

Liquid Structures and Transport Properties of Lithium Bis(fluorosulfonyl)amide/Glyme Solvate Ionic Liquids for Lithium Batteries
Shoshi Terada A , Kohei Ikeda A , Kazuhide Ueno A , Kaoru Dokko A B C and Masayoshi Watanabe A
A B C and Masayoshi Watanabe A
A Department of Chemistry and Biotechnology, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan.
B Unit of Elements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Kyoto 615-8510, Japan.
C Corresponding author. Email: dokko-kaoru-js@ynu.ac.jp
Australian Journal of Chemistry 72(2) 70-80 https://doi.org/10.1071/CH18270
Submitted: 2 June 2018 Accepted: 9 August 2018 Published: 7 September 2018
Journal Compilation © CSIRO 2019 Open Access CC BY-NC-ND
Abstract
The liquid structures and transport properties of electrolytes composed of lithium bis(fluorosulfonyl)amide (Li[FSA]) and glyme (triglyme (G3) or tetraglyme (G4)) were investigated. Raman spectroscopy indicated that the 1 : 1 mixtures of Li[FSA] and glyme (G3 or G4) are solvate ionic liquids (SILs) comprising a cationic [Li(glyme)]+ complex and the [FSA]− anion. In Li[FSA]-excess liquids with Li[FSA]/glyme molar ratios greater than 1, anionic Lix[FSA]y(y – x)– complexes were formed in addition to the cationic [Li(glyme)]+ complex. Pulsed field gradient NMR measurements revealed that the self-diffusion coefficients of Li+ (DLi) and glyme (Dglyme) are identical in the Li[FSA]/glyme = 1 liquid, suggesting that Li+ and glyme diffuse together and that a long-lived cationic [Li(glyme)]+ complex is formed in the SIL. The ratio of the self-diffusion coefficients of [FSA]− and Li+, DFSA/DLi, was essentially constant at ~1.1–1.3 in the Li[FSA]/glyme < 1 liquid. However, DFSA/DLi increased rapidly as the amount of Li[FSA] increased in the Li[FSA]/glyme > 1 liquid, indicating that the ion transport mechanism in the electrolyte changed at the composition of Li[FSA]/glyme = 1. The oxidative stability of the electrolytes was enhanced as the Li[FSA] concentration increased. Furthermore, Al corrosion was suppressed in the electrolytes for which Li[FSA]/glyme > 1. A battery consisting of a Li metal anode, a LiNi1/3Mn1/3Co1/3O2 cathode, and Li[FSA]/G3 = 2 electrolyte exhibited a discharge capacity of 105 mA h g−1 at a current density of 1.3 mA cm−2, regardless of its low ionic conductivity of 0.2 mS cm−1.
Introduction
Solvate ionic liquids (SILs) consisting of glymes (Gn, CH3–O–(CH2–CH2–O)n–CH3) and Li salts show unique physicochemical properties and have therefore been investigated as promising electrolytes for Li batteries.[1,2] Glymes are well known solvents that have the ability to dissolve Li salts at high concentrations.[3] The coordination number of Li+ ions in liquid electrolytes is typically 4–5. Triglyme (G3) and tetraglyme (G4) have 4 and 5 ether oxygen atoms, respectively, and can coordinate to Li+ in a 1 : 1 ratio owing to the strong electrostatic and induction interactions between the ether oxygen atoms and the Li+ ion.[4] In certain 1 : 1 mixtures of glyme (G3 or G4) and Li salts, almost all glyme molecules participate in the formation of cationic [Li(glyme)]+ complexes, with very little free (uncoordinated) glyme molecules existing in the liquids.[5] Such SILs are composed of cationic [Li(glyme)]+ complex ions and counter anions, denoted as [Li(glyme)]X, where X is the counter anion. Representative SILs are composed of [Li(G3 or G4)]+ and amide-type anions such as bis(trifluoromethanesulfonyl)amide ([TFSA]−: N(SO2CF3)2−), bis(pentafluoroethanesulfonyl)amide ([BETA]−: N(SO2C2F5)2−), and bis(fluorosulfonyl)amide ([FSA]−: N(SO2F)2−).[6] Because amide-type anions possess low Lewis basicity, the interaction between them and Li+ is relatively weak, resulting in the high dissociativity of Li-amide salts in electrolyte solutions.[7] Moreover, the low Lewis basicity of amide-type anions is favourable for stabilising the [Li(glyme)]+ complex, as the weak interaction between Li+ and the amide-type anion results in a long lifetime of [Li(glyme)]+.[5,6] This long lifetime and the absence of free glyme impart SILs with low volatility, low flammability, and high thermal stability, making them attractive as thermally stable electrolytes for Li batteries.[8–10]
The liquid structures, transport properties, thermal properties, and battery applications of [TFSA]-based SILs have been studied intensively.[9–12] Regarding [FSA]-based systems, the thermal properties and battery applications of [Li(glyme)][FSA] have been reported previously;[13,14] however, the liquid structures and transport properties of Li[FSA]/glyme electrolytes have not yet been fully elucidated. In this study, the liquid structures and transport properties of electrolytes composed of Li[FSA] and glyme (G3 or G4) mixed in various molar ratios were investigated in detail. Raman spectra and pulsed field gradient (PFG) NMR data suggested that electrolytes with Li[FSA]/glyme molar ratios ≥ 1 acquired characteristic properties of SILs. In electrolytes with excess Li[FSA] (i.e. Li[FSA]/glyme > 1), Li+ ions exist in the forms of cationic [Li(glyme)]+ and anionic Lix[FSA]y(y – x)– complexes, which induced a unique charge transport mechanism in the electrolytes – ion transport through ligand exchange. Finally, the charge–discharge behaviours of Li/LiNi1/3Mn1/3Co1/3O2 cells with Li[FSA]/G3 electrolytes were examined. The cell with an excess of Li[FSA] as Li[FSA]/G3 = 2 exhibited a discharge capacity of 105 mA h g−1 at a current density of 1.3 mA cm−2, regardless of the low ionic conductivity of the electrolyte (0.2 mS cm−1).
Experimental
Materials
Lithium bis(fluorosulfonyl)amide (Li[FSA]) was purchased from Kishida Chemical and used as received. Triglyme (G3) and tetraglyme (G4) were kindly supplied from Nippon Nyukazai and used as received. Li[FSA] was mixed with G3 or G4 in various molar ratios to prepare electrolyte solutions (Table S1, Supplementary Material). The water content of the solutions (less than 50 ppm) was measured by Karl Fischer titration. LiNi1/3Mn1/3Co1/3O2 (NMC) was supplied from AGC Seimi Chemical. Acetylene black (AB, Denka Black) was supplied by Denki Kagaku Kogyo. Li metal was purchased from Honjo Metal. Poly(vinylidene fluoride) (PVDF) and N-methyl-2-pyrrolidinone (NMP) were purchased from Kishida Chemical and Kanto Chemical, respectively.
Measurements
Raman spectra of the samples were measured using a Raman spectrometer equipped with a 785 nm laser (NRS-4100, JASCO). The instrument was calibrated using a polypropylene standard. The spectroscopic resolution was 2.4 cm−1. The sample temperature was adjusted to 30 ± 0.1°C using a Peltier microscope stage (TS62, INSTEC) with a temperature controller (mk1000, INSTEC).
The ionic conductivities (σ) of the Li[FSA]/G3 and Li[FSA]/G4 solutions were determined by the complex impedance method using an impedance analyzer (VMP3, Biologic) in the frequency range of 500 kHz–1 Hz with a sinusoidal alternating voltage amplitude of 10 mV root-mean-square (rms). A cell equipped with two platinized platinum electrodes (CG–511B, TOA Electronics) was utilised for the conductivity measurements, and the cell constant was determined using a 0.01 mol dm−3 KCl aqueous solution at 25°C before the measurements. The cell was placed in a temperature-controlled chamber and conductivity was measured at 30°C. The liquid density and viscosity were determined using a viscometer (SVM 3000, Anton Paar).
PFG-NMR measurements were carried out to determine the self-diffusion coefficients of glyme, Li+, and [FSA]−. A JEOL-ECX 400 NMR spectrometer with a 9.4 T narrow-bore super-conducting magnet equipped with a pulsed-field gradient probe and current amplifier was used for the measurements. Self-diffusion coefficients were calculated with the Hahn spin-echo sequence using the following signals: 1H of the terminal methyl group of glyme, 7Li of Li+, and 19F of [FSA]−. The detailed experimental procedures have been reported elsewhere.[15] The diffusion echo signal attenuation, E, is related to the experimental parameters by the Stejskal equation (Eqn 1) with a sinusoidal pulsed-field gradient:[16]
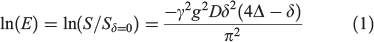
where S is the spin-echo signal intensity, δ is the duration of the field gradient with magnitude g, γ is the gyromagnetic ratio, and Δ is the interval between the two gradient pulses. The values of Δ and δ were set to 50 and 5 ms, respectively, and g was set to 0.01–10 T m−1 depending on the electrolyte. The sample was inserted into an NMR microtube to a height of 3 mm to exclude convection. All measurements were conducted at 30°C. Each sample was placed in a sample tube (BMS-005J Shigemi) with a 4 mm outer diameter; this tube was then inserted into a 5 mm standard NMR sample tube.
Cyclic voltammetry (CV) and linear sweep voltammetry (LSV) were performed with a three-electrode cell using an electrochemical analyzer (VMP3, Biologic). Cu and Pt disk electrodes (3 mm in diameter) were used as the working electrodes for the CV and LSV measurements, respectively, and a Pt wire was used as the counter electrode. The reference electrode was Li metal soaked in a 1 mol dm−3 Li[TFSA]/G3 solution, confined in a glass tube with a Pt liquid junction. The transference number of Li+ in 1 mol dm−3 Li[TFSA]/G3 was ~0.5,[11] therefore, the liquid junction potential was considered to be negligible. All electrochemical measurements were performed at 30°C in an Ar-filled glove box.
Aluminium corrosion in the electrolytes was investigated using a two-electrode cell. A coin-type cell with Al foil as the working electrode, Li metal as the counter electrode, and a glass fibre filter separator (GA-55, Advantec) was used for the measurements. The cell was polarized at 4.8 V for 24 h at 30°C. The cell was then disassembled in an Ar-filled glove box after the polarization, and the Al electrode was observed with a scanning electron microscope (SU8010, Hitachi High-Technologies).
Interfacial charge transfer at the Li metal electrode/electrolyte interface was investigated using a symmetrical coin-type [Li metal | Li[FSA]/glyme electrolyte | Li metal] cell. A glass fibre filter was used to separate the two Li metal electrodes. Electrochemical impedance measurement of the cell was performed using an impedance analyzer (VMP3, Biologic) under the same conditions as the ionic conductivity measurements.
Battery tests were carried out using the Li[FSA]/G3 electrolyte. A slurry containing NMC was prepared by thoroughly mixing NMC, AB, and PVDF in a weight ratio of 80 : 10 : 10 with NMP as the solvent. The slurry was pasted onto Al foil to prepare a composite sheet and dried overnight at 80°C. The composite sheet was then cut into a disk (16 mm diameter) and compressed at 50 MPa followed by drying in a vacuum at 120°C for 12 h. The surface area and thickness of the electrode active materials were 2.0 cm2 and 30 μm, respectively. The loading of NMC was ~4.5 mg cm−2. The coin-type cells were fabricated with a NMC composite cathode, Li metal anode, and Li[FSA]/G3 electrolyte. A glass fibre filter (GA-55, Advantec) was used as the separator. Galvanostatic charge–discharge tests were carried out using an automatic charge/discharge instrument (HJ1001SD, Hokuto Denko) in the voltage range of 2.5–4.2 V at 30°C.
Results and Discussion
Liquid Structures of Li[FSA]/Glyme Electrolytes
The solvation structures of the Li[FSA]/glyme mixtures were elucidated using Raman spectroscopy. It is known that glyme (G3 or G4) coordinates to Li+ as a crown ether-like structure to form a 1 : 1 cationic [Li(glyme)]+ complex.[17–21] This crown ether-like structure is sensitive to Raman spectroscopy, and a strong breathing mode band at ~870 cm−1 is observed when the [Li(glyme)]+ complex is formed in the liquid.[3] Raman bands in the range of 780–900 cm−1 reflect the CH2 rocking and C–O–C stretching vibrations of the glyme molecule.[22–25] Mixtures of Li[FSA] and glyme (G3 and G4) were prepared in a wide concentration range from 0.1 to 5.5 mol dm−3, corresponding to Li[FSA]/glyme (cLi/cglyme) molar ratios of 0.125–2. Note that all the mixtures remained as liquids at room temperature. Fig. 1 shows the Raman spectra of Li[FSA]/G3 and Li[FSA]/G4 at 30°C. All the spectra were normalized by the bands at ~1400–1500 cm−1, which are assigned to CH2 bending/scissoring modes of the glymes. The Raman intensity of the breathing mode at ~870 cm−1 became pronounced as the Li[FSA] concentration increased in both G3 and G4 mixtures. The Raman spectra of the Li[FSA] : G3 = 1 : 1 and Li[FSA] : G4 = 1 : 1 samples in the range of 780–900 cm−1 are similar to those of the SILs [Li(G3)][TFSA] and [Li(G4)][TFSA].[5]

|
For further analysis of the solvation structures, Raman spectra in the range of 680–900 cm−1 were deconvoluted into six bands using the Gaussian–Lorentzian function, as shown in Fig. S1 (Supplementary Material). The fraction of the integrated intensity of the breathing mode (I870) to the total integral intensity of the glyme bands (I870 + I840 + I830 + I810) was calculated. Fig. 2a shows the fraction of I870 plotted against the molar ratio of Li/glymes (cLi/cglyme). For molar ratios less than 1 (cLi/cglyme < 1), the fraction of I870 increases with the Li concentration due to the increase of the concentration of [Li(glyme)]+ in the liquid. However, the fraction of I870 becomes almost constant and saturated at molar ratios higher than 1 (cLi/cglyme ≥ 1). This indicates that almost all glyme molecules form complexes with Li+ (as [Li(glyme)]+) and uncoordinated glyme scarcely exists in mixtures of cLi/cglyme ≥ 1. In fact, the Li[FSA] : G3 = 1 : 1 and Li[FSA] : G4 = 1 : 1 complexes can be crystallized and have melting points of 46 and 23°C, respectively.[13] Although these melting points are close to room temperature, the complexes remain in a supercooled liquid state at room temperature for several months. Therefore, the molten complexes of Li[FSA] : G3 = 1 : 1 and Li[FSA] : G4 = 1 : 1 can be regarded as SILs consisting of a [Li(glyme)]+ cation and a [FSA]− anion. Indeed, the Li[FSA] : G3 = 1 : 1 and Li[FSA] : G4 = 1 : 1 possess good thermal stability, similar to the [Li(glyme)][TFSA] SILs, and do not decompose up to ~180°C.[6] This suggests that the structure of the [Li(glyme)]+ solvate cations are maintained up to 180°C, and the desolvation of Li+ and the evaporation of released glyme do not occur owing to the strong Li+–glyme interaction in the liquids. Consequently, the Li[FSA]-excess liquids of cLi/cglyme > 1 can be considered as mixtures of Li[FSA] and a [Li(glyme)][FSA] SIL.

|
The Raman band assigned to S–N vibrations of the [FSA]− anion appears at ~700–790 cm−1. This band shifts to higher wavenumber when [FSA]− is bound to Li+.[26,27] In this study, Raman spectroscopy was not able to clearly distinguish between bound and free [FSA]−; however, the peak position of [FSA]− shifted depending on the composition of the liquids. Fig. 2b shows the peak top position of the [FSA]− anion band as a function of cLi/cglyme. The peak of the anion band for liquids of cLi/cglyme ≤ 0.25 was ~720 cm−1 and gradually shifted to higher wavenumber as the molar ratio of Li[FSA] increased, and the peak shifted to ~750 cm−1 at cLi/cglyme = 2. For cLi/cglyme < 1, excess glyme molecules were present in the liquids, which are assumed to comprise uncoordinated glyme, [Li(glyme)]+, and [FSA]−. Therefore, the peak shift observed for the cLi/cglyme ≤ 1 liquids is ascribed to the formation of a [Li(glyme)]+–[FSA]− contact ion pair (CIP) in the liquids. By comparison, the cLi/cglyme > 1 liquids can be regarded as mixtures of Li[FSA] and a [Li(glyme)][FSA] SIL. The relatively large Raman shift of [FSA]− (~750 cm−1) observed for the cLi/cglyme > 1 liquids suggests the formation of anionic Lix[FSA]y(y – x)– complexes.[26,27] Yoon et al. reported a similar large Raman shift for a [FSA]-based ionic liquid, N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)amide (C3mpyr[FSA]), mixed with a high concentration of Li[FSA].[27] They attributed the large Raman shift to the formation of Li[FSA]2− in the liquid. Raman spectra suggested that the glyme molecules form a 1 : 1 complex of [Li(glyme)]+ even in cLi/cglyme > 1 mixtures (Figs 1 and 2), and there are an insufficient amount of glyme molecules in the liquids to solvate excess Li+ ions. The excess Li+ ions are presumably stabilised by [FSA]− anions and form anionic complexes of Lix[FSA]y(y – x)– in the cLi/cglyme > 1 mixtures. The large shift of [FSA]− implies that the population of Li[FSA]2− increased with the molar ratio of Li[FSA] in the cLi/cglyme > 1 liquid.
Transport Properties of Li[FSA]/Glyme Electrolytes
Fig. 3 shows the concentration dependencies of ionic conductivity and viscosity for the Li[FSA]/G3 and Li[FSA]/G4 electrolytes. Glyme solvents can dissolve Li[FSA] up to ~5.5 mol dm−3 and maintain a liquid state at room temperature. The viscosity increased as the Li[FSA] concentration increased in both electrolytes, because more of the bulky cationic [Li(glyme)]+ complex was present in the mixtures, as revealed by Raman spectroscopy. The ionic conductivity reached a maximum at ~1 mol dm−3 in each mixture. For salt concentrations higher than 1 mol dm−3, the ionic conductivity decreased gradually due to the increase of the liquid viscosity. The viscosity and conductivity of the Li[FSA]/glyme mixtures are lower and higher than those of Li[TFSA]/glyme mixtures, respectively.[11] This difference can be attributed to the smaller ionic size of [FSA]− than that of [TFSA]−, resulting in a higher mobility of [FSA]− compared with [TFSA]− in the liquids. The electrolyte Li[FSA]/G3 = 1 exhibited an ionic conductivity of 1.65 mS cm−1, which was slightly lower than that of Li[FSA]/G4 = 1 (1.98 mS cm−1). This was attributed to the slightly higher viscosity of Li[FSA]/G3 = 1 (139.0 mPa s) than that of Li[FSA]/G4 = 1 (94.0 mPa s). However, in the concentration range > 3.5 mol dm−3, the Li[FSA]/G4 electrolyte became more viscous than Li[FSA]/G3 (Table S1, Supplementary Material), resulting in the lower ionic conductivity of Li[FSA]/G4. As shown in Fig. 3, the electrolytes became extremely viscous, ~1000 mPa s, when the Li[FSA] concentration was higher than 5 mol dm−3, for the molar ratio of Li[FSA]/glyme > 1. In addition to the formation of the cationic [Li(glyme)]+ complex, formation of the anionic Lix[FSA]y(y – x)– complex (see above) in the Li[FSA]/glyme > 1 liquids may be responsible for the extremely high viscosity.

|
To further understand the transport properties of the electrolytes, the self-diffusion coefficients of glyme (Dglyme), Li+ (DLi), and [FSA]− (DFSA) were measured by PFG-NMR spectroscopy. Fig. 4 shows the concentration dependencies of the self-diffusion coefficients in Li[FSA]/glyme mixtures at 30°C. In both G3 and G4 mixtures, the self-diffusion coefficients decrease as the Li concentration increases due to the increase in viscosity. Fig. 5a shows the self-diffusion coefficient Dglyme/DLi ratio, which decreases as the Li[FSA] content increases and becomes unity at an equimolar composition. When there is an excess of glyme relative to Li+ (cLi/cglyme < 1), uncoordinated glyme exists in the mixture. However, the NMR spectrometer could not distinguish the uncoordinated glyme from the coordinated one, suggesting that the ligand exchange of [Li(glyme)]+ occurs at a rate higher than the time scale of the NMR measurement. Therefore, the measured value of Dglyme is an average of the self-diffusion coefficients of both coordinated and uncoordinated glymes. The value of Dglyme is larger than DLi for cLi/cglyme < 1, suggesting that uncoordinated glymes diffuse faster than coordinated ones. The identical diffusion coefficients of Li+ and glyme in the 1 : 1 mixture suggest that these species diffuse together in the form of [Li(glyme)]+. In this regard, it has been shown previously that the lifetime of [Li(glyme)]+ is relatively long in the liquid state.[5,6,28] However, when Li[FSA] is in excess relative to glyme (cLi/cglyme > 1), the Dglyme/DLi ratio becomes slightly lower than unity. In the cLi/cglyme > 1 mixtures, Li+ ions exist in the forms of cationic [Li(glyme)]+ and anionic Lix[FSA]y(y – x)– complexes (see above). NMR spectroscopy could not distinguish Li+ ions in [Li(glyme)]+ and those in Lix[FSA]y(y – x)–, suggesting that ligand exchange between these complexes (glyme and [FSA]−) occurs at a high rate in the cLi/cglyme > 1 liquids, and this ligand exchange may induce Li+ to diffuse faster than glyme in the cLi/cglyme > 1 liquids.

|

|
Fig. 5b shows the self-diffusion coefficient DFSA/DLi ratio, which was essentially constant at ~1.1–1.3 in the cLi/cglyme ≤ 1 mixtures. By comparison, DFSA/DLi sharply increased in the Li[FSA]-excess mixtures (cLi/cglyme > 1), indicating that [FSA]− diffuses much faster than Li+. NMR spectroscopy could not distinguish the [FSA]− in the anionic Lix[FSA]y(y – x)– complex from free [FSA]−, suggesting that Li+ in the anionic complex rapidly exchanges its ligand (i.e. [FSA]−). This may cause the faster diffusion of [FSA]− compared with Li+ in the Li[FSA]-excess electrolytes. Overall, the charge transport mechanism in Li[FSA]/glyme electrolytes changes distinctly at cLi/cglyme = 1, and the ligand exchange in the Li[FSA]-excess electrolytes (cLi/cglyme > 1) provokes a unique anionic conduction in the liquid.
Electrochemical Properties of Li[FSA]/G3 Electrolytes
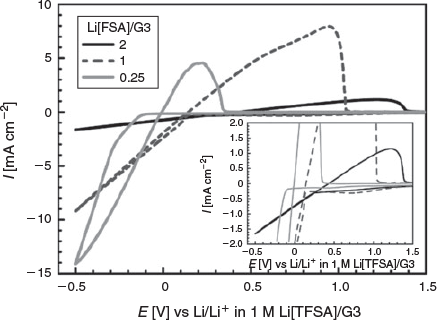
Li[FSA]/G3 and Li[FSA]/G4 electrolytes exhibited very similar liquid structures and transport properties. However, as shown in Table S1 (Supplementary Material), in the high concentration range > 3.5 mol dm−3, Li[FSA]/G3 electrolytes showed slightly higher ionic conductivity than Li[FSA]/G4 electrolytes. For this reason, Li[FSA]/G3 electrolytes were studied further to discern their suitability towards battery application. Fig. 6 shows cyclic voltammograms of Li[FSA]/G3 mixtures measured at 30°C. CV profiles for electrolyte compositions of Li[FSA]/G3 = 1 and 2 presented in Fig. 6 are the first cycle data, and the CV for the Li[FSA]/G3 = 0.25 electrolyte is the third cycle data. The cyclic voltammograms of the initial two cycles with Li[FSA]/G3 = 0.25 are shown in Fig. S2 (Supplementary Material). In Fig. 6, a reduction current at electrode potentials less than 0 V, which arises because of Li deposition, was observed during the cathodic scan, and an oxidation current, which arises because of dissolution of Li metal, was observed during the anodic scan in all electrolytes tested. The cross point of the cathodic and anodic currents shifted to positive potential as the Li[FSA] concentration increased due to the change of the Li/Li+ redox potential.[12] The cathodic current observed at −0.5 V (versus Li/Li+ in 1 M Li[TFSA]/G3) decreased as the Li[FSA] concentration increased, because the ionic conductivity of the electrolyte decreases as the Li[FSA] molar ratio increases (Fig. 3). Although the ionic conductivity of Li[FSA]/G3 = 0.25 was the highest among the examined electrolytes, the anodic peak current for dissolution of Li in this electrolyte was lower than one in Li[FSA]/G3 = 1. Clearly, the coulombic efficiency of Li stripping/plating in Li[FSA]/G3 ≥ 1 electrolytes was higher than that in Li[FSA]/G3 = 0.25. The difference in the reversibility of Li stripping/deposition between the electrolytes might be due to the nature of the passivation layer formed on the deposited Li metal. Several researchers reported that an effective passivation film is formed on the Li metal in the highly concentrated electrolytes containing Li-amide salts.[29–32] According to these reports, the passivation film derived from the amide-type anion are formed on the Li metal before the decomposition of solvents in the electrolytes when the Li-amide salt concentration is higher than 3 mol dm−3. The anion-derived passivation films possess low interfacial resistance but effectively prevent the further decomposition of electrolytes. The molar concentration of Li[FSA] in the Li[FSA]/G3 = 0.25 electrolyte was 1.25 mol dm−3 (Table S1, Supplementary Material), therefore, the FSA-derived passivation film might not be effectively formed on the electrode. Actually, as shown in Fig. S2 (Supplementary Material), an irreversible cathodic current due to the electrolyte decomposition was observed in the first CV cycle of Li[FSA]/G3 = 0.25 measured in the potential range 2.5–0 V versus Li/Li+. The anodic current due to the Li stripping observed in the third cycle was larger than that in the second cycle (Fig. S2, Supplementary Material). This suggests that a passivation film grew on the electrode during the first and second cycles, and suppressed the decomposition of the electrolyte to some extent in the subsequent cycle. Therefore, the irreversible behaviour of the cyclic voltammogram for Li[FSA]/G3 = 0.25 in Fig. 6 is partially attributed to the decomposition of the electrolyte.
|
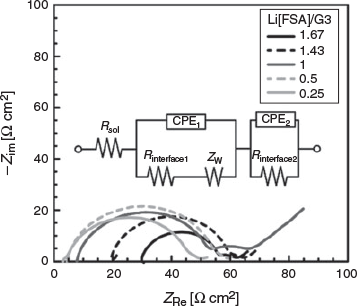
To investigate the effect of the Li[FSA] concentration on the interfacial charge transfer reaction at the Li metal electrode/electrolyte interface, electrochemical impedance measurements for the symmetrical cell [Li metal | electrolyte | Li metal] were performed. Fig. 7 shows Nyquist plots for the cells with Li[FSA]/G3 electrolytes containing various concentrations of Li[FSA]. An equivalent circuit model used to fit the impedance spectra is also shown in Fig. 7; Rsol is the bulk solution resistance for ionic conduction between two electrodes; Rinterface1 is the electrolyte/electrode interfacial resistance for the charge transfer reaction; Rinterface2 is the resistance of the solid–electrolyte interphase (SEI), which is the passivation layer formed on the Li metal electrode by the reductive decomposition of the electrolyte;[33] CPE1 and CPE2, which are constant phase elements, are used instead of electric double-layer capacitance and capacitance of the SEI, respectively; and ZW is the Warburg impedance for ion diffusion in the diffusion layer. The intercept of the real part of the impedance (ZRe) at the highest frequency corresponds to Rsol. The two semicircles observed in the high frequency range are assigned to the charge transfer resistance (Rinterface1) and the SEI resistance (Rinterface2), and the straight line observed at low frequency corresponds to ZW. The Rsol increased as the Li[FSA] concentration increased, which agrees well with the decrease of ionic conductivity at high concentration (Fig. 3). In the present study, we could not distinguish which of the two semicircles observed in the high frequency range was due to Rinterface1. Instead, we discuss here the magnitude of the total interfacial resistance (Rinterface1 + Rinterface2). The total interfacial resistance decreased as the concentration of Li[FSA] increased. The charge transfer resistance can be described as Rinterface1 = RT/(i0AF), where R is the gas constant, T is the absolute temperature, i0 is the exchange current density, A is the electrode area of each Li metal, and F is the Faraday constant. The exchange current density can be described as i0 = k0FcLi, where k0 is the standard heterogeneous rate constant for the electrochemical Li deposition and dissolution at the interface.[34] The increase of the Li+ concentration leads to lower charge transfer resistance (Rinterface1). In addition, the [FSA]-derived passivation layer formed in the highly concentrated electrolytes with composition Li[FSA]/G3 > 1 might result in a smaller Rinterface2 than the SEI formed in the Li[FSA]/G3 < 1 electrolytes. Qian et al. reported that a compact SEI layer mainly comprised of inorganic components was formed on Li metal in the electrolytes of 4 mol dm−3 Li[FSA]/monoglyme, resulting in a low interfacial resistance.[32] In any case, as shown in Fig. 7, the total interfacial resistance of Li/electrolyte (Rinterface1 + Rinterface2) decreases in the Li[FSA]/G3 > 1 electrolytes upon further increasing the molar ratio of Li[FSA]/G3. Consequently, a highly concentrated electrolyte is favourable for high rate interfacial charge transfer reactions, even though the ionic conductivity of the electrolyte is low and the solution resistance is relatively large.
|
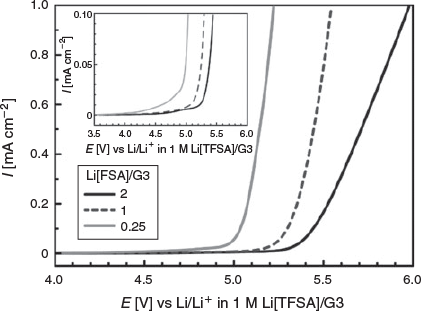
The oxidative stability of the Li[FSA]/G3 electrolytes was investigated by linear sweep voltammetry. Fig. 8 shows linear sweep voltammograms measured with a Pt electrode. The anodic current starts to flow at ~4.0 V in the Li[FSA]/G3 = 0.25 mixture due to the oxidative decomposition of the electrolyte. The onset of the anodic decomposition potential shifted to 4.5 V in the Li[FSA]/G3 = 1 and 2 electrolytes. The electrolyte oxidation is assumed to originate from the decomposition of G3. That is, the lone pairs of the ether oxygen atoms of G3 are attracted to the Li+ ions and they coordinate to form the cationic complex. Following formation of this complex, the highest occupied molecular orbital (HOMO) energy level of G3 is lowered and its oxidative stability is enhanced.[8] The population of G3 coordinated to Li+ increases as the Li[FSA] concentration increases, and the uncoordinated G3 negligibly exists in the [Li(G3)][FSA] SIL and the Li[FSA]-excess mixture. The onset potential for oxidative decomposition of the Li[FSA]-excess mixture (Li[FSA]/G3 = 2) was similar to that of the equimolar mixture; however, the oxidative current was further suppressed by the excess Li[FSA]. Because the concentration of G3 in the liquid decreases as the Li[FSA] concentration increases, the anodic current may become lower due to oxidation of G3 in the Li[FSA]-excess liquid.
|
The effect of the Li[FSA] concentration on Al corrosion was investigated by chronoamperometry, as shown in Fig. 9a. The two-electrode cell consisting of the Li[FSA]/G3 electrolyte, Al foil as the working electrode, and Li metal as the counter electrode was polarized at 4.8 V for 24 h. A large anodic current was observed in the Li[FSA]/G3 = 0.25 electrolyte owing to the oxidative decomposition of the electrolyte and/or Al corrosion. The anodic current was suppressed in the Li[FSA]/G3 = 1 electrolyte, but a current of over 5 μA cm−2 was continuously observed. The anodic current was further suppressed in the Li[FSA]/G3 = 2 electrolyte and decreased to below 1 μA cm−2 after 24 h. The Al electrodes after polarization at 4.8 V were observed by SEM (Fig. 9b–d). Clear holes with diameters of ~100 μm were evident for the Li[FSA]/G3 = 0.25 sample, suggesting that severe pitting caused by the corrosion of Al occurred in the electrolyte. The corrosion increased the surface area of the Al electrode, and the consequent anodic current increased gradually during the measurement. Although the corrosion of Al in the Li[FSA]/G3 = 1 electrolyte was not as severe as that in the Li[FSA]/G3 = 0.25 electrolyte, pitting corrosion did occur to some extent (Fig. 9c). In contrast, a negligible morphology change was observed for the Li[FSA]/G3 = 2 sample, indicating that Al corrosion was inhibited in this electrolyte.

|
There are numerous reports on the corrosion of Al in electrolytes containing Li-amide salts such as Li[TFSA] and Li[FSA].[35–39] The corrosion of Al occurs with concomitant dissolution of Al3+ into the electrolytes. In the case of electrolytes containing excess solvent, Al3+ ions, which are produced by the electrochemical oxidation of Al, are solvated and diffuse into the electrolyte bulk, and the Al corrosion proceeds. When the Al is positively polarized, the surface of Al is expected to be covered with solvent and anions. According to the results of molecular dynamics simulations reported by several groups, a positively polarized electrode surface in a highly concentrated electrolyte is effectively covered solely by anions, i.e. solvent exclusion occurs.[35,36] Solvent exclusion hinders the solvation of Al3+ at the surface of Al. In addition, almost all solvents coordinate to Li+ and free solvent that can coordinate to Al3+ negligibly exists. In the case of glyme-based SILs, the negative charges of ether oxygen atoms are attracted to the Li+ cation,[4] and free glyme negligibly exists in the liquids. The glyme molecules coordinated to Li+ in SILs might show little attraction to the positively polarized Al surface. Instead, the Al3+ ion is anticipated to form complexes with amide anions, such as Al[TFSA]x and Al[FSA]x, at the Al surface. For the dissolution of Al-amide complexes, solvation is also needed to dissociate the complexes into Al(solvent)x3+ and anions. The solvent exclusion and the lack of free solvent would suppress the dissolution of Al-amide complexes, resulting in the suppression of Al corrosion.
As shown in Fig. 9, the corrosion of Al was also suppressed in Li[FSA]/G3 electrolyte as the Li[FSA] concentration increased. However, Al corrosion could not be suppressed completely in the Li[FSA]/G3 = 1, even though free G3 is barely present in this liquid. By comparison, Al corrosion negligibly occurs at 4.8 V in the case of Li[TFSA]/G3 = 1.[9] The difference between the Li[FSA]/G3 = 1 and Li[TFSA]/G3 = 1 electrolytes may originate from the size differences of the [FSA]− and [TFSA]− anions, as it is known that the size of amide-type anions affects Al corrosion. Theivaprakasam et al. also reported the difference of corrosion of Al in electrolytes containing [TFSA]− and [FSA]− anions.[37] They suggested that the [TFSA]-based electrolyte is less corrosive compared with the [FSA]-based electrolyte. It is known that Al corrosion in electrolyte solutions containing Li[BETA] is suppressed relative to that in Li[TFSA]-containing systems.[38,39] As mentioned before, Al3+ ions would form complexes with the anions at the surface of Al. Larger amide-type anions may effectively prevent the approach of solvent molecules to the Al3+ complexes, and so the [BETA]− anions are less corrosive towards Al. Conversely, the smaller size of the [FSA]− anion may facilitate the solvent molecules to contact Al3+ at the Al surface. In other words, the solvent exclusion effect would be weaker as the size of the anion is smaller. This may be a cause of the gradual progression of Al corrosion in the Li[FSA]/G3 = 1 electrolyte. However, Al corrosion can be suppressed in Li[FSA]/G3 = 2, probably because [FSA]− anions cover the Al surface completely, and the solvent (G3) molecules cannot approach Al3+ ions.
Battery Application
Li[FSA]/G3 electrolytes were tested as electrolytes for lithium batteries. Fig. 10a shows the charge–discharge behaviours of [Li | Li[FSA]/G3 | NMC] cells measured at a current density of 65 μA cm−2 (15 mA g−1 based on the NMC mass). In the case of Li[FSA]/G3 = 1, the cell showed charge and discharge capacities of ~140 mA h g−1 (based on the NMC mass) in the initial cycles. However, the charge and discharge capacities increased and decreased, respectively, with cycle number (Fig. 10b), suggesting that an irreversible side reaction occurs in the cell. This irreversible behaviour was attributed to the corrosion of the Al current collector of the NMC cathode. It is assumed that the extent of Al corrosion increased the charging capacity due to oxidation of Al and decreased the discharge capacity because of deterioration of the electronic conduction path of the NMC cathode. In the case of Li[FSA]/G3 = 2 (Fig. 10c), the cell showed stable charge–discharge behaviour, indicating that Al corrosion was effectively suppressed. However, the coulombic efficiency of (discharge capacity)/(charge capacity) was ~96 % throughout the cycling test (Fig. 10d). Because an excess amount of Li metal was used as the anode, the cell charge and discharge capacities were limited by the NMC cathode, suggesting that some irreversible side reactions occurred at the cathode during charging and discharging. If the low coulombic efficiency was caused by degradation of the NMC cathode, the cell capacity should decay much faster. The precise nature of the side reactions in the cell are not clear at present. There is a possibility that oxidative decomposition of the electrolyte occurred slowly at the NMC composite cathode. Indeed, the NMC cathode has a porous structure and its surface area is much larger than that of a planar Pt electrode. In addition, the transition metal oxide, NMC, may catalyse oxidative decomposition of the electrolyte.[40] However, further investigations are necessary to clarify the nature of such side reactions in the cell.

|
Although the coulombic efficiency of the [Li | Li[FSA]/G3 = 2 | NMC] cell was not sufficient for practical applications, a rate capability test was conducted. Fig. 11 shows the discharge curves of the cell measured at various current densities. The discharge capacity gradually decreased with increasing current density. The cell delivered a discharge capacity of 105 mA h g−1 at 1.3 mA cm−2 and the capacity decayed to 60 mA h g−1 at 3.25 mA cm−2. Despite the low ionic conductivity (of 0.22 mS cm−1) of the cell in Li[FSA]/G3 = 2, its rate capability is comparable to that of a cell with the [Li(G4)][TFSA] SIL electrolyte, which has a higher ionic conductivity (of 1.6 mS cm−1).[10] The smaller charge transfer resistance at the electrode–electrolyte interface in Li[FSA]/G3 = 2 (Fig. 7) is presumed to contribute to the relatively good rate capability of the cell. In addition, the extremely high Li+ concentration (5.5 mol dm−3) in the electrolyte may be effective in supplying a sufficient number of Li+ ions to the cathode and anode even at high current densities.

|
Conclusions
The liquid structures and transport properties of electrolytes composed of Li[FSA] and glyme (G3 or G4) mixed in various molar ratios were investigated. Glyme solvents can dissolve Li[FSA] up to ~5.5 mol dm−3 and the highly concentrated electrolytes remained liquid at room temperature. The viscosity of the electrolytes increased as the Li[FSA] concentration increased. The electrolytes became extremely viscous, ~1000 mPa s, when the Li[FSA] concentration was higher than 5 mol dm−3, where the molar ratio of Li[FSA]/glyme > 1. Raman spectra and PFG-NMR data suggested that electrolytes with Li[FSA]/glyme molar ratios ≥ 1 acquired the characteristic properties of SILs. In the Li[FSA]-excess Li[FSA]/glyme > 1 electrolytes, Li+ ions exist in the forms of cationic [Li(glyme)]+ and anionic Lix[FSA]y(y – x)– complexes, and this induced a unique charge transport mode in the electrolytes – anion transport through ligand exchange involving [FSA] and Lix[FSA]y(y – x)–. In addition, Li metal plating and its dissolution in Li[FSA]/glyme electrolytes were possible. The cathodic current for the electroplating of Li metal decreased as the Li[FSA] concentration increased because the electrolyte ionic conductivity decreased as the Li[FSA] molar ratio increased. However, the charge transfer resistance for the Li/Li+ redox reaction at the electrode–electrolyte interface became lower as the Li[FSA] concentration increased, and the oxidative stability of the electrolytes was enhanced as the Li[FSA] concentration increased as well. Furthermore, Al corrosion was suppressed in the Li[FSA]-excess Li[FSA]/glyme > 1 electrolytes. A battery consisting of a Li metal anode, LiNi1/3Mn1/3Co1/3O2 cathode, and Li[FSA]/G3 = 2 electrolyte exhibited a discharge capacity of 105 mA h g−1 at a current density of 1.3 mA cm−2, regardless of its low ionic conductivity of 0.2 mS cm−1.
Supplementary Material
Transport properties of Li[FSA]/glyme electrolytes, the deconvoluted Raman spectrum of Li[FSA]/glyme = 1, and cyclic voltammograms of Li[FSA]/G3 = 0.25 are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported in part by the JSPS KAKENHI (Grant Nos 16J11045, 16H06368, 18H03926, and 15H05758) from the Japan Society for the Promotion of Science (JSPS), the MEXT program ‘Elements Strategy Initiative to Form Core Research Center’ of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, and the Advanced Low Carbon Technology Research and Development Program (ALCA) of the Japan Science and Technology Agency (JST).
References
[1] T. M. Pappenfus, W. A. Henderson, B. B. Owens, K. R. Mann, W. H. Smyrl, J. Electrochem. Soc. 2004, 151, A209.| Crossref | GoogleScholarGoogle Scholar |
[2] T. Mandai, K. Yoshida, K. Ueno, K. Dokko, M. Watanabe, Phys. Chem. Chem. Phys. 2014, 16, 8761.
| Crossref | GoogleScholarGoogle Scholar |
[3] D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski, J. E. Desnoyers, Phys. Chem. Chem. Phys. 2002, 4, 6063.
| Crossref | GoogleScholarGoogle Scholar |
[4] S. Tsuzuki, W. Shinoda, S. Seki, Y. Umebayashi, K. Yoshida, K. Dokko, M. Watanabe, ChemPhysChem 2013, 14, 1993.
| Crossref | GoogleScholarGoogle Scholar |
[5] K. Ueno, R. Tatara, S. Tsuzuki, S. Saito, H. Doi, K. Yoshida, T. Mandai, M. Matsugami, Y. Umebayashi, K. Dokko, M. Watanabe, Phys. Chem. Chem. Phys. 2015, 17, 8248.
| Crossref | GoogleScholarGoogle Scholar |
[6] K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko, M. Watanabe, J. Phys. Chem. B 2012, 116, 11323.
| Crossref | GoogleScholarGoogle Scholar |
[7] M. Schmeisser, P. Illner, R. Puchta, A. Zahl, R. van Eldik, Chem. – Eur. J. 2012, 18, 10969.
| Crossref | GoogleScholarGoogle Scholar |
[8] K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko, M. Watanabe, J. Am. Chem. Soc. 2011, 133, 13121.
| Crossref | GoogleScholarGoogle Scholar |
[9] C. Zhang, A. Yamazaki, J. Murai, J.-W. Park, T. Mandai, K. Ueno, K. Dokko, M. Watanabe, J. Phys. Chem. C 2014, 118, 17362.
| Crossref | GoogleScholarGoogle Scholar |
[10] K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko, M. Watanabe, J. Electrochem. Soc. 2012, 159, A1005.
| Crossref | GoogleScholarGoogle Scholar |
[11] K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko, M. Watanabe, J. Phys. Chem. C 2011, 115, 18384.
| Crossref | GoogleScholarGoogle Scholar |
[12] H. Moon, R. Tatara, T. Mandai, K. Ueno, K. Yoshida, N. Tachikawa, T. Yasuda, K. Dokko, M. Watanabe, J. Phys. Chem. C 2014, 118, 20246.
| Crossref | GoogleScholarGoogle Scholar |
[13] K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko, M. Watanabe, MRS Online Proc. Libr. 2012, 1473,
| Crossref | GoogleScholarGoogle Scholar |
[14] S. Seki, K. Takei, H. Miyashiro, M. Watanabe, J. Electrochem. Soc. 2011, 158, A769.
| Crossref | GoogleScholarGoogle Scholar |
[15] K. Hayamizu, Y. Aihara, S. Arai, C. G. Martinez, J. Phys. Chem. B 1999, 103, 519.
| Crossref | GoogleScholarGoogle Scholar |
[16] W. S. Price, NMR Studies of Translational Motion: Principles and Applications 2009 (Cambridge University Press: Cambridge).
[17] W. A. Henderson, N. R. Brooks, W. W. Brennessel, V. G. Young, Chem. Mater. 2003, 15, 4679.
| Crossref | GoogleScholarGoogle Scholar |
[18] W. A. Henderson, N. R. Brooks, V. G. Young, Chem. Mater. 2003, 15, 4685.
| Crossref | GoogleScholarGoogle Scholar |
[19] W. A. Henderson, F. McKenna, M. A. Khan, N. R. Brooks, V. G. Young, R. Frech, Chem. Mater. 2005, 17, 2284.
| Crossref | GoogleScholarGoogle Scholar |
[20] W. A. Henderson, J. Phys. Chem. B 2006, 110, 13177.
| Crossref | GoogleScholarGoogle Scholar |
[21] D. M. Seo, P. D. Boyle, R. D. Sommer, J. S. Daubert, O. Borodin, W. A. Henderson, J. Phys. Chem. B 2014, 118, 13601.
| Crossref | GoogleScholarGoogle Scholar |
[22] X. Yang, Z. Su, D. Wu, S. L. Hsu, H. D. Stidham, Macromolecules 1997, 30, 3796.
| Crossref | GoogleScholarGoogle Scholar |
[23] L. Ducasse, M. Dussauze, J. Grondin, J. C. Lassegues, C. Naudin, L. Servant, Phys. Chem. Chem. Phys. 2003, 5, 567.
| Crossref | GoogleScholarGoogle Scholar |
[24] J. Grondin, J.-C. Lassegues, M. Chami, L. Servant, D. Talaga, W. A. Henderson, Phys. Chem. Chem. Phys. 2004, 6, 4260.
| Crossref | GoogleScholarGoogle Scholar |
[25] R. Frech, W. Huang, Macromolecules 1995, 28, 1246.
| Crossref | GoogleScholarGoogle Scholar |
[26] K. Fujii, H. Hamano, H. Doi, X. Song, S. Tsuzuki, K. Hayamizu, S. Seki, Y. Kameda, K. Dokko, M. Watanabe, Y. Umebayashi, J. Phys. Chem. C 2013, 117, 19314.
| Crossref | GoogleScholarGoogle Scholar |
[27] H. Yoon, A. S. Best, M. Forsyth, D. R. MacFarlane, P. C. Howlett, Phys. Chem. Chem. Phys. 2015, 17, 4656.
| Crossref | GoogleScholarGoogle Scholar |
[28] C. Zhang, K. Ueno, A. Yamazaki, K. Yoshida, H. Moon, T. Mandai, Y. Umebayashi, K. Dokko, M. Watanabe, J. Phys. Chem. B 2014, 118, 5144.
| Crossref | GoogleScholarGoogle Scholar |
[29] Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama, A. Yamada, J. Am. Chem. Soc. 2014, 136, 5039.
| Crossref | GoogleScholarGoogle Scholar |
[30] K. Sodeyama, Y. Yamada, K. Aikawa, A. Yamada, Y. Tateyama, J. Phys. Chem. C 2014, 118, 14091.
| Crossref | GoogleScholarGoogle Scholar |
[31] J. Wang, Y. Yamada, K. Sodeyama, E. Watanabe, K. Takada, Y. Tateyama, A. Yamada, Nat. Energy 2018, 3, 22.
| Crossref | GoogleScholarGoogle Scholar |
[32] J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin, J.-G. Zhang, Nat. Commun. 2015, 6, 6362.
| Crossref | GoogleScholarGoogle Scholar |
[33] D. Aurbach, J. Power Sources 2000, 89, 206.
| Crossref | GoogleScholarGoogle Scholar |
[34] A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn 2000 (John Wiley & Sons: New York, NY).
[35] D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle, W. A. Henderson, Energy Environ. Sci. 2014, 7, 416.
| Crossref | GoogleScholarGoogle Scholar |
[36] Y. Yamada, C. H. Chiang, K. Sodeyama, J. Wang, Y. Tateyama, A. Yamada, ChemElectroChem 2015, 2, 1687.
| Crossref | GoogleScholarGoogle Scholar |
[37] S. Theivaprakasam, G. Girard, P. Howlett, M. Forsyth, S. Mitra, D. MacFarlane, NPJ Materials Degradation 2018, 2, 13.
| Crossref | GoogleScholarGoogle Scholar |
[38] X. Wang, E. Yasukawa, S. Mori, Electrochim. Acta 2000, 45, 2677.
| Crossref | GoogleScholarGoogle Scholar |
[39] L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch, R. Atanasoski, J. Power Sources 1997, 68, 320.
| Crossref | GoogleScholarGoogle Scholar |
[40] R. Jung, M. Metzger, F. Maglia, C. Stinner, H. A. Gasteiger, J. Phys. Chem. Lett. 2017, 8, 4820.
| Crossref | GoogleScholarGoogle Scholar |