

Flexible Analogues of Azaindole DYRK1A Inhibitors Elicit Cytotoxicity in Glioblastoma Cells*
Qingqing Zhou A , Tristan A. Reekie A , Ramzi H. Abbassi B , Dinesh Indurthi Venkata B , Josep S. Font C , Renae M. Ryan C , Louis M. Rendina A , Lenka Munoz B and Michael Kassiou A DA School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia.
B School of Medical Sciences, Discipline of Pathology and Charles Perkins Centre, The University of Sydney, Sydney, NSW 2006, Australia.
C School of Medical Sciences, Discipline of Pharmacology, The University of Sydney, Sydney, NSW 2006, Australia.
D Corresponding author. Email: michael.kassiou@sydney.edu.au
Australian Journal of Chemistry 71(10) 789-797 https://doi.org/10.1071/CH18251
Submitted: 28 May 2018 Accepted: 26 June 2018 Published: 30 July 2018
Abstract
DYRK1A is a novel target for epidermal growth factor receptor (EGFR)-dependent glioblastoma and it represents a promising strategy for cancer therapy. DYRK1A inhibition has been found to promote EGFR degradation in glioblastoma cells by triggering endocytosis and lysosomal degradation, thus reducing the self-renewal ability of tumorigenic cells. Using a deconstruction approach of a DYRK1A lead molecule DANDY (1a), a set of novel ring-opened compounds was prepared. Despite showing no activity towards DYRK1A, a reduction in the viability of glioblastoma cells was observed with some of the compounds. This suggests other mechanistic pathways are leading to the apoptosis of glioblastoma cells.
Introduction
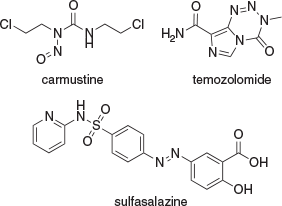
Glioblastoma, the most aggressive subtype of high-grade glioma (HGG), has been studied for decades.[1] Patients with glioblastomas have a median survival time of ~15 months, with an extremely high chance of recurrence after initial therapy.[2] The treatment currently used includes surgical resection, followed by chemo- and/or radiotherapeutic regimens (Fig. 1). For example, the use of 1,3-bis(2-chloroethyl)-1-nitrosourea (carmustine) is very common as an adjuvant concomitant with radiotherapy, which affords significant higher survival rates in comparison with radiotherapy alone.[3] The alkylating agent temozolomide (TMZ) in addition to radiotherapy has notably prolonged survival rates among patients,[4] and the co-administration of TMZ with sulfasalazine (SAS) was also demonstrated to reduce cell viability significantly in both primary glioblastomas and established A172 cells.[5] Among these agents, the monoclonal antibody-derived avastin has been approved as a single drug to treat patients with recurrent HGG in their second-line treatment.[1a] Pozo and coworkers recently reported a promising therapeutic intervention by targeting dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) for epidermal growth factor receptor (EGFR)-dependent glioblastomas.[6] DYRK1A is upregulated in glioblastoma cells.[6] The increased phosphorylation of sprouty2 mediated by DYRK1A blocks EGFR degradation as a result of overexpression of EGFR at the cell surface, and the enhanced EGFR signalling eventually leads to tumour survival. In contrast, DYRK1A inhibition has been found to promote EGFR degradation in glioblastoma cells by triggering endocytosis and lysosomal degradation, thus reducing the self-renewal ability of tumorigenic cells.[6,7] Therefore, DYRK1A as a novel target in EGFR-dependent glioblastoma represents a promising strategy for cancer therapy.
|
7-Azaindole DANDY (1a)[8] is one of the most potent inhibitors of DYRK1A, and we recently reported[7] the pharmacological evaluation of a series of 7-azaindole-based compounds against several kinases (DYRK1A, 1B, 2, and CLK1) belonging to the CMGC family of kinases in glioblastoma. The chemical structures of the 7-azaindole series are related to the adenine core of adenosine triphosphate (ATP), and therefore act as ATP-competitive inhibitors, providing high target selectivity by the engagement of hydrogen bond donors and acceptors. However, the weakness of these inhibitors lies in their high lipophilicity resulting in wider distribution within the body and a higher risk of toxicity. Therefore, we sought to expand on these studies to investigate diverse skeletons with increased hydrophilicity while retaining the hydrogen-bonding characteristics of the lead compounds. To that end, we first examined lead compound 1a, deconstructing it to the 5-membered ring giving rise to the 1-amino pyridine compound 2 with various linker lengths (n = 0 to 3) (Scheme 1). This was then further deconstructed, opening the pyridine ring to produce the secondary or tertiary amine chain compound 3 or 4 respectively, and again examining various linker lengths (n = 0 to 2). We then assessed these compounds in a DYRK1A inhibition assay and also a tumour and non-tumour cell-based viability assay.

|
Results and Discussion
The pyridine diphenol 2c was prepared from commercially available 2,5-dibromopyridine (5), which was treated with p-methoxyphenyletheneamine to give intermediate 6,[9] followed by a Suzuki cross-coupling reaction resulting in aryl-substituted pyridine 7 (Scheme 2). Subsequent demethylation by BBr3 afforded the final compound 2c. The synthetic routes to the remaining analogues of compound 2 were designed from the same starting material 8 (Scheme 2). The intermediate 10 was initially designed by following a similar synthetic route to that for the formation of intermediate 6. Initial attempts employed a nucleophilic aromatic substitution under basic conditions to afford the corresponding secondary amine, but these were unsuccessful. Alternatively, a Cu(OAc)2 Chan–Lam coupling[10] of 8 with tert-butyldimethylsilyl (TBS)-protected phenylboronic acid 9 in air produced 10 in moderate yield. A sequential Suzuki cross-coupling reaction, also with boronic acid 9, was employed to afford compound 11, which, following the deprotection of TBS groups,[11] resulted in the desired compound 2a. The route to compound 2b from the same starting material 8 first employed a reductive amination with 4-hydroxybenzaldehyde and NaBH(OAc)3 in trifluoroethanol at room temperature.[12] A subsequent Suzuki cross-coupling reaction afforded the desired compound 2b in reasonable yield. Reductive amination of 8 with 3-(4-(benzyloxy)phenyl)propanal (13) following similar procedures previously mentioned gave intermediate 14. A Suzuki coupling to give 15 and hydrogenolysis to remove the protecting group afforded the desired compound 2d.

|
Compounds 3a and 3c could be synthesized[13] from p-hydroxybenzyl amine 16a or 16b respectively and glyoxal to give the corresponding Schiff bases, followed by the reduction with NaBH4 to give the secondary amines 3a and 3c (Scheme 3), whereas tertiary amines 4a and 4c could be easily obtained by reductive amination using 37 % formaldehyde and NaBH4 in EtOH. Similarly, condensation of p-hydroxybenzaldehyde (17) with ethylenediamine afforded the diimine intermediate, which, following reduction, gave 3b. An additional reductive amination of 3b produced tertiary amine 4b in comparable yield.

|
The potency of these analogues for DYRK1A inhibition was determined by performing a kinase assay in duplicate with Woodtide peptide as a substrate and an ATP concentration of 100 μM. The lead compound 1a was confirmed to have inhibitory potency against DYRK1A, with a half maximal inhibitory concentration (IC50) value of 14 nM and our improved acetamide 1b[7] showed an IC50 value of 6.6 nM (Table 1). We first assessed the pyrrole ring-opened 2-aminopyridine compounds 2a–d. The diarylamine 2a showed no inhibition at >1000 nM with a greater than 100-fold loss of potency in comparison with lead compound 1a (Table 1). We assumed that the unreasonable distance and angles between the two phenols prevented inhibitory activity. Compounds 2b–d, which avoided this problem by increasing the distance between the two phenols, were also tested, but showed similar results; IC50 values were increased to >1000 nM for all aminopyridine compounds (Table 1).
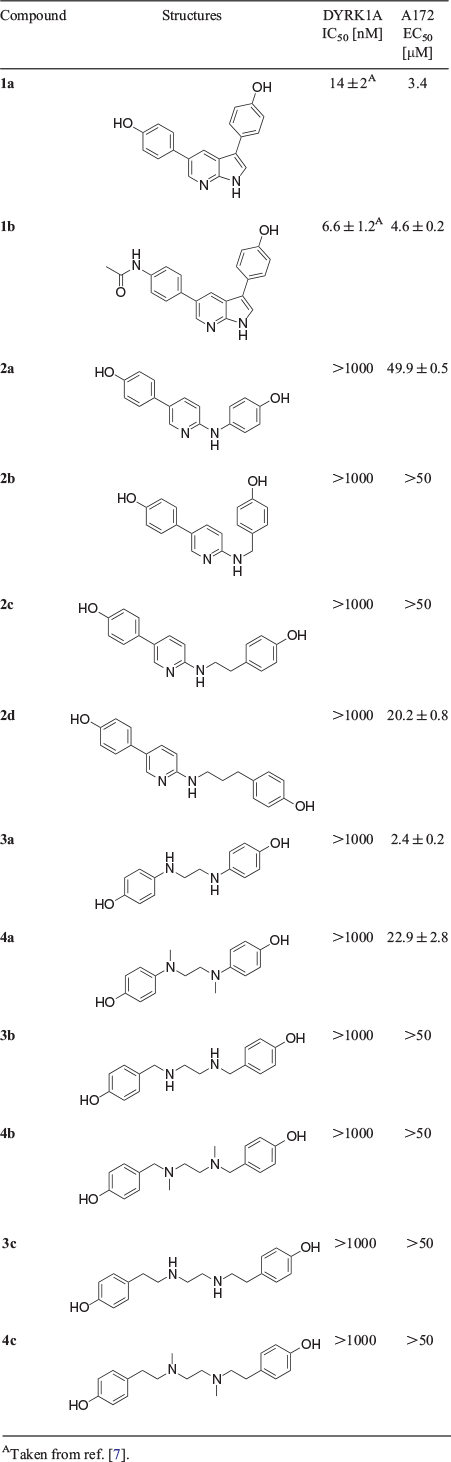
|
Despite these results, we expected that compounds 3a–c and 4a–c, where the pyridine ring was also disconnected, would show better inhibitory effects, which was rationalized in two ways. First, these compounds were now symmetrical, giving greater malleability within the binding pocket. Second, the two nitrogen atoms are essential for inhibition[7] and by increasing the flexibility between these two atoms and not just the phenol groups, we anticipated that enhanced target interactions could be achieved. The hydrogen-bond donor and acceptor type and not just acceptor was also explored with compounds 3 and 4 respectively. Unfortunately, these rationalizations proved to be unfounded, as compounds 3a–c did not improve inhibitory potency against DYRK1A. All IC50 values were shown to be >1000 nM (Table 1). These results were replicated with the methylated tertiary amines 4a–c, with values also >1000 nM (Table 1).
Despite the lack of activity of these compounds towards DYRK1A, we explored their cytotoxicity against the established A172 glioblastoma cell line. In our investigation, lead compound 1a showed cytotoxicity against the A172 cell line with half maximal effective concentration (EC50) values of 3.4 μM, which is in line with results obtained with the improved DYRK1A inhibitor 1b (Table 1). In comparison, analogue 2d, which bears a three-carbon chain linker between the phenol amine and pyridine ring, showed moderate cytotoxic activity (20.2 μM) despite the total loss of DYRK1A inhibition, although analogues 2a–c exhibited EC50 values of more than 50 μM (Table 1). More interestingly, secondary amine 3a with disconnection of both the pyrrole and pyridine rings showed the best cytotoxicity against A172 cells, with an EC50 value of 2.4 μM. This means that although 3a did not show any inhibition against DYRK1A, it exhibited more cytotoxicity than lead compound 1a. When testing methylated tertiary amine 4a, much weaker activity was observed, though moderate cytotoxicity was still present (EC50 22.9 μM). Secondary amines 3b–c or tertiary amines 4b–c with a longer distance between the amine and aromatic groups totally lost their cytotoxic behaviour in this study.
Conclusion
In conclusion, we generated a library of 10 novel compounds and subjected them to inhibition assays against DYRK1A. Unfortunately, all the new compounds reported, 2a–d, 3a–c, and 4a–c, displayed no inhibition of DYRK1A, confirming the importance of the azaindole motif. We can conclude that it is not just the positioning of two nitrogen atoms in a similar spatial arrangement that affords strong potency. Indeed, the importance of the aromatic skeleton with specific spatial arrangements of the two nitrogen atoms is confirmed. These data will be of use for future structure–activity relationship studies to further improve the selective inhibition of DYRK1A.
Additionally, an interesting finding of the present study is that although ring-opened compounds completely lost inhibitory activity against our initial target DYRK1A, some of them exhibit markedly potent cytotoxicity against glioblastoma cells. In particular, analogue 3a with no inhibition against DYRK1A was cytotoxic against cancer cells at higher potencies than the lead compound 1a, with an EC50 value of 2.4 μM. Nevertheless, further investigations are necessary to explore the exact mechanism of how these compounds exhibit their glioblastoma cytotoxicity, and to analyse their effects in vivo.
Experimental
General Chemical Synthesis Details
Unless noted otherwise, commercially obtained reagents were used as purchased without further purification. Solvents for flash chromatography were distilled before use, or used as purchased for HPLC grade, with the eluent mixture reported as the volume/volume ratio (v/v). Flash chromatography was performed using Merck Kieselgel 60 (230–400 mesh) silica gel. Analytical thin-layer chromatography was performed using Merck aluminium-backed silica gel 60 F254 (0.2 mm) plates (Merck, Darmstadt, Germany), which were visualized using shortwave (254 nm) ultraviolet fluorescence. Melting points were measured with a rate of 6°C min−1 and are uncorrected. Infrared absorption spectra are reported as vibrational frequency (cm−1). Nuclear magnetic resonance spectra were recorded at 300 K using a 200, 300, 400 or 500-MHz spectrometer. The data are reported as chemical shift (δ, ppm) relative to the residual protonated solvent resonance, relative integral, multiplicity (s, singlet; br s, broad singlet; d, doublet; dd, doublet of doublets; t, triplet; m, multiplet, etc.), and coupling constants (J, Hz). Low-resolution mass spectra (LRMS) were obtained from a ThermoQuest Finnigan LCQ Deca ion-trap mass spectrometer with electrospray ionization in positive (+ESI) mode. Data are expressed as observed mass (m/z), assignment (M = molecular ion) and relative intensity (%). High-resolution mass spectroscopy was performed on a Bruker Apex Qe 7T Fourier-transform ion cyclotron resonance mass spectrometer equipped with an Apollo Π ESI dual source. Samples were run with syringe infusion at 150 μL h−1 on a Cole Palmer syringe pump into ESI. High-performance liquid chromatography (HPLC) analysis of organic purity was conducted on a Waters Alliance 2695 instrument using a SunFireTM C18 column (5 μm, 2.1 × 150 mm) and detected using a Waters 2996 photodiode array (PDA) detector set at 254 nm. Separation was achieved using water (solvent A) and acetonitrile (solvent B) at flow rate of 0.2 mL min−1 and a gradient of 0 to 100 % B (Method A) or 0 to 80 % B (Method B) or 0 to 40 % B (Method C) over 30 min. HPLC data are reported as percentage purity and retention time (RT) in minutes.
General Procedure A for Suzuki Coupling Reaction
To a solution of aryl bromide (1.0 equiv.) in 1,4-dioxane (0.05 M) were added arylboronic acid (1.0 equiv.), aq. K2CO3 (2 M, 2.0 equiv.), and Pd(PPh3)4 (2 mol-%), and the reaction was heated to 90°C for 5 h under argon. The reaction mixture was cooled to room temperature, solvent was removed under reduce pressure, and the product then partitioned between water and ethyl acetate. The aqueous layer was extracted with ethyl acetate, and the combined organic layers were dried over MgSO4 and concentrated under vacuum.
General Procedure B for Reductive Aminations[12]
To a solution of 4-hydroxybenzaldehyde or 13 (1.0 equiv.) in 2,2,2-trifluoroethanol (0.04 M) was added 5-bromopyridin-2-amine 8 (1.0 equiv.), and the mixture was stirred at room temperature (rt). After 30 min, NaBH(OAc)3 (1.5 equiv.) was added and the mixture stirred for 12 h at rt. The solvent was removed under reduced pressure, and the residue was purified by flash chromatography to give the products.
General Procedure C for the Synthesis of Secondary Amines
Compounds were prepared according to the literature.[14] To a solution of glyoxal (40 % in ethanol, 0.9 equiv.) in ethanol (0.10 M) was added amine 16a–b (1.1 equiv.), and the mixture was stirred at rt for 4 h, filtered and washed with hexane, and dried under vacuum to get the yellow solid intermediate diimine without further purification. To a solution of the intermediate diimine in CH2Cl2 and methanol (1 : 1 v/v, 0.03 M) was added NaBH4 (1.0 equiv.) at 0°C, and the mixture stirred at room temperature for 30 min. The solvent was removed under reduced pressure, and water (10 mL) was added, affording a white precipitate, which was filtered and washed with hexane, recrystallized in ethanol, and dried under vacuum to get products 3a and 3c respectively.
General Procedure D for Methylation of Secondary Amines to Tertiary Amines
To a solution of secondary amine-based diphenol (1.0 equiv.) in acetic acid (0.04 M) was added formaldehyde (37 % aqueous, 10.0 equiv.), and the mixture was stirred at room temperature. After 30 min, NaBH4 (1.0 equiv.) was added and the mixture was stirred for another 30 min. The solvent was removed under reduced pressure and the residue was taken up in Na2CO3(aq.) and extracted with ethyl acetate (3 × 15 mL), and the combined organic layers were dried over MgSO4 and concentrated under vacuum.
5-Bromo-N-(4-methoxyphenethyl)pyridin-2-amine (6)
To a solution of 5-bromo-N-(4-methoxyphenethyl)pyridin-2-amine 5 (200 mg, 0.65 mmol) in DMF (15 ml) was added K2CO3 (270 mg, 1.95 mmol) and 2-(4-methoxyphenyl)ethan-1-amine (148 μL, 0.78 mmol), and the mixture was stirred at 120°C for 8 h. The mixture was quenched with water and extracted with ethyl acetate. The combined organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified using flash chromatography (hexane/ethyl acetate 10 : 1 → 4 : 1) to give the product as a brown solid (125 mg, 63 % yield). Mp. 154–156°C. Rf (hexane/ethyl acetate 2 : 1) 0.40. δH (500 MHz, CDCl3) 8.09 (1H, dd, J 0.4, 2.4), 7.44 (1H, dd, J 2.4, 8.8), 7.12 (2H, ddd, J 2.0, 2.9, 8.6), 6.84 (2H, ddd, J 2.0, 2.9, 8.6), 6.25 (1H, dd, J 0.4, 8.8), 4.60 (1H, br s), 3.78 (3H, s), 3.48 (2H, dd, J 6.9, 12.8), 2.84 (2H, t, J 6.9). δC (125 MHz, CDCl3) 158.4, 157.3, 148.8, 139.8, 131.0, 129.8, 114.2, 108.4, 107.0, 55.4, 43.6, 34.6. m/z (HRMS ESI+) 307.0443 and 309.0423; [M + H]+ requires 307.0441 and 309.0420 for C14H15N2OBr. νmax (neat)/cm−1 3334, 2954, 2930, 2835, 1635, 1502, 1241, 1026, 816, 528.
N-(4-Methoxyphenethyl)-5-(4-methoxyphenyl)pyridin-2-amine (7)
This compound was prepared according to General Procedure A. The residue was purified by flash chromatography (hexane/ethyl acetate 3 : 1 → 1 : 1) to give the product as an off-white solid (99 mg, 90 % yield). Mp 236–240°C. Rf (hexane/ethyl acetate 4 : 1) 0.40. δH (400 MHz, CDCl3) 8.30 (1H, d, J 2.0), 7.61 (1H, dd, J 2.5, 8.6), 7.42 (2H, dd, J 2.1, 6.7), 7.16 (2H, d, J 8.6), 6.96 (2H, dd, J 2.1, 6.7), 6.86 (2H, dd, J 2.1, 6.7), 6.43 (1H, d, J 8.6), 4.57 (1H, t, J 5.6), 3.84 (3H, s), 3.80 (3H, s), 3.56 (2H, dd, J 6.9, 12.9), 2.89 (2H, t, J 6.9). δC (100 MHz, CDCl3) 158.8, 158.4, 157.7, 146.1, 136.0, 131.3, 129.9, 127.3, 126.1, 114.5, 114.2, 106.8, 55.5, 55.4, 43.8, 34.9. m/z (HRMS ESI+) 357.1575; [M + Na]+ requires 357.1573 for C21H22N2O2. νmax (neat)/cm−1 3204, 2978, 2956, 1610, 1478, 1098, 876, 789.
4-(6-((4-Hydroxyphenethyl)amino)pyridin-3-yl)phenol (2c)
To a solution of 7 (1.0 equiv.) in dry CH2Cl2 (0.05 M) was added BBr3 (1 M solution in CH2Cl2, 6.0 equiv.) under nitrogen. The reaction mixture was stirred at room temperature for 15 h, then quenched at 0°C with MeOH and concentrated under vacuum. The residue was purified by flash chromatography (CH2Cl2/MeOH 100 : 1 → 40 : 1) to give the product as a white solid (54 mg, 59 % yield). Mp 156–158°C. Rf (CH2Cl2/MeOH 20 : 1) 0.45. δH (500 MHz, [D6]DMSO) 9.70 (1H, s), 9.23 (1H, s), 8.69 (1H, br s), 8.18 (1H, dd, J 2.1, 9.3), 8.01 (1H, s), 7.48 (2H, ddd, J 2.0, 2.9, 8.6), 7.13 (1H, s), 7.10 (2H, d, J 8.6), 6.87 (2H, ddd, J 2.0, 2.9, 8.6), 6.71 (2H, ddd, J 2.0, 2.9, 8.6), 3.55 (2H, dd, J 7.4, 11.5), 2.82 (2H, t, J 7.4). δC (125 MHz, [D6]DMSO) 158.2, 156.4, 151.7, 141.8, 131.9, 130.2, 128.7, 127.8, 125.5, 125.2, 116.4, 115.6, 114.1, 43.9, 33.6. m/z (HRMS ESI+) 329.1262; [M + Na]+ requires 329.1260 for C19H18N2O2. νmax (neat)/cm−1 3513, 3256, 3009, 1662, 1514, 833. HPLC 98.4 % (Method A), RT 16.5 min.
(4-((tert-Butyldimethylsilyl)oxy)phenyl)boronic Acid (9)
To a solution of 4-hydroxyphenylboronic acid (1.0 equiv., 5 mmol) in dry DMF (0.05 M) was added imidazole (5.0 equiv.) at rt, followed by the addition of TBSCl (3.5 equiv.). The reaction mixture was stirred at rt for 12 h; after completion monitored by TLC, the mixture was extracted with ethyl acetate (3 × 30 mL) and water (50 mL), and the organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1 → 3 : 1) to give the product as a white solid (2.27 g, 90 % yield). Rf (hexane/ethyl acetate 1 : 1) 0.30. δH (300 MHz, CDCl3) 8.11 (2H, d, J 8.4), 6.96 (2H, d, J 8.4), 1.02 (9H, s), 0.26 (6H, s). δC (75 MHz, CDCl3) 159.9, 137.6, 119.9, 111.0, 25.8, 18.4, −4.2. The spectroscopic data matched those reported in the literature.[15]
5-Bromo-N-(4-((tert-butyldimethylsilyl)oxy)phenyl)pyridin-2-amine (10)
To a solution of 5-bromopyridin-2-amine 8 (1.0 equiv., 1 mmol) in MeOH (0.05 M) was added 9 (1.2 equiv., 1.2 mmol) at rt, followed by the addition of Cu(OAc)2 (0.1 equiv., 0.1 mmol). The reaction mixture was stirred for 12 h under air at rt. After completion monitored by TLC, the metal solid was filtered through Celite®, and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1) to give the product as a white solid (227 mg, 60 % yield). Mp 131–132°C. Rf (hexane/ethyl acetate 10 : 1) 0.40. δH (400 MHz, CDCl3) 8.16 (1H, d, J 2.0), 7.51 (1H, dd, J 2.0, 8.8), 7.14 (2H, dd, J 2.0, 6.4), 6.83 (2H, dd, J 2.0, 6.4), 6.60 (1H, d, J 8.8), 6.55 (1H, s), 0.99 (9H, s), 0.20 (6H, s). δC (100 MHz, CDCl3) 155.8, 152.8, 148.4, 140.5, 133.0, 124.2, 121.0, 109.0, 108.4, 25.8, 18.4, −4.3. m/z (HRMS ESI+) 401.0659 and 403.0639; [M + Na]+ requires 401.0655 and 403.0635 for C17H23BrN2OSi. νmax (neat)/cm−1 3244, 2947, 2855, 1605, 1525, 1251, 904, 778.
N,5-Bis(4-((tert-butyldimethylsilyl)oxy)phenyl)pyridin-2-amine (11)
This compound was prepared according to General Procedure A. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1 → 5 : 1) to give the product as a white solid (203 mg, 80 % yield). Mp 135–136°C. Rf (hexane/ethyl acetate 5 : 1) 0.45. δH (300 MHz, CDCl3) 8.26 (1H, s), 7.71 (1H, d, J 9.0), 7.37–7.34 (3H, m), 7.18 (2H, d, J 8.1), 6.90 (2H, d, J 8.1), 6.86–6.79 (3H, m), 1.00 (18H, s), 0.22 (6H, s), 0.21 (6H, s). δC (75 MHz, CDCl3) 155.6, 155.1, 153.0, 142.9, 137.8, 132.7, 130.5, 127.5, 127.4, 124.4, 121.1, 120.8, 108.3, 25.8 (two overlapping signals), 18.4 (two overlapping signals), −4.2 (two overlapping signals). m/z (HRMS ESI+) 507.2861; [M + H]+ requires 507.2858 for C29H42N2O2Si2. νmax (neat)/cm−1 3232, 2962, 2856, 1603, 1508, 1249, 911, 822, 775.
4-(6-((4-Hydroxyphenyl)amino)pyridin-3-yl)phenol (2a)
To a solution of 11 (1.0 equiv., 0.1 mmol) in dry THF (5 mL) was added tetra-n-butylammonium fluoride (TBAF) (1.5 equiv., 0.15 mmol) at rt dropwise, and the reaction mixture was stirred for 10 min under a nitrogen atmosphere and the solvent was removed under reduced pressure. The residue was washed with H2O (10 mL) and extracted with ethyl acetate (3 × 10 mL), the organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified by flash chromatography (hexane/ethyl acetate 3 : 1 → 1 : 1) to give the product as a pale yellow solid (53 mg, 95 % yield). Mp 199–200°C. Rf (hexane/ethyl acetate 1 : 1) 0.30. δH (400 MHz, [D4]MeOD) 8.18 (1H, dd, J 0.8, 2.8), 7.70 (1H, dd, J 2.8, 8.8), 7.37 (2H, ddd, J 2.8, 4.8, 9.6), 7.21 (2H, ddd, J 3.2, 5.6, 10.0), 6.84 (2H, ddd, J 2.8, 4.8, 9.6), 6.78 (2H, ddd, J 3.2, 5.6, 10.0), 6.74 (1H, dd, J 0.8, 8.8). δC (100 MHz, [D4]MeOD) 157.9, 157.4, 154.5, 145.5, 137.3, 134.1, 130.8, 128.4, 128.1, 124.5, 116.8, 116.7, 110.1. m/z (HRMS ESI+) 279.1131; [M + H]+ requires 279.1128 for C17H14N2O2. νmax (neat)/cm−1 3217, 2953, 1607, 1490, 1218, 815. HPLC >99.9 % (Method A), RT 16.2 min.
4-(((5-Bromopyridin-2-yl)amino)methyl)phenol (12)
This compound was prepared according to General Procedure B. The residue was purified by flash chromatography (hexane/ethyl acetate 5 : 1 → 3 : 1) to give the product as a white solid (190 mg, 68 % yield). Mp 125–128°C. Rf (hexane/ethyl acetate 4 : 1) 0.30. δH (400 MHz, [D4]MeOD) 7.98 (1H, d, J 2.4), 7.49 (1H, dd, J 2.4, 8.8), 7.17 (2H, dd, J 2.0, 6.8), 6.75 (2H, dd, J 2.0, 6.8), 6.48 (1H, d, J 8.8), 4.36 (2H, s). δC (100 MHz, [D4]MeOD) 159.0, 157.6, 148.5, 140.9, 131.3, 129.8, 116.2, 111.4, 106.9, 46.0. m/z (HRMS ESI+) 300.9950 and 302.9930; [M + Na]+ requires 300.9947 and 302.9926 for C12H11BrN2O. νmax (neat)/cm−1 3218, 1594, 1343, 815.
4-(6-((4-Hydroxybenzyl)amino)pyridin-3-yl)phenol (2b)
This compound was prepared according to General Procedure A. The residue was purified by flash chromatography (hexane/ethyl acetate 3 : 1 → 1 : 1) to give the product as a pale yellow solid (51 mg, 70 % yield). Mp 193–196°C. Rf (hexane/ethyl acetate 1 : 1) 0.35. δH (300 MHz, [D4]MeOD) 8.13 (1H, s), 7.65 (1H, d, J 8.7), 7.34 (2H, d, J 8.1), 7.20 (2H, d, J 8.1), 6.85 (2H, d, J 8.1), 6.76 (2H, d, J 8.1), 6.58 (1H, d, J 8.7), 4.41 (2H, s). δC (75 MHz, [D4]MeOD) 158.9, 157.7, 157.5, 145.2, 137.2, 131.6, 131.0, 129.7, 128.1, 127.1, 116.7, 116.2, 109.6, 46.3. m/z (HRMS ESI+) 607.2321; [M + Na]+ requires 607.2316 for (C18H16N2O2)2. νmax (neat)/cm−1 3211, 3025, 1613, 1508, 1247, 822, 529. HPLC >99.9 % (Method B), RT 16.2 min.
3-(4-(Benzyloxy)phenyl)propanal (13)
To a solution of 4-(3-hydroxypropyl)phenol (1.0 equiv., 1 mmol) in acetone (0.05 M) was added K2CO3 (1.5 equiv., 1.5 mmol), followed by the addition of benzyl bromide (1.2 equiv., 1.2 mmol) dropwise at rt, and the mixture was stirred at rt for 12 h. After completion, the solvent was removed under reduced pressure, and the residue was washed with H2O (30 mL) and extracted with ethyl acetate (3 × 20 mL); the organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1 → 5 : 1) to give the product 3-(4-(benzyloxy)phenyl)propan-1-ol as a white solid (230 mg, 95 % yield). Rf (hexane/ethyl acetate 4 : 1) 0.30. δH (400 MHz, CDCl3) 7.48–7.33 (5H, m), 7.15 (2H, ddd, J 2.8, 5.2, 9.6), 6.95 (2H, ddd, J 2.8, 5.2, 9.6), 5.06 (2H, s), 3.66 (2H, t, J 6.4), 2.70–2.66 (2H, m), 2.19 (1H, s), 1.92–1.85 (2H, m). δC (100 MHz, CDCl3) 157.1, 137.3, 134.3, 129.4, 128.6, 127.9, 127.5, 114.9, 70.1, 62.1, 34.4, 31.2. The spectroscopic data matched those reported in the literature.[16]
To a solution of 3-(4-(benzyloxy)phenyl)propan-1-ol (1.0 equiv., 0.5 mmol) in dry CH2Cl2 (10 mL) was added Dess–Martin periodinane (DMP, 1.5 equiv., 0.75 mmol), and the mixture was stirred at rt for 2 h. The mixture was quenched with a mixture of saturated NaHCO3 (2 mL) and Na2S2O3 (2 mL), and the resulting mixture was extracted with CH2Cl2 (3 × 10 mL), and the organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1) to give the product 13 as a white solid (114 mg, 95 % yield). Rf (hexane/ethyl acetate 10 : 1) 0.50. δH (300 MHz, CDCl3) 9.81 (1H, s), 7.44–7.32 (5H, m), 7.11 (2H, d, J 8.1), 6.91 (2H, d, J 8.1), 5.05 (2H, s), 2.91 (2H, t, J 7.2), 2.75 (2H, t, J 7.2). δC (75 MHz, CDCl3) 201.9, 157.5, 137.2, 132.8, 129.4, 128.7, 128.1, 127.6, 115.1, 70.2, 45.6, 27.4. The spectroscopic data matched those reported in the literature.[17]
N-(3-(4-(Benzyloxy)phenyl)propyl)-5-bromopyridin-2-amine (14)
This compound was prepared according to General Procedure B. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1) to give the product as a white solid (353 mg, 89 % yield). Mp 97–98°C. Rf (hexane/ethyl acetate 10 : 1) 0.40. δH (400 MHz, CDCl3) 8.09 (1H, d, J 2.4), 7.47–7.31 (6H, m), 7.10 (2H, ddd, J 2.8, 5.2, 9.6), 6.91 (2H, ddd, J 2.8, 5.2, 9.6), 6.23 (1H, d, J 8.8), 5.05 (2H, s), 4.65 (1H, br.s), 3.24 (2H, t, J 6.4), 2.66 (2H, t, J 7.6), 1.91 (2H, tt, J 7.6, 14.4). δC (100 MHz, CDCl3) 157.5, 157.3, 148.7, 139.9, 137.3, 133.9, 129.4, 128.7, 128.0, 127.6, 115.0, 108.0, 106.8, 70.2, 41.9, 32.4, 31.2. m/z (HRMS ESI+) 397.0915 and 399.0894; [M + H]+ requires 397.0910 and 399.0890 for C21H21BrN2O. νmax (neat)/cm−1 3247, 2937, 1586, 1243, 815, 726.
4-(6-((3-(4-(Benzyloxy)phenyl)propyl)amino)pyridin-3-yl)phenol (15)
This compound was prepared according to General Procedure A. The residue was purified by flash chromatography (hexane/ethyl acetate 10 : 1 → 3 : 1) to give the product as a pale yellow solid (154 mg, 75 % yield). Mp 138–140°C. Rf (hexane/ethyl acetate 6 : 1) 0.30. δH (300 MHz, CDCl3) 8.26 (1H, s), 7.63 (1H, d, J 8.7), 7.44–7.34 (7H, m), 7.09 (2H, d, J 8.1), 6.93–6.88 (4H, m), 6.41 (1H, d, J 8.7), 5.03 (2H, s), 4.68 (1H, br s), 3.30–3.28 (2H, m), 2.67 (2H, t, J 7.2), 1.93 (2H, tt, J 7.2, 13.8). δC (75 MHz, CDCl3) 157.5, 157.3, 155.9, 145.3, 137.3, 136.6, 133.9, 130.5, 129.5, 128.7, 128.0, 127.6 (two overlapping signals), 126.3, 116.2, 115.0, 106.5, 70.2, 42.0, 32.4, 31.3. m/z (HRMS ESI+) 411.2071; [M + H]+ requires 411.2067 for C27H26N2O2. νmax (neat)/cm−1 3409, 3029, 2928, 2856, 1606, 1501, 1232, 811, 517.
4-(6-((3-(4-Hydroxyphenyl)propyl)amino)pyridin-3-yl)phenol (2d)
To a solution of 15 (1.0 equiv., 0.1 mmol) in MeOH (5 mL) was added Pd/C (10 wt-%, 5 mg) under a nitrogen atmosphere, and the mixture was stirred at room temperature for 2 h under 1 atm of H2. After completion, the Pd/C was filtered through Celite®, and the solvent of the filtrate was removed under reduced pressure. The residue was purified by flash chromatography (hexane/ethyl acetate 1 : 1) to give the product as a pale yellow solid (69 mg, 90 % yield). Mp 217–219°C. Rf (hexane/ethyl acetate 1 : 1) 0.40. δH (400 MHz, [D6]DMSO) 9.37 (1H, br s), 9.11 (1H, br s), 8.18 (1H, d, J 2.4), 7.57 (1H, dd, J 2.4, 8.8), 7.34 (2H, dd, J 2.0, 6.4), 7.00 (2H, dd, J 2.0, 6.4), 6.79 (2H, dd, J 2.0, 6.4), 6.67 (2H, dd, J 2.0, 6.4), 6.52–6.48 (2H, m), 3.25–3.20 (2H, m), 2.54 (2H, t, J 7.6), 1.77 (2H, tt, J 7.6, 14.8). δC (100 MHz, [D6]DMSO) 157.7, 156.1, 155.2, 144.7, 134.5, 131.9, 129.1 (two overlapping signals), 126.5, 123.7, 115.7, 115.0, 107.9, 40.5, 31.9, 31.2. m/z (HRMS ESI+) 321.1601; [M + H]+ requires 321.1598 for C20H20N2O2. νmax (neat)/cm−1 3403, 3150, 2922, 2852, 1609, 1508, 1450, 1234, 813. HPLC 99.0 % (Method B), RT 18.3 min.
4,4′-(Ethane-1,2-diylbis(azanediyl))diphenol (3a)
This compound was prepared according to General procedure C to get the product as an off-white solid (220 mg, 90 % yield). Mp 170–172°C. Rf (CH2Cl2/MeOH 20 : 1) 0.35. δH (500 MHz, [D6]DMSO) 8.38 (2H, s), 6.54 (4H, d, J 8.8), 6.44 (4H, d, J 8.8), 4.90 (2H, s), 3.09 (4H, t, J 2.5). δC (125 MHz, [D6]DMSO) 148.3, 141.7, 115.7, 113.5, 43.4. m/z (HRMS ESI+) 267.1104; [M + Na]+ requires 267.1104 for C14H16N2O2. νmax (neat)/cm−1 3283, 3066, 2970, 1507, 641, 533. HPLC 96.5 % (Method A), RT 10.9 min.
4,4′-((Ethane-1,2-diylbis(azanediyl))bis(ethane-2,1-diyl))diphenol (3c)
This compound was prepared according to General procedure C to afford the product as an off-white solid (129 mg, 86 % yield). Mp 205–209°C. Rf (CH2Cl2/MeOH 20 : 1) 0.30. δH (500 MHz, [D6]DMSO) 6.96 (4H, d, J 8.1), 6.66 (4H, d, J 8.1), 2.65–2.62 (4H, m), 2.55–2.52 (8H, m), NH and OH signals not observed. δC (125 MHz, [D6]DMSO) 155.9, 130.9, 129.8, 115.5, 51.9, 49.4, 35.6. m/z (HRMS ESI+) 301.1910; [M + H]+ requires 301.1910 for C18H24N2O2. νmax (neat)/cm−1 3179, 3019, 2814, 1671, 1182, 1131. HPLC >99.9 % (Method C), RT 16.2 min.
4,4′-(Ethane-1,2-diylbis(methylazanediyl))diphenol (4a)
This compound was prepared according to General Procedure D. The residue was purified by flash chromatography (CH2Cl2/MeOH 100 : 1 → 80 : 1) to give the product as a white solid (60 mg, 88 % yield). Mp 174–176°C. Rf (CH2Cl2/MeOH 20 : 1) 0.45. δH (400 MHz, [D4]MeOD) 6.70–6.34 (8H, m), 3.33 (4H, s), 2.81 (6H, s), OH signals not observed. δC (100 MHz, [D4]MeOD) 150.6, 144.7, 116.9, 116.8, 52.4, 40.2. m/z (HRMS ESI+) 273.1597; [M + H]+ requires 273.1597 for C16H20N2O2. νmax (neat)/cm−1 3489, 3377, 3100, 1662, 1192, 545. HPLC 99.7 % (Method C), RT 16.7 min.
4,4′-((Ethane-1,2-diylbis(methylazanediyl))bis(ethane-2,1-diyl))diphenol (4c)
This compound was prepared according to General Procedure D. The residue was purified by flash chromatography (CH2Cl2/MeOH 100 : 1 → 80 : 1) to give the product as a pale brown solid (73 mg, 89 % yield). Mp 229–230°C. Rf (CH2Cl2/MeOH 20 : 1) 0.40. δH (400 MHz, [D6]DMSO) 9.07 (2H, br s), 6.96 (4H, d, J 8.4), 6.62 (4H, d, J 8.4), 2.56–2.52 (4H, m), 2.48–2.43 (4H, m), 2.40 (4H, s), 2.17 (6H, s). δC (100 MHz, [D6]DMSO) 155.8, 131.0, 129.9, 115.4, 60.2, 55.4, 42.6, 32.6. m/z (HRMS ESI+) 329.2224; [M + H]+ requires 329.2223 for C20H28N2O2. νmax (neat)/cm−1 3201, 2939, 2622, 1513, 1200, 830, 659. HPLC 95.7 % (Method A), RT 12.3 min.
4,4′-((Ethane-1,2-diylbis(azanediyl))bis(methylene))diphenol (3b)
This compound was prepared according to the literature method.[18] To a solution of ethylenediamine (55 μL, 0.82 mmol) in toluene (20 mL) was added 4-hydroxybenzaldehyde 17 (200 mg, 1.64 mmol), and the mixture was stirred at 110°C for 20 min, forming a yellow precipitate, which was filtered without further purification; the solid was dissolved in methanol (20 mL), followed by treatment with NaBH4 (15.9 mg, 0.42 mmol) at 0°C, and heated at reflux for 20 min. The solvent was removed under reduced pressure. The mixture was quenched with NH4Cl(aq) and extracted with ethyl acetate (3 × 15 mL) and the combined organic layers were dried over MgSO4 and concentrated under vacuum. The residue was purified by flash chromatography (CH2Cl2/MeOH 100 : 1 → 80 : 1) to give the product as off-white solid 3b (207 mg, 93 % yield). Mp 139–141°C. Rf (CH2Cl2/MeOH 20 : 1) 0.35. δH (500 MHz, [D6]DMSO) 9.17 (2H, s), 7.07 (4H, d, J 8.4), 6.67 (4H, d, J 8.4), 3.52 (4H, s), 2.53 (4H, s). δC (125 MHz, [D6]DMSO) 155.9, 131.2, 128.9, 114.7, 52.5, 48.2. m/z (HRMS ESI+) 273.1598; [M + H]+ requires 273.1597 for C16H20N2O2. νmax (neat)/cm−1 3254, 3018, 2856, 1611, 1512, 876. HPLC >99.9 % (Method A), RT 10.8 min.
4,4′-((Ethane-1,2-diylbis(methylazanediyl))bis(methylene))diphenol (4b)
This compound was prepared according to General Procedure D. The residue was purified by flash chromatography (CH2Cl2/MeOH 100 : 1 → 80 : 1) to give the product as a white solid (67 mg, 90 % yield). Mp 158–159°C. Rf (CH2Cl2/MeOH 20 : 1) 0.45. δH (500 MHz, [D6]DMSO) 9.21 (2H, s), 7.03 (4H, d, J 8.3), 6.67 (4H, d, J 8.3), 3.32 (4H, s), 2.42 (4H, s), 2.06 (6H, s). δC (100 MHz, [D4]MeOD) 158.0, 132.0, 129.3, 116.1, 62.8, 54.8, 42.7. m/z (HRMS ESI+) 301.1910; [M + H]+ requires 301.1910 for C18H24N2O2. νmax (neat)/cm−1 3269, 3009, 1671, 1126. HPLC >99.9 % (Method C), RT 14.3 min.
Purification of His-DYRK1A
Human DYRK1A Kinase domain (126–490aa) with an N-terminal histidine tag was expressed in Escherichia coli BL21 (DE3) cells. A 10 mL overnight (O/N) culture (1 mL) containing 50 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol was used to inoculate 1 L of Luria–Bertani (LB) media supplemented with the same antibiotics. The culture was grown at 37°C until an optical density 600 (OD600) of 0.5 was reached; the temperature was then reduced to 18°C. Expression was induced with the addition of 1 mM IPTG (isopropyl B-d-thiogalactoside) and incubated O/N at 18°C. The cells were harvested by centrifugation, 7459 g for 10 min, and resuspended in lysis buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5 % glycerol, 5 mM imidazole, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP)). The cell pellet was lysed using an Emulsiflex C5 high-pressure homogenizer (Avestin) in the presence of protease inhibitors. The insoluble debris was removed by centrifugation at 10980 g for 30 min. The supernatant was bound to Ni-NTA resin (Ni2+-nitriloacetate, Qiagen) and washed with 30 column volumes (CV) of lysis buffer and 5 CV of wash buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5 % glycerol, 25 mM imidazole, and 0.5 mM TCEP). The purified protein was finally eluted from the resin with 5 CV of elution buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5 % glycerol, 250 mM imidiazole, and 0.5 mM TCEP). The histidine tag was cleaved with the addition of tobacco etch virus (TEV) protease (1 mg mL−1) and incubated at 4°C O/N. The eluted proteins were further purified by gel-filtration chromatography using an S200 16/60 column (GE Healthcare) in 25 mM HEPES pH 7.5, 500 mM NaCl, 5 mM dithiothreitol (DTT).
Kinase Inhibition Assay
Active DYRK1A was assayed in TRIS buffer (50 mM TRIS-HCl, pH 7.5) containing 0.1 mM egtazic acid (EGTA), 15 mM DTT, MgAc/ATP cocktail (0.5 mM HEPES pH 7.4; 10 mM Mg(CH3COO)2; 0.1 mM ATP), [γ-32P]-ATP 100–300 cpm pmol−1, and test compounds diluted in deionized water. As substrate, Woodtide (50 µM, Genscript) was used in the DYRK1A assay. The reaction was initiated with 1 ng µL−1 DYRK1A. The reaction mixture was incubated at 30°C for 10 min. Reaction was stopped by pipetting 10 µL of the reaction mixture onto P81 paper (Reaction Biology) and washing with 0.75 % w/v H3PO4 and acetone. P81 papers were transferred to sample bags containing Optiphase Supermix scintillation cocktail (PerkinElmer) and radioactivity (cpm) was measured with a MicroBeta Trilux 2 counter (PerkinElmer). Compounds were tested in duplicate at 1 and 10 µM for their ability to inhibit DYRK1A activity.
Supplementary Material
1H and 13C NMR spectra of new compounds and HPLC chromatograms of final compounds are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the National Health & Medical Research Council of Australia (grant no. APP1106145).
References
[1] (a) M. C. Chamberlain, Cancer 2010, 116, 3988.| Crossref | GoogleScholarGoogle Scholar |
(b) S. K. Singh, C. Hawkins, I. D. Clarke, J. A. Squire, J. Bayani, T. Hide, R. M. Henkelman, M. D. Cusimano, P. B. Dirks, Nature 2004, 432, 396.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. Galli, E. Binda, U. Orfanelli, B. Cipelletti, A. Gritti, S. De Vitis, R. Fiocco, C. Foroni, F. Dimeco, A. Vescovi, Cancer Res. 2004, 64, 7011.
| Crossref | GoogleScholarGoogle Scholar |
[2] E. T. Wong, K. R. Hess, M. J. Gleason, K. A. Jaeckle, A. P. Kyritsis, M. D. Prados, V. A. Levin, W. K. A. Yung, J. Clin. Oncol. 1999, 17, 2572.
| Crossref | GoogleScholarGoogle Scholar |
[3] M. D. Walker, E. Alexander, W. E. Hunt, C. S. MacCarty, M. S. Mahaley, J. Mealey, H. A. Norrell, G. Owens, J. Ransohoff, C. B. Wilson, E. A. Gehan, T. A. Strike, J. Neurosurg. 1978, 49, 333.
| Crossref | GoogleScholarGoogle Scholar |
[4] R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. B. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer, R. O. Mirimanoff, N. Engl. J. Med. 2005, 352, 987.
| Crossref | GoogleScholarGoogle Scholar |
[5] R. S. Ignarro, G. Facchini, A. S. Vieira, D. R. De Melo, I. Lopes-Cendes, R. F. Castilho, F. Rogerio, Mol. Cell. Biochem. 2016, 418, 167.
| Crossref | GoogleScholarGoogle Scholar |
[6] N. Pozo, C. Zahonero, P. Fernandez, J. M. Linares, A. Ayuso, M. Hagiwara, A. Pérez, J. R. Ricoy, A. Hernández-Laín, J. M. Sepúlveda, P. Sánchez-Gómez, J. Clin. Invest. 2013, 123, 2475.
| Crossref | GoogleScholarGoogle Scholar |
[7] Q. Zhou, A. F. Phoa, R. H. Abbassi, M. Hoque, T. A. Reekie, J. S. Font, R. M. Ryan, B. W. Stringer, B. W. Day, T. G. Johns, L. Munoz, M. Kassiou, J. Med. Chem. 2017, 60, 2052.
| Crossref | GoogleScholarGoogle Scholar |
[8] S. Gourdain, J. Dairou, C. Denhez, L. C. Bui, F. Rodrigues-Lima, N. Janel, J. M. Delabar, K. Cariou, R. H. Dodd, J. Med. Chem. 2013, 56, 9569.
| Crossref | GoogleScholarGoogle Scholar |
[9] R. J. Ife, K. W. Catchpole, G. J. Durant, C. Robin Ganellin, C. A. Harvey, M. L. Meeson, D. A. A. Owen, M. E. Parsons, B. P. Slingsby, C. J. Theobald, Eur. J. Med. Chem. 1989, 24, 249.
| Crossref | GoogleScholarGoogle Scholar |
[10] D. N. Rao, S. Rasheed, S. Aravinda, R. A. Vishwakarma, P. Das, RSC Adv. 2013, 3, 11472.
| Crossref | GoogleScholarGoogle Scholar |
[11] G. D. Kishore Kumar, A. Natarajan, Tetrahedron Lett. 2008, 49, 2103.
| Crossref | GoogleScholarGoogle Scholar |
[12] M. Tajbakhsh, R. Hosseinzadeh, H. Alinezhad, S. Ghahari, A. Heydari, S. Khaksar, Synthesis 2011, 490.
| Crossref | GoogleScholarGoogle Scholar |
[13] M. B. Abrams, B. L. Scott, R. T. Baker, Organometallics 2000, 19, 4944.
| Crossref | GoogleScholarGoogle Scholar |
[14] H. Türkmen, B. Çetinkaya, J. Organomet. Chem. 2006, 691, 3749.
| Crossref | GoogleScholarGoogle Scholar |
[15] M. A. van der Horst, T. P. Stalcup, S. Kaledhonkar, M. Kumauchi, M. Hara, A. Xie, K. J. Hellingwerf, W. D. Hoff, J. Am. Chem. Soc. 2009, 131, 17443.
| Crossref | GoogleScholarGoogle Scholar |
[16] B. Chinnababu, S. P. Reddy, C. B. Rao, K. Rajesh, Y. Venkateswarlu, Helv. Chim. Acta 2010, 93, 1960.
| Crossref | GoogleScholarGoogle Scholar |
[17] C. G. Frost, B. C. Hartley, J. Org. Chem. 2009, 74, 3599.
| Crossref | GoogleScholarGoogle Scholar |
[18] S.-F. Hsu, B. Plietker, ChemCatChem 2013, 5, 126.
| Crossref | GoogleScholarGoogle Scholar |
* Michael Kassiou is the recipient of the 2017 RACI Adrian Albert Award.