci. 1997, 39, 969.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjtVCktr8%3D&md5=23fe3d20666004fa7948c5c065950fafCAS |
[30] B. S. Yeo, A. T. Bell, J. Am. Chem. Soc. 2011, 133, 5587.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsVGjur8%3D&md5=7ff3d9cd48960667239b185426b940a3CAS |
[31] A. C. C. Tseung, S. Jasem, Electrochim. Acta 1977, 22, 31.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2sXhs1altrg%3D&md5=1cfe03fb6c9cb67c7fcaa6d46d519888CAS |
[32] D. A. Lutterman, Y. Surendranath, D. G. Nocera, J. Am. Chem. Soc. 2009, 131, 3838.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisVKrt7Y%3D&md5=367b335a4c2d892733ee6093f9476d76CAS |
[33] A. Izgorodin, B. Winther-Jensen, D. R. MacFarlane, Catalysts and Methods of Use. PCT patent application 2012.