

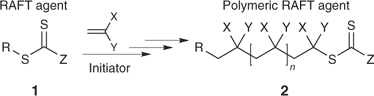
Living Radical Polymerization by the RAFT Process
Graeme Moad A , Ezio Rizzardo A and San H. Thang AA CSIRO Molecular Science, Bag 10, Clayton South VIC 3169, Australia.
Email: graeme.moad@csiro.au; ezio.rizzardo@csiro.au; san.thang@csiro.au

Graeme Moad obtained his B.Sc.(Hons1) in 1974 and Ph.D. in 1977 from the University of Adelaide in the field of organic free radical chemistry. Between 1977 and 1979 he undertook postdoctoral research at Pennsylvania State University in the field of biological organic chemistry. He joined CSIRO as a research scientist in 1979 and is currently a chief research scientist. Dr Moad is coauthor of the book ‘The Chemistry of Free Radical Polymerization’ which is about to go to a second edition. His research interests lie in the fields of polymer design and synthesis (free radical polymerization, reactive extrusion), polymerization kinetics and mechanism, and most recently polymer nanocomposites. |

Ezio Rizzardo is a graduate of the University of New South Wales and received his Ph.D. from the University of Sydney for his studies on the photochemistry of nitro compounds. He joined CSIRO in 1976 after a postdoc at Rice University, RIMAC, and the Australian National University. His CSIRO research has focussed on developing methods for controlling free radical polymerization. For this he has received a number of awards including the RACI Australian Polymer Medal and the CSIRO Chairman’s Gold Medal. Currently he is a CSIRO Fellow and a Fellow of the Australian Academy of Science. |

San H. Thang was born in Saigon, Vietnam, in 1954 and came to Australia in 1979 as a refugee. He completed his B.Sc.(Hons) degree in 1983 and Ph.D. in 1987 from Griffith University. He joined CSIRO in 1986 as a research fellow. He then moved to ICI Australia in late 1987 to undertake the challenge of industrial research. He returned to CSIRO in late 1990, and in 1995 he was co-inventor of the RAFT Process. He is currently a senior principal research scientist at CSIRO Molecular Science where his research focusses on the interface between organic and polymer chemistry. |
Australian Journal of Chemistry 58(6) 379-410 https://doi.org/10.1071/CH05072
Submitted: 18 March 2005 Accepted: 3 May 2005 Published: 14 June 2005
Abstract
This paper presents a review of living radical polymerization achieved with thiocarbonylthio compounds [ZC(=S)SR] by a mechanism of reversible addition–fragmentation chain transfer (RAFT). Since we first introduced the technique in 1998, the number of papers and patents on the RAFT process has increased exponentially as the technique has proved to be one of the most versatile for the provision of polymers of well defined architecture. The factors influencing the effectiveness of RAFT agents and outcome of RAFT polymerization are detailed. With this insight, guidelines are presented on how to conduct RAFT and choose RAFT agents to achieve particular structures. A survey is provided of the current scope and applications of the RAFT process in the synthesis of well defined homo-, gradient, diblock, triblock, and star polymers, as well as more complex architectures including microgels and polymer brushes.
Introduction
Radical polymerization is one of the most widely used processes for the commercial production of high-molecular-weight polymers.[1] The main factors responsible for the pre-eminent position of radical polymerization are that: (a) it can be used with a large variety of monomers including (meth)acrylates, styrene, (meth)acrylamides, butadiene, and vinyl acetate; (b) it is tolerant to a wide range of functional groups (e.g. OH, NR2, COOH, CONR2) and reaction conditions (bulk, solution, emulsion, miniemulsion, suspension); and (c) it is simple to implement and inexpensive in relation to competitive technologies. However, the conventional process has some notable limitations with respect to the degree of control that can be asserted over macromolecular structure, in particular, the molecular weight distribution, composition, and architecture.
The recent emergence of techniques for implementing living radical polymerization has provided a new set of tools for polymer chemists that allow very precise control over the polymerization process while retaining much of the versatility of conventional radical polymerization.[2–8] It is no longer a formidable task to apply radical polymerization to the synthesis of blocks, stars, or other polymers of complex architecture. New materials that have the potential of revolutionizing a large part of the polymer industry are beginning to appear. Possible applications range from novel surfactants, dispersants, coatings, and adhesives, to biomaterials, membranes, drug delivery media, and materials for microelectronics.
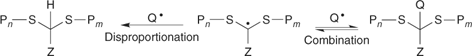
To understand how RAFT and other forms of living radical polymerization work, we first need to consider the mechanism of the conventional process.[1] Radical polymerization is a chain reaction. The chains are initiated by radicals (formed from an initiator) adding to monomer. Chain propagation then involves the sequential addition of monomer units to the radical (Pn•) so formed. Chain termination occurs when the propagating radicals react by combination or disproportionation. A much simplified mechanism is shown in Scheme 1.
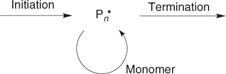
|
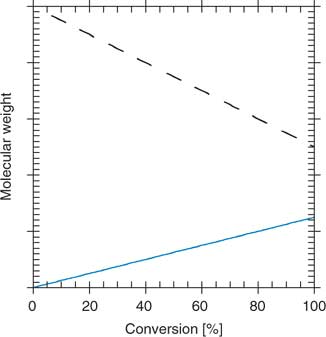
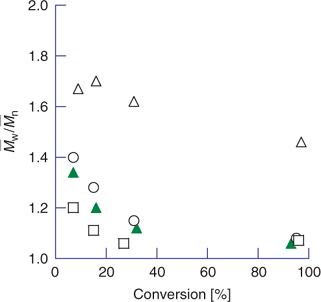
In conventional radical polymerization, the steady-state concentration of propagating species is about 10−7 M, and individual chains grow for 5–10 s before terminating. Chains are continuously formed, propagate, and are terminated by radical–radical reaction. The molecular weight of chains formed in the early stages of polymerization is high and will reduce with conversion because of monomer depletion (Fig. 1). The breadth of the molecular-weight distribution and polydispersity is governed by statistical factors. The polydispersity, expressed in terms of the ratio of weight to number average molecular weights,* is broad (
|
 ).
).In an ideal living polymerization, all chains are initiated at the beginning, grow at the same rate, and survive the polymerization (there is no termination). The propensity of free radicals to undergo radical–radical termination means that, for the case of radical polymerization, all chains cannot be simultaneously active. To confer living character on a radical polymerization, it is necessary to suppress or render insignificant all processes that terminate chains irreversibly. Thus, living radical polymerization only becomes possible in the presence of reagents that react with the propagating radicals (Pn•) by reversible deactivation (Scheme 2) or reversible chain transfer (Scheme 3) so that the majority of chains are maintained in a dormant form (Pn–X). The average concentration of the active propagating species in a living radical polymerization may be similar to that for the conventional process although the cumulative lifetime of an individual chain as an active species will be lower. Rapid equilibration between the active and dormant forms ensures that all chains possess an equal chance for growth and that all chains will grow, albeit intermittently. Under these conditions, the molecular weight increases linearly with conversion (Fig. 1) and the molecular weight distribution can be very narrow (e.g.
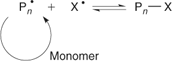
|
|
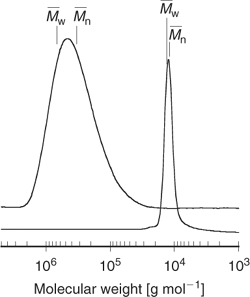
|
The living radical polymerization techniques that have recently received greatest attention are nitroxide-mediated polymerization (NMP), atom-transfer radical polymerization (ATRP), and reversible addition–fragmentation chain transfer (RAFT). The NMP technique was devised in our laboratories in the early 1980s,[10] and in recent years has been exploited extensively for the synthesis of narrow molecular-weight distribution homopolymers and block copolymers of styrene and acrylates.[2,11,12] Recent developments have made NMP applicable to a wider, though still restricted, range of monomers.[2] ATRP is substantially more versatile;[3,4] however, it requires unconventional initiating systems that often have poor compatibility with polymerization media. RAFT polymerization, also devised in our laboratories,[13,14] is one of the most recent entrants in this field and arguably the most convenient and versatile.[5–7]
In this paper, we report on the development, scope and mechanism of living radical polymerization with reversible addition–fragmentation chain transfer. The paper will emphasize research carried out at CSIRO but is augmented by the work of other groups, where appropriate, to provide a more complete picture.
RAFT Polymerization
The utility of the RAFT process is illustrated by the following example of RAFT polymerization of methyl methacrylate (MMA). A series of MMA polymerizations were carried out at 90°C with 1,1′-azobis(1-cyclohexanenitrile) initiator, and using an approximate 60-fold range of concentrations of S-dodecyl S-(2-cyano-4-carboxy)but-2-yl trithiocarbonate 64 (see Table 5).[15] The molecular weight distributions observed after six hours are shown in Fig. 3. The molecular weights, ranging from 2870 to 126000, agree with expectation based on the concentrations of RAFT agent and initiator used (Table 1). All samples have narrow molecular weight distributions (
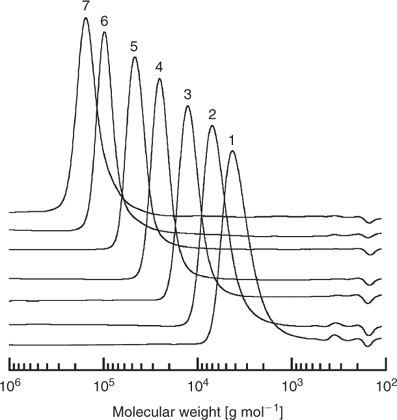
|
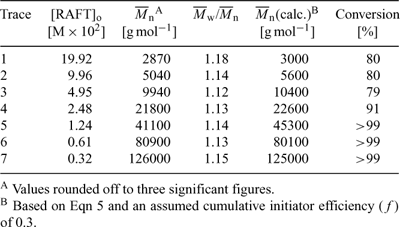
|
The main features of the ideal RAFT polymerization can be summarized as follows.
-
RAFT polymerization (Scheme 4) can be performed by simply adding a chosen quantity of an appropriate RAFT agent to a conventional free-radical polymerization. The same monomers, initiators, solvents, and temperatures are used.
-
RAFT polymerization possesses the characteristics usually associated with living polymerization. All chains begin growth at the commencement of polymerization and continue to grow until the monomer is consumed. Molecular weights increase linearly with conversion (Fig. 1). Active chain ends are retained.
-
Molecular weights in RAFT polymerization can be predicted using Eqn 1, where [M]o – [M]t is the monomer consumed and mM is the monomer molecular weight.
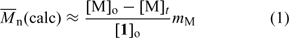
-
Narrow molecular weight distributions are achievable (Fig. 2).
-
Blocks, stars, and complex molecular architectures are accessible (Fig. 4).
|

|
With appropriate selection of the RAFT agent for the monomers used and the reaction conditions, all or most of the above can be routinely achieved. An understanding of the mechanism of RAFT polymerization provides insight and allows the formulation of some simple guidelines for successfully implementing RAFT polymerization.
History of RAFT
Although the acronym RAFT[13] can be used in a more general sense, it has come to be closely associated with radical polymerizations carried out in the presence of thiocarbonylthio compounds which react by reversible addition–fragmentation chain transfer.
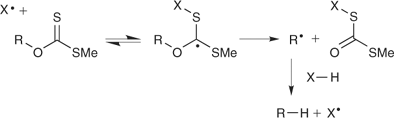
The same process when it involves xanthate RAFT reagents is sometimes also called MADIX (macromolecular design by interchange of xanthate).[16] Reversible addition–fragmentation chemistry involving xanthate esters has been known to organic chemists for some time. It forms the basis of the Barton–McCombie process for deoxygenation of alcohols (Scheme 5).[17–19] Note that the process relies on selective cleavage of the R–O bond. The deoxygenation pathway is facilitated by formation of a strong C=O bond, but it is also important that the group on sulfur is chosen to be a poor homolytic leaving group (typically methyl or ethyl). X–H in Scheme 5 is usually tri-n-butyl stannane, but other free-radical reducing agents[20] may also be used. When the group on sulfur is a good homolytic leaving group (a propagating radical), chemistry similar to that involved in the Barton–McCombie reaction can be used to desulfurize thiols and to cleave RAFT end groups (see later).
|
In 1988, Zard and coworkers[21] first proposed xanthate esters and reversible chain transfer as a convenient source of alkyl radicals and applied this chemistry in the synthesis of monoadducts to a monomer (a maleimide). Numerous applications of xanthate addition fragmentation chemistry in organic synthesis have been described in papers and reviews by the same group.[22,23]
We first reported the use of addition–fragmentation chain transfer agents to control polymerization in the mid 1980s.[7,32,33]. The agents include macromonomers,[24–26] allyl sulfides,[27] allyl bromides,[28] allyl peroxides,[29] vinyl ethers,[30] and thionoesters.[31] We also reported that living characteristics (narrow polydispersities, block synthesis) could be achieved with the use of macromonomer RAFT agents in emulsion polymerization of methacrylate monomers in 1995 (the acronym RAFT was not used at that time).[34,35]
Living radical polymerization using thiocarbonylthio RAFT agents was first described in a patent published in 1998.[14] The first paper and conference reports describing the process also appeared in 1998.[13] Other patents[36–38] and papers soon followed. This method, along with NMP and ATRP, now dominates the literature on living radical polymerization and radical polymerization generally.[39,40] As of January 2005, the first paper on RAFT[13] had received more than 500 citations (Scifinder). The RAFT patent[14] was the ninth most cited in the field of Chemistry and Related Science in 2003 and the most cited of those published within the last ten years (Chemical Abstracts Service).[41]
Mechanism of RAFT
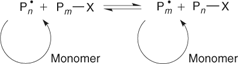
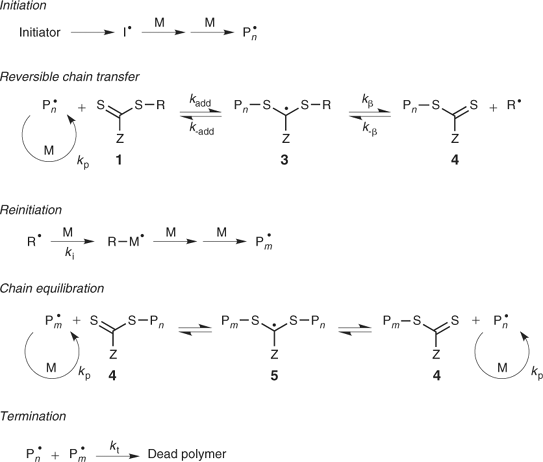
The key feature of the mechanism of RAFT polymerization is a sequence of addition–fragmentation equilibria as shown in Scheme 6.[5,13] Initiation and radical–radical termination occur as in conventional radical polymerization. In the early stages of the polymerization, addition of a propagating radical (Pn•) to the thiocarbonylthio compound [RSC(Z)=S, 1] followed by fragmentation of the intermediate radical gives rise to a polymeric thiocarbonylthio compound [PnS(Z)C=S, 4] and a new radical (R•). Reaction of the radical (R•) with monomer forms a new propagating radical (Pm•). Rapid equilibrium between the active propagating radicals (Pn• and Pm•) and the dormant polymeric thiocarbonylthio compounds 4 provides equal probability for all chains to grow, and allows for the production of polymers with narrow polydispersity. When the polymerization is complete (or stopped), most of chains retain the thiocarbonylthio end group and can be isolated as stable materials.
|
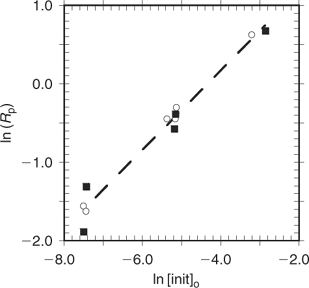
The mechanism for achieving control in RAFT polymerization differs significantly from that involved in NMP and ATRP. The latter processes involve reversible deactivation of propagating radicals by radical–radical reaction (NMP) or atom transfer (ATRP). The dormant species (the alkoxyamine in NMP or the halo-compound in ATRP) is also the source of radicals. The position of the deactivation–activation equilibria and the ‘persistent radical effect’ determine the rate of polymerization.[42,43] In RAFT polymerization, the deactivation–activation equilibria are chain-transfer reactions. Radicals are neither formed nor destroyed in these steps and an external source of free radicals is required to initiate and maintain polymerization. Notwithstanding effects of molecular weight and chain length distribution on radical–radical termination and any side reactions (discussed later), the polymerization kinetics should be similar to those found for conventional radical polymerization. Thus the rate of polymerization should be half order with respect to initiator and independent of the RAFT agent. Such behaviour has been demonstrated for MMA polymerization in the presence of dithiobenzoate RAFT agents over an approximate fivefold range of RAFT agent concentrations (0.006–0.03 M) and an approximate 100-fold range of initiator concentrations (0.0005–0.045 M; Fig. 5).[44] Significant retardation of polymerization rate is only evident for the highest concentration of cumyl dithiobenzoate 22.
|
Choice of RAFT Agents
A wide variety of thiocarbonylthio RAFT agents (ZC(=S)SR, 1) have now been reported. Our initial communication on this form of RAFT polymerization[13] focussed on the utility of dithiobenzoate (Z = Ph) and other dithioesters. However, our patents[14,38,45] and subsequent papers demonstrate that a wide range of thiocarbonylthio compounds can be used. These include certain trithiocarbonates, xanthates, dithiocarbamates, and other compounds. The effectiveness of the RAFT agent depends on the monomer being polymerized and depends strongly on the properties of the free-radical leaving group R and the group Z which can be chosen to activate or deactivate the thiocarbonyl double bond and modify the stability of the intermediate radicals (Fig. 6).[6,9,44,46] For an efficient RAFT polymerization (Scheme 6):
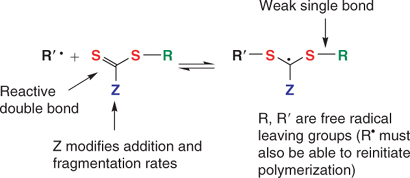
|
-
The RAFT agents 1 and 4 should have a reactive C=S double bond (high kadd).
-
The intermediate radicals 3 and 5 should fragment rapidly (high kβ, weak S–R bond) and give no side reactions.
-
The intermediate 3 should partition in favour of products (kβ ≥ k−add).
-
The expelled radicals (R•) should efficiently re-initiate polymerization.
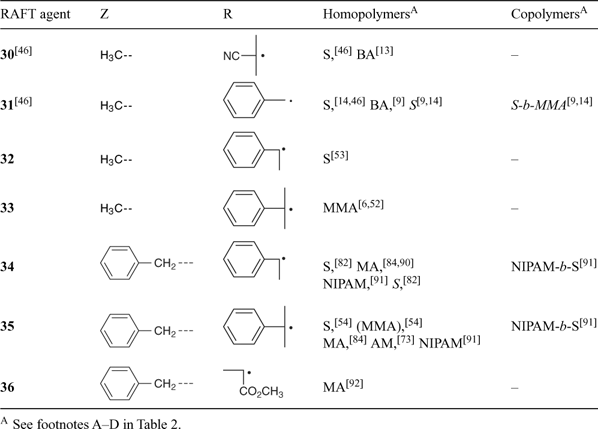
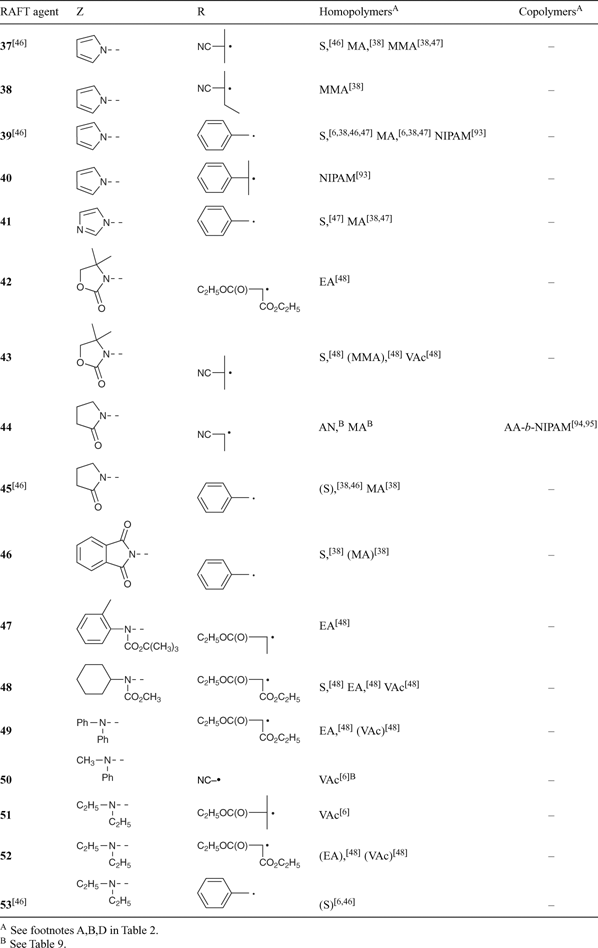
A non-exhaustive summary of RAFT agents and the polymerizations in which they have been applied is provided in Tables 2–7. These tables include some RAFT agent/monomer combinations which provide poorer molecular weight control and/or polydispersity > 1.4. These are indicated by the monomer being in parentheses. Often these have been investigated in order to provide an understanding of the mechanism and to allow construction of guidelines for the choice of RAFT agent. Fig. 7 provides a summary of how to select the appropriate RAFT agent for particular monomers.

|
|
|

|

|
|

|
Several papers have been published on the effects of the substituents R and Z on the effectiveness and transfer coefficients of RAFT agents.[6,9,44,46] The rate of addition of radicals to the C=S double bond is strongly influenced by the substituent Z. This rate is higher when Z = aryl, alkyl (dithioesters), or S-alkyl (trithiocarbonates), and lower when Z = O-alkyl (xanthates) or N,N-dialkyl (dithiocarbamates).
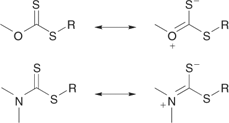
We performed a series of styrene polymerizations (110°C, thermal initiation) for two series of RAFT agents which differ in the activating group Z [S=C(Z)S–CH2Ph and S=C(Z)S–C(Me)2CN].[46] The chain transfer coefficients were found to decrease as shown in Fig. 7. Only the first five in this series provide narrow-polydispersity polystyrene (<1.2). More generally, chain-transfer coefficients decrease in the series dithiobenzoates > trithiocarbonates ≈ dithioalkanoates > dithiocarbonates (xanthates) > dithiocarbamates. The relatively low activity of O-alkyl xanthates and N,N-dialkyl dithiocarbamate derivatives can be qualitatively understood in terms of the importance of the zwitterionic canonical forms (Scheme 7), which arise through interaction between the O or N lone pairs and the C=S double bond. Electron-withdrawing substituents on Z can enhance the activity of RAFT agents to modify the above order.[38,46–49] Thus, xanthate and dithiocarbamate RAFT agents where the oxygen or nitrogen lone pair is less available for delocalization with the C=S by virtue of being part of an aromatic ring or by possessing an adjacent electron-withdrawing substituent are substantially more effective. For examples see Table 6 (xanthates) or Table 4 (dithiocarbamates). The trend in relative effectiveness of RAFT agents with varying Z is rationalized in terms of interaction of Z with the C=S double bond to activate or deactivate that group towards free-radical addition.
|
For ring-substituted cyanoisopropyl dithiobenzoate RAFT agents in MMA polymerization, electron-withdrawing groups, which render the thiocarbonyl sulfur more electrophilic, enhance the rate of addition to the C=S double bond and provide narrower polydispersities from the early stages of polymerization (Fig. 8).[50] The opposite is true for electron-donating substituents. Cyanoisopropyl 2,5-dimethyldithiobenzoate gives poor control which is attributed to the ortho-substituents sterically inhibiting conjugation of the aromatic ring with the C=S double bond.
|
 ), and 2,5-di(trifluoromethyl)dithiobenzoate (□).[
), and 2,5-di(trifluoromethyl)dithiobenzoate (□).[For fragmentation to occur efficiently in the desired direction, the substituent R must be a good homolytic leaving group, relative to the attacking radical Pn•. For example, the RAFT agent where R = CH2Ph e.g. benzyl dithiobenzoate 20 functions as a suitable chain-transfer agent in polymerization with styryl and acrylyl propagating radicals but not in those with methacrylyl propagating radical. The benzyl radical is a reasonable leaving group with respect to the styryl and acrylyl propagating radicals but is a poor leaving group with respect to the methacrylyl propagating radical. In MMA polymerization, RAFT agents such as benzyl dithiobenzoate can appear almost inert because R is a poor leaving group with respect to the PMMA propagating radical.[44] The poor control seen with benzyl dithiobenzoate in α-methylstyrene copolymerizations probably has a similar explanation.[51]
The rate of fragmentation of intermediate 3 is enhanced by increasing steric hindrance, by the presence of electron-withdrawing groups, and by radical-stabilizing groups on R.
The suggestion that R might be selected to be a monomeric analogue of the propagating radical is flawed since penultimate unit effects can be substantial, particularly when R is tertiary. Thus, RAFT agent 18 (R = 2-ethoxycarbonylprop-2-yl), where R may be considered as a monomeric propagating radical, is a poor RAFT agent in the polymerization of MMA and other methacrylates because R is a poor leaving group with respect to the PMMA propagating radical.[44] Analogous behaviour is observed for macromonomer RAFT agents.[25,26] For similar reasons, RAFT agent 6 (R = t-butyl) is poor with respect to RAFT agent 7 (R = t-octyl).[44] These differences in RAFT agent activity are attributed to steric factors. During chain extension, the attacking and leaving propagating radicals (Pn• and Pm•, n, m > 2) are, in essence, identical in all respects other than chain length, and therefore should have equivalent rates in addition to RAFT agent, fragmentation, and propagation. When R is secondary, penultimate unit effects on addition and fragmentation reactions are likely to be smaller but still should not be ignored.
The leaving ability of the substituent R must also be balanced with the ability of the radical R• to re-initiate polymerization. The triphenylmethyl radical, for instance, would be an excellent leaving group but would be a poor re-initiator of chains and its use would result in retardation of polymerization. For similar reasons, one should also not choose benzylic species (cumyl, 1-phenylethyl, benzyl) for vinyl monomer (VAc) polymerization, since rate of re-initiation would be very low.
In many studies, the effectiveness of RAFT agents has been estimated qualitatively by observing the correspondence between found and calculated molecular weights and the narrowness of the molecular weight distribution. A lesser number of papers have made an attempt to evaluate the transfer coefficients of the RAFT agents or the individual rate constants for addition and fragmentation. In this context, the methods used for determining the transfer coefficients of RAFT agents deserve mention. Conventional methods such as the Mayo method should not be used in the case of the more active RAFT agents. The Mayo method is difficult to apply when transfer coefficients are high. The reversibility of chain transfer means that apparent chain-transfer coefficients are dependent on the transfer agent concentration and on conversion of monomer and the transfer agent. One of the ways of estimating the transfer coefficient is to determine the rates of consumption of RAFT agent and monomer. In the case of RAFT polymerization, it can be shown that the rate of consumption of the transfer agent depends on two transfer coefficients, Ctr (=ktr/kp) and C−tr (=k−tr/ki), which describe the reactivity of the propagating radical (Pn•), and the expelled radical (R•) respectively (see Eqn 2).[44,46,52]
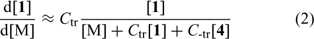
Note that in chain transfer by addition–fragmentation (Scheme 6), the rate coefficient for chain transfer (ktr) is given by Eqn 3:
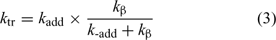
We similarly define k-tr as shown in Eqn 4.
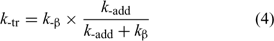
These relationships (Eqns 2–4) are derived by application of a steady-state approximation and assume that the adduct radical 3 undergoes no reactions other than fragmentation. Eqn 2 can be solved numerically to estimate transfer coefficients.[46,52] Transfer coefficients in RAFT polymerization can also be estimated by analyzing the dependence of the molecular-weight distribution on monomer/RAFT agent conversion.[43,46,53–55] We have shown[46] apparent transfer constants determined with neglect of the back reaction of R• with RAFT agent (i.e. by assuming C−tr = 0) can underestimate the actual transfer constant by more than an order of magnitude in the case of the more active RAFT agents.
The dependence of RAFT agent activity on the substituents R and Z can be qualitatively predicted using low-level molecular orbital calculations, and these also provide a guide to the relative importance of the various factors.[44,46,56] There also appear to be good prospects for more quantitative predictions using high-level ab initio calculations.[46,50,57–62] These studies are able to predict dependence of RAFT agent activity on the Z and R substituents and, by providing some understanding of the reason for this dependence, may prove extremely useful in RAFT agent design. However, this work is still in its infancy, and the use of these methods to predict absolute values of rate constants or equilibrium constants associated with RAFT must still be treated with caution.
Even though there is a large range of potential RAFT agents, it should be stated that a majority of polymerizations could be conducted with just two RAFT agents (Fig. 7); one suited to (meth)acrylate, (meth)acrylamide, and styrenic monomers, for example, a tertiary cyanoalkyl trithiocarbonate, and another suited to vinyl monomers such as VAc, for example, a cyanoalkyl xanthate. Ease of preparation, compatibility with reaction media, desired end-group functionality, and polymer architecture might, however, dictate the use of other RAFT agents. In these circumstances, Fig. 7 will provide some guidance. For example, in RAFT polymerization of a methacrylate, one should choose a RAFT agent with the Z group from aryl, S-alkyl, or pyrrole and the R group from tertiary cyanoalkyl or cumyl.
Synthesis of RAFT Agents
Currently, few RAFT agents are commercially available. However, RAFT agents are available in moderate-to-excellent yields by a variety of methods, and syntheses are generally straightforward.
Some of the methods exploited in recent work include:
-
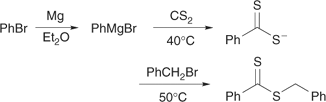
Reaction of a carbodithioate salt with an alkylating agent (Scheme 8).[14,44,46,56,113] Often this will involve sequential treatment of an anionic species with carbon disulfide and an alkylating agent in a one-pot reaction. For example, we have used this process to prepare benzyl dithiobenzoate,[46] 2-(ethoxycarbonyl)prop-2-yl dithiobenzoate,[46] and 2-cyanoprop-2-yl dithiobenzoate.[9] Similar chemistry is used in the synthesis of unsymmetrical trithiocarbonates (Scheme 9).[114,115] Yields are generally high (>70%) for substitution of primary and secondary alkyl halides, but can be low for tertiary halides (5–40%).
-
Addition of a dithio acid across an olefinic double bond.[44,116,117] This procedure has been used to prepare cumyl dithiobenzoate (Scheme 10).[9,14] Electron-rich olefins (S, AMS, isooctene, VAc) give Markownikov addition (sulfur at substituted position) and useful RAFT agents, while electron-deficient olefins (MMA, MA, AN) unfortunately give Michael-like addition (sulfur at unsubstituted position).
-
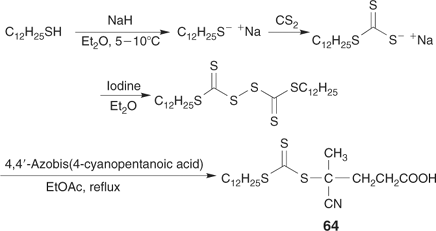
Radical-induced decomposition of a bis(thioacyl) disulfide (e.g. Scheme 11).[15,45,46,118,119] This is probably the most used method for the synthesis of RAFT agents requiring tertiary R groups. It is also possible to use this chemistry to generate a RAFT agent in situ during polymerization.[45]
-
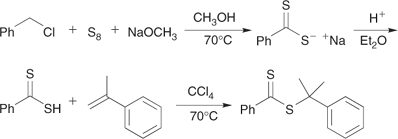
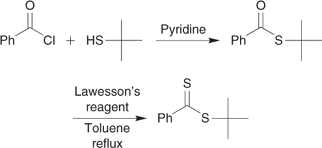
Sulfuration of a thioloester,[14,44] a carboxylic acid plus an alcohol[120] or a carboxylic acid with a halide or olefin[121] with P4S10, or Davey or Lawesson’s reagent (Scheme 12).
-
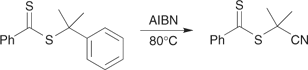
Radical-induced ester exchange.[9,44,46,122] For this method to be effective, the R group of the precursor RAFT agent should be a good free-radical leaving group with respect to that of the product RAFT agent. For example, the cyanoisopropyl radical generated from azobis(isobutyronitrile) (AIBN) can replace the cumyl group of cumyl dithiobenzoate (Scheme 13).[9]
|
|
|
|
|
|
|
In the synthesis of dithiobenzoates (and related dithioesters) by the above processes, either dithiobenzoic acid or its salts will often be formed in situ by various routes. These include the reaction of benzyl chloride[9,123] or a benzyl sulfone[124] with elemental sulfur in the presence of base, reaction of phenyl Grignard reagent with carbon disulfide,[9] and treatment of benzoic acid with P4S10 or Davey reagent.[121,125]
Evidence in Support of the RAFT Mechanism
Evidence in support of the overall RAFT reaction (Scheme 4) is the retention of the RAFT end groups; this can be demonstrated by NMR spectroscopy and mass spectrometry. Many papers on RAFT provide NMR spectra of RAFT-made polymers. Fig. 9 shows the expected structure and the 1H NMR spectrum of a PMMA of

|
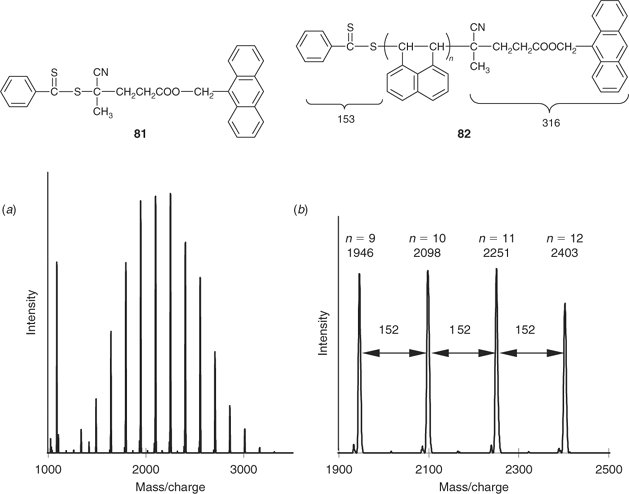
Additional evidence for the retention of the RAFT end groups and the quality of RAFT polymerization under various conditions is provided by matrix-assisted laser desorption ionization time-of-flight (MALDI–TOF) or electrospray ionization (ESI) mass spectrometry. Systems studied by mass spectrometry include (polymer/RAFT agents): PNIPAM/20,22,[78] PNIPAM/39,40,[93] PVAc/49,[48] PS/21,[126] PS/69,[106] PMMA/8,11,[100] PBA/57,63,[127] PMA/21,22,24,[81,83] PEA/79,[49] and poly(acenaphthalene)/81.[128] ESI is a softer ionization technique than MALDI–TOF and is more likely to leave the end groups intact to be observed in the mass spectrum.[100,129] MALDI–TOF analysis was successfully carried out on the poly(acenaphthalene) 82 prepared with RAFT agent 81.[128] The major peaks in the spectrum (Fig. 10) correspond to: m81 + nmAcN + mAg = (153 + 316) + (152n) + 108; consistent with the polymer being capped with PhC(=S)S and anthracenyl moieties as expected for structure 82.
|
Interaction chromatography (liquid chromatography at the critical point of adsorption; LC-CC) and two-dimensional chromatography (2D GPC/LC-CC) have also been applied to establish the presence of RAFT end groups and the quality of RAFT polymerization. Pasch et al.[130] used 2D GPC/LC-CC to examine a PMMA-b-PS prepared in our laboratories by RAFT with cumyl dithiobenzoate as the RAFT agent and were able to ‘confirm the remarkable molar mass and compositional homogeneity’ of the sample. Jiang et al. have determined the end-group composition (fraction of RAFT agent and initiator-derived chain ends) of PMMA[100] and PBA[127] prepared by RAFT with functional RAFT agents by LC-CC coupled with mass spectrometry.
We obtained the electron paramagnetic resonance (EPR) spectrum of intermediate radical 83 shown in Fig. 11 during BA polymerization at 90°C with cumyl dithiobenzoate 22 as RAFT agent.[131] This provided the first direct evidence for the addition–fragmentation mechanism and the involvement of species such as 3 and/or 5 as discrete intermediates (Scheme 6). The intermediates have been observed directly by EPR in RAFT polymerizations of styrene and acrylate esters and in model reactions with dithiobenzoate and dithiophosphonate RAFT agents.[112,131–136] The intermediates 3 or 5 are not detectable during MMA polymerizations or in polymerizations with aliphatic dithioester, trithiocarbonate, xanthate, or dithiocarbamate RAFT agents. This is attributed to the greater rate of fragmentation and the correspondingly shorter lifetime of the adduct in these polymerizations.

|
Side Reactions in RAFT
A variety of side reactions can potentially complicate the RAFT mechanism causing retardation, by-products, and anomalies in the molecular-weight distributions.[137] Whether these occur depends on the particular RAFT agent/monomer combination and the reaction conditions.
Retardation is observed in RAFT polymerizations when high concentrations of RAFT agent are used and/or an inappropriate choice of RAFT agent is made. Some decrease in polymerization rate is clearly attributable to a mitigation of the gel (or Trommsdorf) effect.[44,46] However, it is also clear that other effects are important in some circumstances.
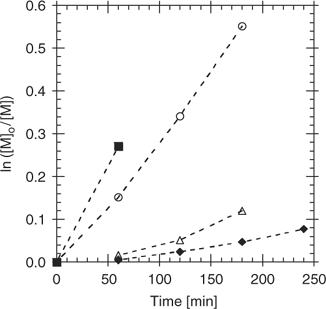
For example, there is significant retardation in the polymerization of acrylate esters in the presence of dithiobenzoate esters.[9,44,52,64,84,136–138] For polymerization of MA with benzyl 20 or cyanoisopropyl dithiobenzoate 8 as RAFT agent at 60°C, we found retardation from the onset of polymerization. The retardation appeared not to be directly related to consumption of the initial RAFT agent (which was rapid), with the dithioester being completely consumed at the first time/conversion point. An aliphatic dithioester, benzyl dithioacetate 31, was found to give substantially less retardation under the same reaction conditions (Fig. 12). The observation of less retardation in RAFT polymerization of acrylate esters with aliphatic dithioesters and trithiocarbonate RAFT agents than is seen with dithiobenzoate RAFT agents has also been reported under other circumstances.[9,52,64,84,137] Quinn et al.[90] observed that 1-phenylethyl dithiophenylacetate 34 enabled RAFT polymerization of MA at ambient temperature, whereas 1-phenylethyl dithiobenzoate 21 strongly retarded polymerization under the same conditions. McLeary et al.[84] observed that RAFT polymerization of MA with cumyl dithiophenylacetate 35 was subject to an inhibition period corresponding to the consumption of the initial RAFT agent. This was attributed to slow re-initiation by the cumyl radical during what was called the initialization period. Available data indicate that the rate constant for addition of cumyl radicals to MA is slow with respect to that for propagation.[44,139] The reported rate constants for benzyl and cyanoisopropyl radicals adding to MA is similarly slow or slower with respect to propagation, yet no substantial inhibition period is seen with these RAFT agents.[44,139] We propose that the inhibition period relates not to slow re-initiation by cumyl radical in itself, rather to the importance of the back reaction of cumyl radicals with the RAFT agent.[44] Cumyl radicals add to RAFT agents at close to diffusion controlled rates which has the effect of magnifying any problems associated with slow re-initiation and also leads to concentration dependence of the apparent transfer constants. This is also a known issue in MMA and styrene polymerization with cumyl dithiobenzoate (see below).[44]
|
Retardation has also been observed in polymerizations of styrene and methacrylates, and is pronounced when high concentrations of dithiobenzoate RAFT agents are used.[9,52–55,133,137,140,141] With lower concentrations of RAFT agents such as cyanoisopropyl dithiobenzoate 8 and cumyl dithiobenzoate 22 (<0.03 M), we have shown that rates of polymerization for styrene and MMA are little different from that expected in the absence of RAFT agent and appear independent of the particular dithiobenzoate.[9,44,52] Inconsistencies in reported rates of polymerization suggest that, in some cases, lower rates may in part be attributed to extraneous factors such as impurities in the RAFT agent or incomplete degassing. For higher RAFT agent concentrations, the extent of retardation becomes markedly dependent on which initial RAFT agent is used and its concentration. Much less retardation is seen with cyanoisopropyl dithiobenzoate 8 than is seen with cumyl dithiobenzoate 22. The retardation is in part manifested as an inhibition period which corresponds to the time taken to convert that RAFT agent into the polymeric RAFT agent. The apparent transfer constant measured by the rate of utilization of the RAFT agent is strongly concentration dependent.[9,44,52]
There is currently some controversy surrounding the stability and possible alternate fates of the intermediate radical in RAFT polymerization. These adducts might, in principle, undergo various side reactions. They include:
-
Reaction of the intermediate radicals 3 or 5 with other radical species such as an initiator-derived or a propagating radical.[140] In the case of 5, coupling with a propagating radical leads to a three-armed star (Scheme 14).[53,126,133,141]
-
Reaction of the intermediate radicals 3 or 5 with another such radical. In the case of self-reaction of 5, this pathway leads to a four-armed star.
-
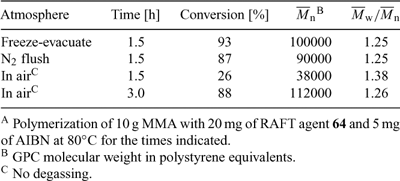
Reaction with oxygen and impurities. RAFT polymerization can show sensitivity to air and impurities in the monomer. Results for polymerizations carried out with unpurified commercial MMA at 80°C with thorough degassing by freeze–evacuate–thaw cycles, a simple nitrogen flush, and no degassing are shown in Table 8. All polymerizations provide narrow molecular-weight distributions and some molecular weight control. However, polymerizations with no degassing give substantial retardation. All polymerizations yield a lower-than-expected molecular weight which we attribute to the use of unpurified monomer.
-
For some RAFT agents there are multiple pathways for β-scission involving cleavage of a bond within the ‘Z’-group. For unsymmetrical trithiocarbonates[96] and xanthates[57] two possible fragmentation pathways are possible.
-
Reaction with monomer to re-initiate polymerization. Copolymerization of macromonomer RAFT agents has been observed;[32] however, the pathway is currently unknown for thiocarbonylthio RAFT agents.
|
|
Slow fragmentation, by itself, is an unlikely cause for retardation in RAFT polymerization of styrene and methacrylates. This would require that the rate of fragmentation be very low and that the concentrations of the adduct radicals 3 or 5 be very high (approx. 10−4 M for styrene polymerization with cumyl dithiobenzoate); much higher than is observed experimentally by EPR (< 10−7 M).[131] However, the adduct radical formed by addition to dithiobenzoate RAFT agents should be significantly more stable than that formed by addition to aliphatic dithioesters or trithiocarbonates. This expectation is supported by molecular orbital calculations.[58] Slower fragmentation should then mean that this radical is more likely to be involved in side reactions such as coupling with radical species. There is currently vigorous debate with respect to the significance of coupling reactions involving the adduct radicals 3 or 5 during polymerization. For dithiobenzoate RAFT agents, the pathway has been observed with model systems under conditions of high radical flux.[126,133,140,141] However, there is as yet no direct evidence that the process occurs during polymerization even under conditions where significant retardation is observed. We have proposed that inhibition in vinyl acetate polymerization is caused by side reactions of the adduct radical.[6]
Initiation and Termination in RAFT Polymerization
Since radicals are neither formed nor destroyed during reversible chain transfer, RAFT polymerization must be initiated by a source of free radicals. RAFT polymerization is usually carried out with conventional radical initiators. In principle, any source of free radicals can be used,[14] but most often thermal initiators (e.g. AIBN, ACP, K2S2O8) are used. Styrene polymerization may be initiated thermally between 100 and 120°C. Polymerizations initiated with UV irradiation,[142,143] a γ-source,[144–149] or a plasma field[150] have been reported. In these polymerizations, radicals may be generated directly from the RAFT agent and these may be responsible for initiation. It was suggested by Pan and coworkers that the mechanism for molecular weight control in UV[143] and γ-initiated[147] processes might involve reversible coupling and be similar to that proposed by Otsu et al.[151] to describe the chemistry of dithiocarbamate photoiniferters. However, Quinn et al.[142,145] have demonstrated that the living behaviour observed in these polymerizations can be attributed to the standard RAFT mechanism (Scheme 6).
The initiator concentration and rate of radical generation in RAFT polymerization are chosen to provide a balance between an acceptable rate of polymerization and an acceptable level of dead chains (radical–radical termination). One useful guideline is to choose conditions so that the target molecular weight is about 10% of that which would have been obtained in the absence of RAFT agent. One common misconception is that it is necessary to use very low rates of polymerization in order to achieve narrow molecular weight distributions. Sometimes, using a high rate of polymerization and a correspondingly short reaction time can provide better results. It is very important not to use prolonged reaction times when retention of the RAFT functionality is important. Once the monomer is fully converted, continued radical generation may not change the molecular weight distribution, but it can lead to formation of dead chains.
Side reactions of the initiator or initiator-derived radicals with the RAFT agent are possible. However, these are not always readily discernable because of the high RAFT agent-to-initiator ratios used in well designed experiments. It follows from the mechanism of the RAFT process that there should be a fraction of dead chains formed relating directly to the number of chains initiated. Ideally, this fraction should be taken into account when calculating the molecular weights of polymers formed by the RAFT process.[44] The molecular weight of the polymer formed can be estimated knowing the concentration of the monomer consumed and the initial concentration of RAFT agent by using the simple relationship shown in Eqn 1. Positive deviations from Eqn 1 indicate incomplete usage of RAFT agent. Negative deviations indicate that other sources of polymer chains are significant. These include the initiator-derived chains.
If initiator-derived chains are significant, Eqn 5 should be used to calculate molecular weights:
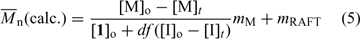
where mM and mRAFT are molecular weights of the monomer and the RAFT agent respectively, d is the number of chains produced from radical–radical termination (d ≈ 1.67 in MMA and d ≈ 1.0 in styrene polymerization), [I]o – [I]t is the concentration of initiator consumed, and f is the initiator efficiency.
If the initiator decomposition rate constant is known, the initiator consumption can be estimated using Eqn 6:
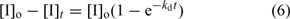
The fraction of living chains (L) in RAFT polymerization (assuming no other side reactions) is given by Eqn 7:
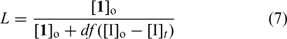
In Fig. 13 we show the usual plot of the dependence of molecular weight and polydispersity on conversion for a RAFT polymerization of MMA. The initiator concentration was about sixfold lower than the RAFT agent concentration. The polydispersity observed at high conversion is very low (
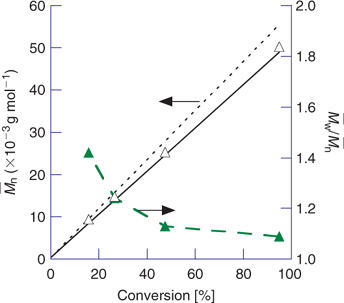
|
 ); line of best fit (
); line of best fit ( ).
).It is also possible to calculate the fraction of living chains based on the difference between the experimental molecular weight and that predicted by Eqn 1 by using Eqn 8. Fig. 14 shows the dependence of the fraction of living chains on RAFT agent concentration for a series of polymerizations carried out with the same reaction times and initiator levels. All polymerizations gave narrow polydispersity products (
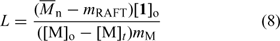
One method for gaining further insight into the RAFT mechanism is through the use of kinetic simulation. Our first paper on kinetic simulation of the RAFT process was published in 1998.[152] Many papers have now been written on kinetic simulation of the RAFT process. Zhang and Ray,[153] and Wang and Zhu[154,155] have used a method of moments. Shipp and Matyjaszewski,[156] and Barner-Kowollik and coworkers[54,55,138,157] have used a commercial software package (Predici) to evaluate complete molecular-weight distributions. Peklak et al.[158] have used a coarse-graining approach to give molecular-weight distributions. We have applied a hybrid scheme in which the differential equations are solved directly to give the complete molecular-weight distribution to a finite limit (DP < 500), and a method of moments is then used to provide closure to the equations, accurate molecular weights, and polydispersities.[44,46,152] Much of the research in this area has been carried out with a view to understanding the factors which influence retardation.[159,160] Unfortunately, many studies have made use of a simplified mechanism which in some cases may compromise the findings. The main difficulty in modelling RAFT lies in choosing values for the various rate constants.
Reaction Conditions (Temperature, Pressure, Solvent, Lewis Acids)
There have been no comprehensive studies of the effect of temperature on the course of RAFT polymerization. Temperatures reported for RAFT polymerization range from ambient to 140°C. There is evidence with dithiobenzoates that retardation is less at higher temperatures and also data that show narrower molecular-weight distributions can be achieved at higher temperatures.[7] For MMA polymerization with trithiocarbonate 64 there appears to be no dramatic effect of temperature on the molecular-weight distribution (Fig. 15). It should be noted, however, that higher temperatures offer higher rates of polymerization allowing a given conversion to be achieved in a shorter reaction time.
RAFT polymerizations under very high pressure (5 kbar) have been reported.[161–163] At high pressure, radical–radical termination is slowed and this allows the formation of higher-molecular-weight polymers and higher rates of polymerization than are achievable at ambient pressure.
The RAFT process is compatible with a wide range of reaction media including protic solvents such as alcohols and water[13,14,164,165] and less conventional solvents such as ionic liquids[166] and supercritical carbon dioxide.[167] RAFT polymerization has been successfully carried out in aqueous media. However, care should be taken because certain RAFT agents show some hydrolytic sensitivity.[243] We have found that this roughly correlates with activity of the RAFT agent (dithiobenzoates > trithiocarbonates ≈ aliphatic dithioesters).
RAFT polymerization can be conducted in the presence of Lewis acids. There are reports of attempted tacticity control of homopolymers[91,168–170] (to enable the synthesis of stereoblock copolymers[171]) and control of the alternating tendency for copolymerizations[172,173] through the use of Lewis acids as additives. For MMA polymerization, the addition of the Lewis acid scandium triflate, Sc(OTf)3, increases the fraction of isotactic triads and enhances the rate of polymerization in conventional radical[174] and RAFT processes.[168,170,171] Polymerizations with dithiobenzoate RAFT agents 8[170] or 22[168,170,171] and Sc(OTf)3 gave poor control over molecular weight and polydispersity. We[170] have shown by NMR analysis that the poor results can be attributed to the Lewis acid causing degradation of the dithiobenzoate RAFT agents; and further we have shown that trithiocarbonates are substantially more stable. Thus, polymerizations with the trithiocarbonate RAFT agent 58 provided polymer with narrow molecular-weight distributions (
RAFT in Heterogeneous Media
Much has now been written on RAFT polymerization in emulsion and mini-emulsion conditions. Our first communication on RAFT polymerization briefly mentions the successful emulsion polymerization of butyl methacrylate with cumyl dithiobenzoate as a table entry.[13] Additional examples and brief discussion of some of the important factors for successful use of RAFT polymerization in emulsion and mini-emulsion were provided in a subsequent paper.[9] Much research has shown that the success in RAFT emulsion polymerization depends strongly on the choice of RAFT agent and polymerization conditions.[9,175–183] Most work has focussed on styrene polymerization,[9,176,177,183] although RAFT emulsion polymerizations of BA[178] and methcrylates[9,13] have also been reported. Use of cumyl dithiobenzoate 22 as RAFT agent in ab initio emulsion polymerization of styrene is not recommended.[9]
The emulsion recipes we reported[9,13] were feed processes in which conversion of monomer into polymer was maintained at a high level (often >90%). In a first ab initio step, a low-molecular-weight polymeric RAFT agent was prepared. Control during this stage was not always good. However, the poorer polydispersity obtained in this step does not substantially affect control exerted during the later stages of polymerization.
A novel approach to RAFT emulsion polymerization has been reported recently.[103,181] In a first step, a water-soluble monomer (AA) was polymerized in the water phase to a low degree of polymerization to form a macro-RAFT agent. A hydrophobic monomer (BA) was then added under controlled feed to give oligomers that form rigid micelles. These constitute a RAFT-containing seed. Continued controlled feed of hydrophobic monomer may be used to continue the emulsion polymerization. The process appears directly analogous to the ‘self-stabilizing lattices’ approach we have previously used in macro-monomer RAFT polymerization which involves sequential polymerization of methacrylic acid and non-polar methacrylates.[184] Both processes allow emulsion polymerization without added surfactant.
RAFT in mini-emulsion has also been reported.[9,68,82,102,185–195] We showed that RAFT in mini-emulsion can be used to produce polystyrene of narrow polydispersity in a batch process.[9] Some retardation is observed with dithiobenzoate RAFT agents.[9,82] However, this is markedly reduced when aliphatic dithioesters[82] or trithiocarbonate RAFT agents are used.[102] One of the issues with traditional mini-emulsion polymerization is the high level of surfactant and co-stabilizer that is typically employed. Pham et al.[102] have recently described surfactant-free mini-emulsion polymerization. Amphipathic macro-RAFT agents synthesized in situ by polymerization of AA were used as the sole stabilizers. This process eliminated secondary nucleation of new particles and led to a latex with no labile surfactant and good particle size control.
RAFT Polymer Syntheses
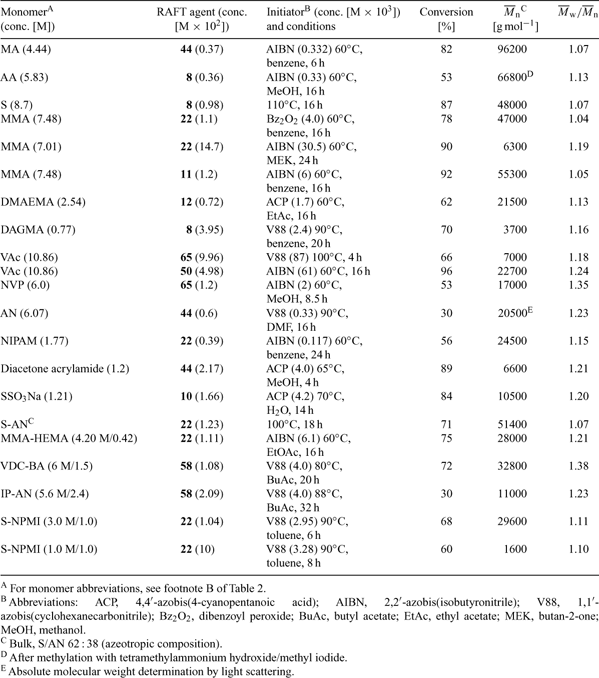
Some results for RAFT polymerizations taken from our own work are summarized in Table 9. Conversions listed are those obtained for the conditions and times stated. They are not limiting conversions or optimized reaction conditions. The data are intended to demonstrate the applicability of RAFT polymerization to a wide range of monomers under a wide range of reaction conditions. The process has been used to prepare both high- and low-molecular-weight polymers and copolymers with narrow-molecular-weight distributions. The results in Table 9 also demonstrate the compatibility of RAFT polymerization with various functional monomers.
|
End-Functional Polymers and End-Group Transformations
One significant advantage of RAFT over other processes for living polymerization is its compatibility with protic and other functionality (e.g. OH, SO3−, CO2H) present in the monomer or the RAFT agent. This makes the technique eminently suitable for the synthesis of end-functional polymers by incorporating the functionality into the Z or R groups of the RAFT agent. Functional RAFT agents include 11, 14 (–OH); 12, 15, 57, 59, 61, 63, 64 (–CO2H); 10 (–CO2Na); and 13 (–SO3Na). References to the synthesis and use of these RAFT agents can be found in Tables 2 and 5. Additional examples are provided in Table 9.
The synthesis of polymers with primary or secondary amine functionality presents a problem since these amines undergo facile reaction with thiocarbonylthio compounds. Thus, amine end-functional polymers cannot be prepared directly. Such polymers can be indirectly prepared by using RAFT agents with latent amine functionality, such as the phthalimido group in RAFT agents 56 and 60,[15] which can be subsequently deprotected.
A key feature of RAFT is that the thiocarbonylthio group(s), present in the initial RAFT agent, is(are) retained in the polymeric product(s). The retention of these groups is responsible for the polymers’ living character. The products are themselves RAFT agents. However, the presence of the thiocarbonylthio groups also means that the polymers synthesized by RAFT polymerization are usually coloured. This colour may range from violet through red to pale yellow depending on the absorption spectrum of the particular thiocarbonylthio chromophore. The presence of colour can be disadvantageous in some applications. Even though colour may be modified by appropriate selection of the initial RAFT agent, there has nonetheless been some incentive to develop effective methods for treatment of RAFT-made polymer to cleave the thiocarbonylthio end groups post polymerization.
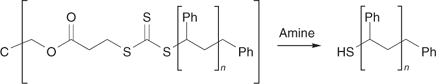
In some circumstances, it is also necessary or desirable to deactivate thiocarbonylthio groups because of their reactivity or to transform them for use in subsequent processing. For certain applications, it is desirable to have polymers possessing thiol functionality. These applications include the use of bisthiols in the synthesis of condensation polymers such as polythiourethanes or polythioesters. Thiol functionality may also be used to form crosslinks. Other applications of thiol functional polymers relate to the property of thiols to complex metals and to form conjugates with biological polymers, such as proteins.[95,196]
Some of the processes that have been used for thiocarbonylthio group removal/transformation are shown in Scheme 15. They include hydrolysis/aminolysis, various radical induced reactions, and thermal elimination.[15,101] The RAFT end-group is also light sensitive and can be removed under UV irradiation;[70] it may be oxidized with reagents such as peroxides or sodium hypochlorite.[13]
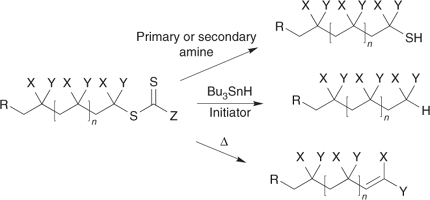
|
The kinetics and mechanism of the reaction of compounds containing thiocarbonyl groups with nucleophiles has been reviewed by Castro.[197] It is well known that thiocarbonylthio groups can be transformed into thiols by reaction with nucleophiles which include pyridines, primary and secondary amines, ammonia, other thiols, and hydroxide. They may also be reduced to thiols with hydride reducing agents such as sodium borohydride, lithium aluminum hydride, or with zinc in acetic acid. The thiocarbonylthio groups in RAFT-synthesized polymers are subject to the same reactions. The potential of this chemistry to cleave end groups and decolourize polymers and produce polymers with thiol end groups was cited in our initial communication on RAFT polymerization.[13] Examples of end-group cleavage with nucleophiles such as amines,[15,96,198,199] hydroxide,[93] and borohydride[164,200] can be found in recent publications. Radical induced reduction with, for example, tri-n-butylstannane[15,201,202] can be used to replace the thiocarbonylthio group with hydrogen.
RAFT end groups are known to be unstable at very high temperatures. At temperatures >200°C, thermal elimination has been used as a means of trithiocarbonate end-group removal from polystyrene or poly(butyl acrylate).[15,101] Thermal C–S bond homolysis is also possible. Depending on the propagating chain and the RAFT group, these reactions may also occur at lower temperatures and will set an upper temperature limit for the RAFT process.
Gradient Copolymers
In most copolymerizations, the monomers are consumed at different rates dictated by the steric and electronic properties of the reactants. Consequently, both the monomer feed and copolymer composition will drift with conversion. Thus conventional copolymers are generally not homogeneous in composition at the molecular level. In RAFT polymerization processes, where all chains grow throughout the polymerization, the composition drift is captured within the chain structure. All chains have similar composition and are called gradient or tapered copolymers.
Copolymers (e.g. S–AN, MMA–HEMA, see Table 9) when formed in the presence of a RAFT agent have the same overall composition and sequence distribution (NMR) as those formed in the absence of a RAFT agent. Reactivity ratios are unaffected by the RAFT process. However, for very low conversions when molecular weights are low, copolymer composition may be different for that seen in conventional copolymerization because of specificity shown by the initiating species (R).[203]
A difference between copolymers synthesized by RAFT copolymerization and those synthesized by conventional radical copolymerization is that copolymers formed by RAFT copolymerization are gradient copolymers whereas those formed by the conventional process are blends. It is possible to synthesize block polymers in a batch polymerization by taking advantage of disparate reactivity ratios between particular monomer pairs (BA-MMA,[5,7] tBA-VAc,[110] S-MAH[51,70,79,80]). The composition can be further tailored by use of suitable monomer feed protocols. In general, it is important in designing copolymerizations to choose a RAFT agent that is compatible with all the monomers in the feed. RAFT copolymerization can be successful (provide molecular weight control and narrow molecular weight distributions) even when one of the monomers is not amenable to direct homopolymerization using a particular RAFT agent. RAFT polymerization of MMA with benzyl dithiobenzoate provides very poor control yet copolymerization of S with MMA succeeds while the medium contains some (approx. 5%) styrene.
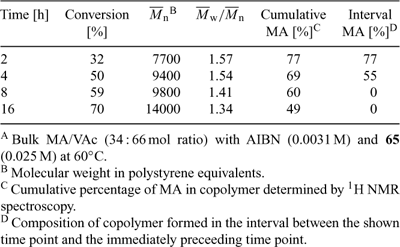
An example of gradient copolymer formation is the batch copolymerization of MA and VAc. The reactivity ratios (rMA ≈ 9, rVAc ≈ 0.1) are such that both propagating radicals prefer to add to MA. Thus, MA-VAc copolymerization in the presence of a xanthate RAFT agent provides copolymer chains that are rich in MA at one end and rich in VAc at the other. The results of one such experiment are shown in Table 10. When a mixture of MA and VAc (34 mol % M) was copolymerized with AIBN in the presence of O-ethyl S-cyanomethyl xanthate 65 at 60°C, the first-formed polymer (at 2 h) is predominantly composed of MA units, and the product following exhaustion of this monomer is poly(methyl acrylate-co-vinyl acetate)-block-poly(vinyl acetate).
|
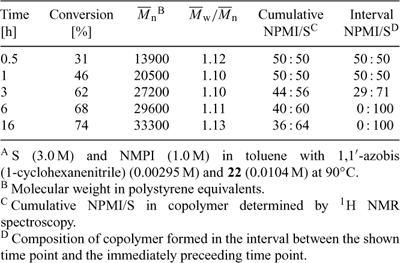
Another example is the copolymerization of styrene (S) with N-phenylmaleimide (NPMI; Table 11). In this case, the reactivity ratios (rS ≈ 0.02, rNPMI ≈ 0.04) are such that the two monomers have a strong tendency to alternate in the chain. When the copolymerization is conducted with an excess of styrene the product is poly(N-phenylmaleimide-alt-styrene)-block-polystyrene. It is thus possible to use RAFT polymerization to prepare block copolymers in a ‘one-pot’ batch polymerization process.
|
Diblock Copolymers
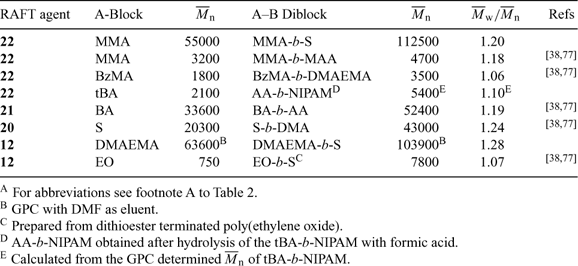
RAFT polymerization is recognized as one of the most versatile methods for block copolymer synthesis and numerous examples of block synthesis have now appeared in the literature. RAFT polymerization proceeds with retention of the thiocarbonylthio group. This allows an easy entry to the synthesis of AB diblock copolymers by the simple addition of a second monomer (Scheme 16).[77] Higher order (ABA, ABC, etc.) blocks are also possible by sequential addition of further monomer(s). The results of several such experiments are summarized in Table 12. References to other diblock syntheses are provided in Tables 2–6.
|
|
|
Of considerable interest is the ability to make hydrophilic–hydrophobic block copolymers where the hydrophilic block is composed of unprotected polar monomers such as acrylic acid, dimethylaminoethyl methacrylate, or ethylene oxide. A doubly hydrophilic block composed of acrylic acid and N-isopropylacrylamide (NIPAM) segments has also been prepared.[95] Other examples of doubly hydrophilic block copolymers have been reported by McCormick and coworkers.[244–247]
In RAFT polymerization, the order of constructing the blocks of a block copolymer can be very important.[44,77] The propagating radical for the first formed block must be a good homolytic leaving group with respect to that of the second block. For example, in the synthesis of a methacrylate–acrylate or methacrylate–styrene diblock, the methacrylate block should be prepared first.[53,77] The styrene or acrylate propagating radicals are very poor leaving groups with respect to methacrylate propagating radicals.
Block copolymers based on polymers formed by other mechanisms can be prepared by forming a pre-polymer containing thiocarbonylthio groups and using this as a macro-RAFT agent (Scheme 17).[77,110] We first exploited this methodology to prepare poly(ethylene oxide)-block-PS from commercially available hydroxy end-functional poly(ethylene oxide) (last example in Table 12).[77,110] Other block copolymers that have been prepared using similar strategies include poly(ethylene-co-butylene)-block-poly(S-co-maleic anhydride),[70] poly(ethylene oxide)-block-poly(MMA),[204] poly(ethylene oxide)-block-poly(N-vinyl formamide),[205] poly(ethylene oxide)-block-poly(NIPAM),[206] poly(ethylene oxide)-block-poly(1,1,2,2-tetrahydroperfluorodecyl acrylate),[207] poly(lactic acid)-block-poly(MMA),[204] and poly(lactic acid)-block-poly(NIPAM)[208,209]
|
|
Triblock Copolymers
A–B–A triblock copolymers can be prepared by RAFT polymerization in several different ways. One is to chain extend an A–B diblock with monomer A. However, the use of a bis-RAFT agent as a triblock precursor means that triblock synthesis can be accomplished in only two polymerization steps and helps ensure that both arms are of the same length and composition.[14,77,96]
There are two main classes of bis-RAFT agent. With the bis-dithioesters 84[77] or 85,[210] the bis-trithiocarbonate 86,[110] the bis-xanthate 87,[108] or bis-dithiocarbamate 88[195] the polymer is grown out from the core and the RAFT functionality is retained at the chain ends. With symmetrical trithiocarbonates (e.g. 54–57)[96,99] or bis-RAFT agents such as 89[14] or 90,[211] the RAFT functionality remains in the centre of the block (Scheme 18). The merits and demerits of the two approaches will be discussed further in the section on star polymers.
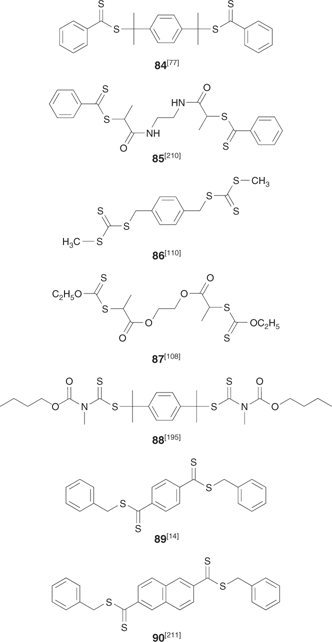
|
Some recent work has been orientated towards the synthesis of triblocks intended as thermoplastic elastomers based on a ‘soft’ acrylate mid-section and ‘hard’ styrene or styrene-co-acrylonitrile outer blocks (Scheme 19).[110] Rather than have labile functionality internal to the triblock, the RAFT agent 86 was chosen so that the thiocarbonylthio functions would remain at the chain ends (methyl is a very poor free-radical leaving group). The α,ω-bis(methylsulfanylthiocarbonylsulfanyl)–poly(butyl acrylate),
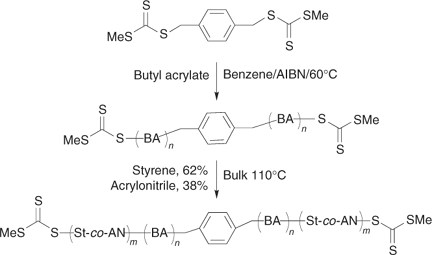
|
|
Star Polymers
There are now a large number of papers on the synthesis of star polymers using the RAFT process. The most common approach begins with a compound containing multiple thiocarbonylthio groups of appropriate design; a multi-RAFT agent. The synthesis of star polymers from multi-RAFT agents can be seen as an extension of the triblock syntheses described above where the number of thiocarbonylthio groups exceeds two. The multi-RAFT agent may be a small organic compound,[6,9,77,110,198,212–214] an organometallic complex,[215] a dendrimer,[216–218] a hyperbranched species,[219] a macromolecular species,[204,220] a particle, or indeed, any moiety possessing multiple thiocarbonylthio groups (though here the distinction between star and graft copolymers becomes blurred). Our first RAFT patent[14] recognized two limiting forms of star growth depending on the orientation of the thiocarbonylthio group with respect to the core. We have discussed the advantages and disadvantages of the ‘propagation away from core’ (Scheme 20) and ‘propagation attached to core’ (Scheme 21) strategies in various publications.[6,9,110,198]
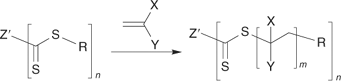
|
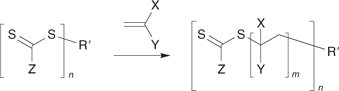
|
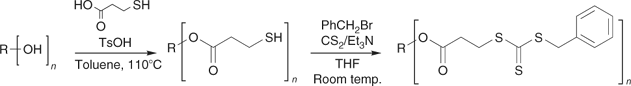
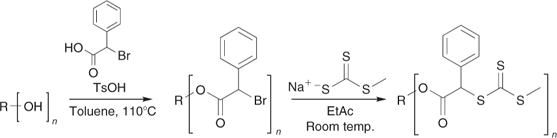
Multi-RAFT agents are readily synthesized from the corresponding multi-thiol, multi-hydroxy, and multi-halo compounds. The challenge in preparing such multifunctional molecules lies with developing reactions that proceed in quantitative yield and produce no by-products. Whereas incomplete reaction or side reactions in the synthesis of a monofunctional compound will give residual starting materials or an easily removed by-product, in the case of multifunctional compounds such process lead to molecules with incomplete functionality. For example, in the case of an eight-armed core, a yield of 95% for an individual functionalization step means that <70% of the product would contain the requisite number of arms. Purification of mixtures of cores with different levels of functionality can be extremely difficult and the process then becomes unviable. We have reported[114] methods to quantitatively (>95% overall yield) convert multi-hydroxy compounds into both classes of multi-RAFT agent (Schemes 22 and 23), and have applied these in making three-, four-, six-, and eight-armed stars with narrow polydispersity based on trimethylolethane, pentaerythritol, dipentaerythritol, and tripentaerythritol cores respectively.
|
|
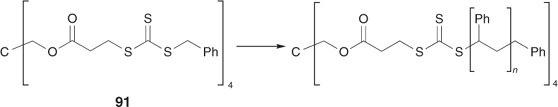
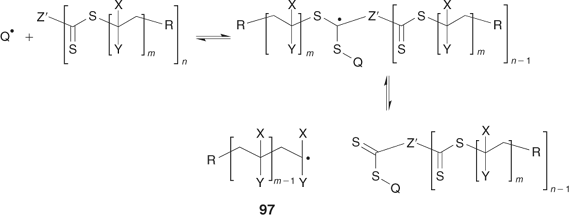
The ‘propagation away from core’ strategy (Scheme 20) is illustrated by the synthesis of a four-arm star from the tetra-trithiocarbonate 91 (Scheme 24, Fig. 17).[198] RAFT agents 93–95 also belong to this class (Scheme 25). The advantage of this strategy is that, since propagating radicals are never directly attached to the core, by-products from star–star coupling are unlikely (Scheme 26). Radical–radical termination will involve linear propagating radicals 97 and will produce a low-molecular-weight by-product that may be observed in high conversion polymerizations (for example, Fig. 17). All of the thiocarbonylthio functionality remains at the core of the star structure. It might be envisaged that steric factors associated with attack of the propagating radical at the core of the star would be an issue particularly at high conversions. There is, however, no direct evidence of problems attributable to this cause in the examples we have reported.
|
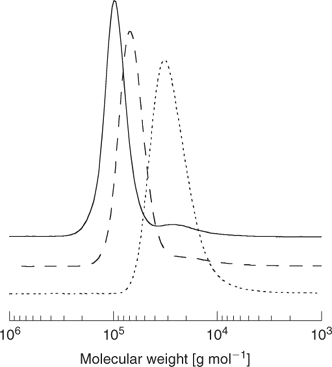
|

|
|
A potential disadvantage of the ‘propagation away from core’ strategy is that any reaction which cleaves the thiocarbonylthio groups (e.g. hydrolysis, thermolysis) results in destruction of the star structure. However, this property may be turned into an advantage in some circumstances; for example, in polymer-supported polymer synthesis. Arm cleavage also enables the molecular weights of the arms and the uniformity of arm growth to be readily verified (Scheme 27).[198]
|
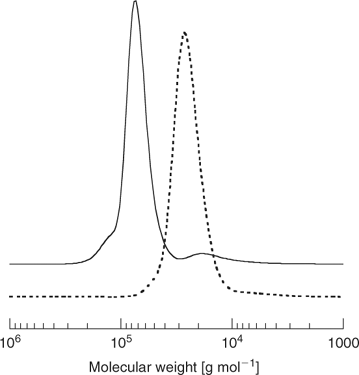
The ‘propagation attached to core’ strategy (Scheme 21) is illustrated by the synthesis of a four-arm star from the tetra-trithiocarbonate 92 (Scheme 28, Fig. 18).[198] RAFT agent 96 also belongs to this class.
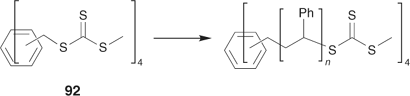
|
|
For these RAFT agents, the thiocarbonylthio functionality remains at the periphery of the star and the majority of propagating radicals will be attached to the core. Star–star coupling by self reaction of radicals 98 is therefore a potential complication (Scheme 29). A fraction of linear dormant chain commensurate with the number of initiator-derived chains will be formed as a by-product (for example, Fig. 18). The likelihood of star–star coupling is expected to increase with the number of arms to the star but can be minimized by controlling the conversion and rate of initiation.
|
The synthesis of stars is readily extended to star-blocks using strategies similar to those described under diblocks.[198]
Microgels
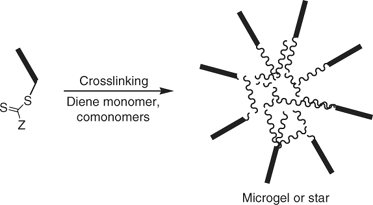
The so called ‘arm-first’ process[221,222] involves making a living polymer then using this to initiate (co)polymerization of a di- (or higher) functional monomer which forms a crosslinked core (Scheme 30). The methodology has been used to make star-shaped molecules using NMP or ATRP. Again, RAFT polymerization offers new scope to this process.[223] The RAFT process usually involves (co)polymerization of an appropriate diene monomer (e.g. divinylbenzene) in the presence of a polymer formed by RAFT polymerization. Lord et al.[224] have also used this strategy to prepare star microgels.
|
Suitably constructed hydrophilic–hydrophobic (or solvophilic–solvophobic) block copolymers possess the ability to self-assemble to form micelles (or other supramolecular structures). Such supramolecular assemblies may only be stable in a particular medium and/or over a specific concentration range. Various crosslinking mechanisms can be used to stitch these structures together to form a stable star polymer or microgel (or other structure). The core, the shell, or mid-block section of the micelle may be crosslinked to stabilize the structure.[110,225,226] Star microgel synthesis would usually require crosslinking of the micelle’s core (Scheme 31). Care is needed in selecting reaction conditions so as to avoid inter-micelle crosslinking, and gel formation. Variants of this methodology have been used to prepare a range of structures.[208,227]

|
Polymer Brushes/Graft Copolymers
The preparation of polymer brushes by controlled radical polymerization from appropriately functionalized polymer chains, surfaces, or particles by a grafting-from approach has recently attracted a lot of attention.[228] Most work has used ATRP or NMP, though papers on the use of RAFT polymerization have begun to appear.[229–235] The first to apply RAFT in this context were Tsujii et al.[229] and Brittain and coworkers.[230,231] Recent papers describe RAFT polymerization from plasma-treated Teflon surfaces[232] and ozonolyzed polyimide films.[235] The approach used in these and most other studies[230–235] has been to immobilize initiator functionality on the surface by chemical or plasma modification and use this to initiate polymerization in the presence of a dithioester RAFT agent. Tsujii et al.[229] have indicated that some difficulties arise in using RAFT for grafting from particles because of an abnormally high rate of radical–radical termination caused by a locally high concentration of RAFT functionality.
Skaff and Emrick[236] bound RAFT agent 99 (Scheme 32) to cadmium selenide nanoparticles by a ligand-exchange process, and grew various narrow molecular-weight distribution polymers (PS, PMA, PBA, PS-MA, PS-AA, PS-IP, PS-b-MA, PS-b-BA) from these particles. Perrier and coworkers[204,237] have attached RAFT moieties to cellulose (cotton) in order to from PS, PMA, or PMMA grafts. This involved derivatization of the cellulosic OH groups with thiocarbonylthio functionality.
|
Grafting-through approaches have also been applied. There have been several studies on the graft polymers by macromonomer (co)polymerization.[238–240] End-functional polymers prepared by RAFT polymerization can be used in ‘grafting-to’ reactions. The property of thiols and dithioesters to bind to heavy metals such as gold and cadmium has been used in preparing brushes based on gold film or nanoparticles[200,241,248] and cadmium selenide nanoparticles.[242] RAFT-synthesized thiols have also been used to make protein conjugates.[95,196]
Summary
RAFT polymerization has emerged as one of the most important methods for living radical polymerization. The method is robust and versatile and can be applied to the majority of polymers prepared by radical polymerization. However, selection of the RAFT agent for the monomers and reaction conditions is crucial for success. Guidelines for selection exist and most monomers can be polymerized with good control by using one of two RAFT agents. One suited to (meth)acrylate, (meth)acrylamide, and styrenic monomers, for example, a tertiary cyanoalkyl trithiocarbonate, and another suited to vinyl monomers such as VAc, for example, a cyanoalkyl xanthate. Monomers that remain difficult to homopolymerize by the RAFT process are the olefins (ethylene, propylene). Notwithstanding these comments, many features of the kinetics of RAFT polymerization with specific RAFT agents remain to be detailed and unravelled.
RAFT polymerization can provide control over the manner of chain initiation and termination in radical polymerization. A remaining challenge in free-radical polymerization is to obtain simultaneous control over the propagation step to achieve greater chemo-, regio-, and stereo-specificity. In RAFT polymerization, all data point to the propagating radicals behaving as conventional propagating radicals. With judicious choice of RAFT agent, the reagents (e.g. Lewis acids) used to control propagation in conventional polymerization can be applied to achieve similar control during the RAFT process.
We have illustrated how the RAFT process is being used in the synthesis of well defined homo-, gradient, diblock, triblock, and star polymers as well as more complex architectures including microgels and polymer brushes. New materials that have the potential of revolutionizing a large part of the polymer industry are beginning to appear. Possible applications range from novel surfactants, dispersants, coatings, and adhesives, to biomaterials, membranes, drug delivery media, and materials for microelectronics.
We are grateful to DuPont Performance Coatings, in particular Charles T. Berge, Michael Fryd, and Robert R. Matheson, for their support of the work carried out at CSIRO Molecular Science. We also thank our many colleagues at CSIRO and other institutions who have contributed to the development of RAFT polymerization; their names are provided in the references.
[1]
[2]
C. J. Hawker,
A. W. Bosman,
E. Harth,
Chem. Rev. 2001, 101, 3661.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |

* The number average molecular weight or molar mass is simply the total weight of the sample divided by the number of molecules in the sample: