

Telomerase Inhibition Studies of Novel Spiroketal-Containing Rubromycin Derivatives
Tsz-Ying Yuen A , Yu-Pong Ng B , Fanny C. F. Ip B , Jack L.-Y. Chen A , Darcy J. Atkinson A , Jonathan Sperry A , Nancy Y. Ip B C and Margaret A. Brimble A CA School of Chemical Sciences, The University of Auckland, 23 Symonds Street, Auckland 1010, New Zealand.
B Division of Life Science, The Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong, China.
C Corresponding authors. Email: boip@ust.hk; m.brimble@auckland.ac.nz
Australian Journal of Chemistry 66(5) 530-533 https://doi.org/10.1071/CH13035
Submitted: 19 January 2013 Accepted: 22 February 2013 Published: 4 March 2013
Abstract
Twenty nine novel spiroketal derivatives related to the rubromycins were evaluated for their anti-telomerase activity using the real-time quantitative telomeric repeat amplification protocol assay. The parent compound γ-rubromycin exhibited the highest potency against human telomerase activity within the series. Modification of the spiroketal motif by the introduction of heteroatoms and substituents at different positions produced analogues with varying bioactivity. Variation at the isocoumarin subunit of the title compound resulted in weaker activity, indicative of its importance in telomerase inhibition.
Introduction
The rubromycins (Fig. 1) belong to a family of structurally related pigments that were first isolated from Streptomyces collinus in the 1950s and 1960s.[1] With the exception of α-rubromycin, this class of compounds possesses a densely oxygenated naphthoquinone moiety and an isocoumarin unit connected through an optically active bisbenzannulated 5,6-spiroketal ring system. The rubromycins initially garnered scientific interest due to their vivid colours but they were later found to exhibit antimicrobial and cytostatic activity against several enzymes including the human telomerase reverse transcriptase, an enzyme strongly associated with cell immortalisation.[2] β-Rubromycin and γ-rubromycin appeared to be the most potent telomerase inhibitors, with IC50 values of ~3 µM. In contrast, the ring-opened α-rubromycin displayed substantially decreased potency (IC50 >200 µM), suggesting the role of the spiroketal core as an essential pharmacophore for telomerase inhibition. To lend further support to this theory, the closely related spiroketal-containing purpuromycin and griseorhodins A and C were also shown to exhibit comparable inhibition of human telomerase (IC50 = 3–12 µM).[2b,3]

|
Given our long standing interest in the total synthesis and biological evaluation of spiroketal-containing natural products,[4] we were attracted to the rubromycins, motivated in no small part by the prominent synthetic challenge posed by the presence of the electron-withdrawing isocoumarin unit in the molecules. Despite substantial efforts towards the synthesis of this family of natural products by several research groups, only a single total synthesis of the heliquinomycin aglycon had been achieved by Danishefsky[5] at the commencement of this project. The low success rate for the total synthesis of the rubromycins was attributed to difficulties assembling the spiroketal core under acidic conditions. Through comprehensive modelling studies, Kozlowski[6] and Reissig[7] established that the presence of electron-withdrawing groups in the isocoumarin moiety markedly diminished the nucleophilicity of the phenolic group by inductive and mesomeric effects, thus reducing the efficiency of the key spiroketalisation step.
Based on the premise that the spiroketal moiety of the rubromycins acts as the crucial pharmacophore for telomerase inhibition,[2b] we focussed our attention on the preparation of γ-rubromycin and a series of modified spiroketal analogues to establish additional insight into the underlying structure-activity relationship and the key pharmacophore for this class of compounds. Recent studies by the Pettus group has provided further mechanistic insight into the role of β-rubromycin in telomerase inhibition and identified the spiroketal scaffold to be the best lead inhibitor.[8] Biological assay results for the parent compound, γ-rubromycin together with our synthetic analogues are reported herein.
Results and Discussion
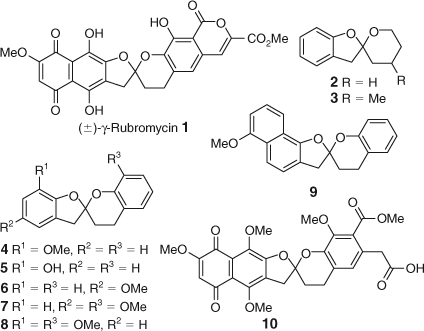
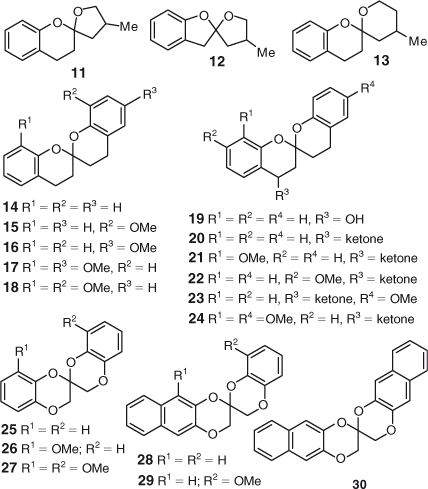
The chemical structures of the rubromycin analogues evaluated in the in vitro telomerase inhibition assay are depicted in Figs 2 and 3. The analogues were designed to establish the role of the spiroketal unit to inhibit telomerase activity. Specifically, two classes of spiroketal compounds with varying ring sizes, bulk and substitution pattern were prepared: (1) closely related analogues of γ-rubromycin;[9] and (2) modified analogues containing a 5,5-, 6,5- or 6,6-spiroketal scaffold.[9b,10] The choice of compounds was also governed by the availability of reagents and their ease of preparation. The synthesis of all spiroketal compounds has been published previously and the original papers should be consulted for detailed experimental procedures.
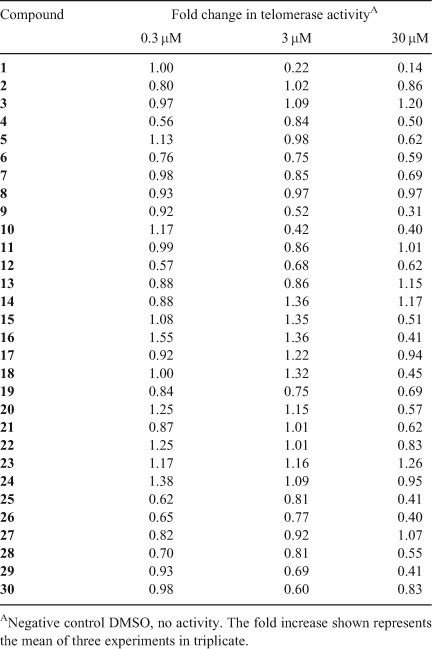
All the synthesised spiroketal derivatives (1–30) were evaluated for their anti-telomerase activity using a real-time quantitative telomeric repeat amplification protocol (RQ-TRAP) assay, with HEKneo cells as a source of telomerase. The results are summarised in Table 1 and the relative anti-telomerase activity at 0.3, 3 and 30 µM are expressed as means of fold change relative to the DMSO-treated control.
|
Amongst the closely related rubromycin-related analogues (Fig. 2), compound 9 displayed moderate levels of telomerase inhibition at 30 µM whereas the parent compound (±)-γ-rubromycin 1 was the most effective inhibitor. Whilst the rubromycins have been shown to competitively bind to the telomerase substrate primer by steady-state kinetic analysis,[2b] little information is known regarding the exact mechanism of telomerase inhibition or the role of functional groups such as the isocoumarin moiety. Thus, ring-opened lactone derivative 10 was subjected to the biological assay with HEKneo cells. Interestingly, 10 showed a significant decrease in anti-telomerase activity in comparison to the reference compound 1, suggesting the possibility of additional synergistic enzyme-substrate interactions between the isocoumarin subunit and the telomerase enzyme.
Next, we investigated the effects of various spiroketal scaffolds and substitution patterns on telomerase activity (Fig. 3). Among the 3,4,3′,4′-tetrahydro-2,2′-spirobis(2H-1-benzopyran) hetero-cyclic compounds (14–24), compounds 15, 16, and 18 promoted telomerase activity at 3 µM, yet surprisingly exhibited modest levels of telomerase suppression at 30 µM. Although the influence of substitution of the phenyl ring on telomerase inhibition is not apparent, these results are intriguing as they suggest the dual ability of some spiroketals to act as telomerase inhibitors or activators, depending on the substrate concentration. Whilst a tremendous amount of research is being conducted in the area of telomerase inhibition for cancer treatment,[11] the impact of telomerase activation in the context of age-related disorders is still in its infancy.[12] Thus, further evaluation of the representative samples will be carried out as part of the next phase of our medicinal chemistry program.
Modification of the spiroketal framework was further undertaken by the introduction of oxygen atoms in the pyran ring or on the carbon β to the spirocenter, as exemplified by purpuromycin and griseorhodins A and C. From Table 1, it can be seen that the presence of the added hydroxyl or ketone functional groups did not significantly influence anti-telomerase activity (compounds 19–24). On the other hand, spiroketals 25, 26, and 29, bearing a 3H,3′H-2,2′-spirobi(benzo[b][1,4]dioxine) ring system, exhibited enhanced telomerase inhibition in the HEKneo cell line.
Conclusion
In order to validate the postulate that the spiroketal system of the rubromycins is the active moiety responsible for inhibition of telomerase, several structurally related analogues of the rubromycins were evaluated for their anti-telomerase activity. Among the 30 compounds tested, reference compound (±)-γ-rubromycin 1 demonstrated the most potent anti-telomerase activity at 3 and 30 µM in the HEKneo cell line, whereas compounds 9, 10, 16, 18, 25, 26, and 29 exhibited modest levels of activities at 30 µM. Furthermore, the RQ-TRAP bioassay revealed that incorporation of the isocoumarin subunit into the title compounds may be of significant importance to inhibit telomerase as ring-opened lactone derivative 10 showed considerably weaker telomerase inhibition properties. Although conclusive structure-activity relationships cannot be constructed, the preliminary results reported herein focusing on several classes of small-molecule aryl spiroketal derivatives are encouraging and warrant further investigation.
Experimental
Cell Culture and Telomerase Activity Assay
Human neonatal keratinocytes (HEKneo) purchased from Invitrogen (Carlsbad, USA) were cultured using Epilife with 1 % HKGS in a T75 flask (BD Biosciences, USA) and maintained at 37°C in a 5 % CO2 incubator following the manufacturer’s protocol. HEKneo cells (PD 6–9) seeded at a density of 1 × 105 cells/well in a 12-well plate (TPP, Switzerland) were treated with the compounds for 24 h. Total cell lysates were collected with a lysis buffer and protein concentrations were quantified by DC protein assay (Bio-Rad, USA). RQ-TRAP assay was then performed according to the manufacturer’s manual to measure the telomerase activity in the lysate through its ability to synthesise telomeric repeats (Quantitative Telomerase Detection Kit, Allied Biotech, USA). The amplified polymerase chain reaction (PCR) products were visualised by DNA fluorochromes SYBR green using 7500 Fast Real-time PCR System (Applied Biosystems, USA). Each sample was analysed at least in triplicate for three independent experiments. Telomerase activity was expressed relative to vehicle control, i.e. the fold change of telomerase activity compared with DMSO treatment.
Acknowledgements
We thank the New Zealand Foundation for Research, Science and Technology (FRST) for financial support through the International Investment Opportunities Fund (IOFF). We would like to thank Ms Estella Tong for her excellent technical assistance. This study was also in part supported by National Key Basic Research Program of China (973 Program) 2013CB530900, Shenzhen Peacock Plan, and Hong Kong Jockey Club Charities Trust.
References
[1] (a) H. Brockmann, K. H. Renneberg, Naturwissenschaften 1953, 40, 59.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG3sXms1Kgtw%3D%3D&md5=e4c66037db0f016f1b52b44a3ab42dd8CAS |
(b) H. Brockmann, K. H. Renneberg, Naturwissenschaften 1953, 40, 166.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Brockmann, W. Lenk, G. Schwantj, A. Zeeck, Tetrahedron Lett. 1966, 7, 3525.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) C. Puder, S. Loya, A. Hizi, A. Zeeck, Eur. J. Org. Chem. 2000, 2000, 729.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. Ueno, H. Takahashi, M. Oda, M. Mizunuma, A. Yokoyama, Y. Goto, Y. Mizushina, K. Sakaguchi, H. Hayashi, Biochemistry 2000, 39, 5995.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) C. Coronelli, H. Pagani, M. R. Bardone, G. C. Lancini, J. Antibiot. (Tokyo) 1974, 27, 161.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2cXktVagtL4%3D&md5=40582b1e2db7f5df859c3e1e1b1ad619CAS |
(b) M. R. Bardone, E. Martinel, L. F. Zerilli, C. Coronell, Tetrahedron 1974, 30, 2747.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. Eckardt, D. Tresselt, W. Ihn, Tetrahedron 1978, 34, 399.
| Crossref | GoogleScholarGoogle Scholar |
(d) K. Eckardt, D. Tresselt, W. Ihn, J. Antibiot. (Tokyo) 1978, 31, 970.
| Crossref | GoogleScholarGoogle Scholar |
[4] (a) J. L. Y. Chen, M. A. Brimble, Chem. Commun. 2010, 46, 3967.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmsFSrsro%3D&md5=94dcb28b32e0ebb9be05538627368a9bCAS |
(b) M. A. Brimble, O. C. Finch, A. M. Heapy, J. D. Fraser, D. P. Furkert, P. D. O’Connor, Tetrahedron 2011, 67, 995.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. C. McLeod, Z. E. Wilson, M. A. Brimble, Org. Lett. 2011, 13, 5382.
| Crossref | GoogleScholarGoogle Scholar |
(d) T. Y. Yuen, S. H. Yang, M. A. Brimble, Angew. Chem. Int. Ed. 2011, 50, 8350.
| Crossref | GoogleScholarGoogle Scholar |
(e) H. M. Geng, J. L. Y. Chen, D. P. Furkert, S. D. Jiang, M. A. Brimble, Synlett 2012, 855.
(f) M. C. McLeod, Z. E. Wilson, M. A. Brimble, J. Org. Chem. 2012, 77, 400.
| Crossref | GoogleScholarGoogle Scholar |
(g) T. Y. Yuen, M. A. Brimble, Org. Lett. 2012, 14, 5154.
| Crossref | GoogleScholarGoogle Scholar |
[5] T. Siu, D. Qin, S. J. Danishefsky, Angew. Chem. Int. Ed. 2001, 40, 4713.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XivFKhtQ%3D%3D&md5=97dda71e5db0f0eb0817b13273da4673CAS |
[6] (a) S. P. Waters, M. W. Fennie, M. C. Kozlowski, Org. Lett. 2006, 8, 3243.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmsVKmsLw%3D&md5=8be8d72d8f5708da81d40e7e2be9ecd7CAS |
(b) R. Bandichhor, A. N. Lowell, M. C. Kozlowski, J. Org. Chem. 2011, 76, 6475.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. N. Lowell, M. W. Fennie, M. C. Kozlowski, J. Org. Chem. 2011, 76, 6488.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) M. Brasholz, S. Sorgel, C. Azap, H. U. Reissig, Eur. J. Org. Chem. 2007, 3801.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXptlahu7k%3D&md5=4a6b3d7fe88f15f3566784cab63c41f7CAS |
(b) C. Venkatesh, H. U. Reissig, Synthesis-Stuttgart 2008, 3605.
[8] E. P. M. T. Cohn, K.-L. Wu, T. R. R. Pettus, N. O. Reich, J. Med. Chem. 2012, 55, 3678.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjs1OitLc%3D&md5=82e34ed9e4c6379c9f4767426821d537CAS |
[9] (a) D. C. K. Rathwell, S. H. Yang, K. Y. Tsang, M. A. Brimble, Angew. Chem. Int. Ed. 2009, 48, 7996.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Kju7zM&md5=7693ce3831230d3e0713b08927704108CAS |
(b) J. Sperry, Y. C. Liu, Z. E. Wilson, J. G. Hubert, M. A. Brimble, Synthesis-Stuttgart 2011, 1383.
(c) K. Y. Tsang, M. A. Brimble, Tetrahedron 2007, 63, 6015.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) M. A. Brimble, C. L. Flowers, M. Trzoss, K. Y. Tsang, Tetrahedron 2006, 62, 5883.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvFyktLo%3D&md5=fc8bd5d79be92b772ec0e7515594c6a8CAS |
(b) P. J. Choi, D. C. K. Rathwell, M. A. Brimble, Tetrahedron Lett. 2009, 50, 3245.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. A. Brimble, Y. C. Liu, M. Trzoss, Synthesis-Stuttgart 2007, 1392.
| Crossref | GoogleScholarGoogle Scholar |
[11] (a) C. M. Buseman, W. E. Wright, J. W. Shay, Mutat. Res. – Fund. Mol. M. 2012, 730, 90.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlsFWksQ%3D%3D&md5=63fbc16b6af46bc291aadf38c56557bdCAS |
(b) A. Agrawal, S. Dang, R. Gabrani, Recent Pat. Anti-Canc. 2012, 7, 102.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Folini, L. Venturini, G. Cimino-Reale, N. Zaffaroni, Expert Opin. Ther. Targets 2011, 15, 579.
(d) J. L.-Y. Chen, J. Sperry, N. Y. Ip, M. A. Brimble, MedChemComm 2011, 2, 229.
| Crossref | GoogleScholarGoogle Scholar |
(e) C. Philippi, B. Loretz, U. F. Schaefer, C. M. Lehr, J. Control. Release 2010, 146, 228.
| Crossref | GoogleScholarGoogle Scholar |
[12] (a) M. Jaskelioff, F. L. Muller, J.-H. Paik, E. Thomas, S. Jiang, A. C. Adams, E. Sahin, M. Kost-Alimova, A. Protopopov, J. Cadinanos, J. W. Horner, E. Maratos-Flier, R. A. DePinho, Nature 2011, 469, 102.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVOrsLnJ&md5=91cb6a636904286184a830d49ae5747bCAS |
(b) A. A. Sprouse, C. E. Steding, B. S. Herbert, J. Cell. Mol. Med. 2012, 16, 1.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. B. de Jesus, K. Schneeberger, E. Vera, A. Tejera, C. B. Harley, M. A. Blasco, Aging Cell 2011, 10, 604.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. Zhu, S. Lee, M. K. C. Ho, Y. Hu, H. Pang, F. C. F. Ip, A. C. Chin, C. B. Harley, N. Y. Ip, Y. H. Wong, Drug Metab. Pharmacokinet. 2010, 25, 477.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. G. Bodnar, M. Ouellette, M. Frolkis, S. E. Holt, C. P. Chiu, G. B. Morin, C. B. Harley, J. W. Shay, S. Lichtsteiner, W. E. Wright, Science 1998, 279, 349.
| Crossref | GoogleScholarGoogle Scholar |
(f) A. Tomas-Loba, I. Flores, P. J. Fernandez-Marcos, M. L. Cayuela, A. Maraver, A. Tejera, C. Borras, A. Matheu, P. Klatt, J. M. Flores, J. Vina, M. Serrano, M. A. Blasco, Cell 2008, 135, 609.
| Crossref | GoogleScholarGoogle Scholar |