

Can Molecular Sieves be Used as Water Scavengers in Microwave Chemistry?
Mostafa Baghbanzadeh A and C. Oliver Kappe A BA Christian Doppler Laboratory for Microwave Chemistry (CDLMC) and Institute of Chemistry, Karl-Franzens-University Graz, Heinrichstrasse 28, A-8010 Graz, Austria.
B Corresponding author. Email: oliver.kappe@uni-graz.at
Australian Journal of Chemistry 62(3) 244-249 https://doi.org/10.1071/CH08450
Submitted: 21 October 2008 Accepted: 18 December 2008 Published: 20 March 2009
Abstract
The use of sealed-vessel microwave-assisted organic synthesis in combination with 4 Å molecular sieves as water scavengers is investigated. Two classical model transformations, namely acetal formation and imine formation, are evaluated under both conventional heating and microwave dielectric heating. The results obtained in these studies indicate that molecular sieves cannot be used effectively as water scavengers in microwave-assisted transformations owing to the fact that these zeolites are generally most effective in adsorbing water at room temperature. For both acetal and imine formation, performing these reversible reactions at a higher temperature in the presence of molecular sieves leads to significantly reduced conversions compared with experiments at room temperature.
Introduction
High-speed synthesis using microwave heating technology has attracted a considerable amount of attention in recent years. More than 3500 articles have been published in the area of microwave-assisted organic synthesis (MAOS) since the first reports on the use of microwave heating to accelerate organic chemical transformations in 1986.[1,2] The efficiency of ‘microwave flash heating’ in dramatically reducing reaction times and increasing product yields/purities is one of the key advantages of this enabling technology. Most of the published applications of controlled MAOS today involve the use of sealed vessel technology in combination with single-mode microwave reactors. Here, the advantages of rapid and direct volumetric microwave heating are combined with the capability to superheat solvents far above their boiling points in a sealed vessel (autoclave).[1,2] This method allows significantly higher reaction temperatures to be reached than using conventional reflux conditions, and therefore often results in considerable rate enhancements when compared with experiments carried out at the boiling point of the solvent.[1,2]
Apart from these important advantages, there are a few key disadvantages of applying pressurized sealed-vessel microwave technology for organic synthesis: first of all, in a closed reactor, the operator is not able to visually follow the progress of the reaction as in a conventional reflux experiment. Recent advances in single-mode cavity design allow the interfacing of digital cameras into the system and therefore a visual inspection of the reaction mixture, eliminating some of the above-mentioned monitoring issues.[3] Similarly, several publications have shown that by using in situ Raman spectroscopy, the progress of a variety of chemical transformations performed under microwave conditions can be directly monitored online.[4] An additional handicap of the sealed vessel technology is that the portion or dropwise addition of a reagent, or the withdrawal of samples during the reaction for reaction monitoring can be troublesome using standard instrumentation.[5] Notably, there are some instances where the use of sealed vessel microwave technology will fail and an open vessel microwave system must be used.[6] Typically, this is the case when one of the (volatile) products or by-products needs to be eliminated from the reaction mixture, for example, in the case of an equilibrium process.[6] In some important types of synthetic transformation (e.g. acetal or imine formation from carbonyl compounds), water is produced as a by-product that must be removed to shift the equilibrium to the product side. In conventional organic synthesis, there are two main physical methods to achieve this aim: azeotropic distillation with an inert solvent (Dean–Stark apparatus), and the use of dehydrating agents, which are generally applied in small-scale experiments. Although it has been recently reported that the use of a Dean–Stark apparatus can be readily combined with microwave heating under open-vessel conditions,[7] to our knowledge no detailed and systematic study on the use of dehydrating agents as water scavengers in conjunction with microwave heating has been published.[8] The latter technique would be particularly valuable because it would allow the successful use of standard sealed-vessel microwave technology on small scale.[1,2]
Among the known dehydrating agents, molecular sieves have a special place because of the high adsorption capacity for water, even at very low temperatures caused by the highly polar surface within the pores.[9] However, the use of these materials in conjunction with microwave heating is further complicated because molecular sieves are known to be very strong microwave absorbers.[10] In fact, one of the best ways to dry/activate molecular sieves is to irradiate them with microwaves.[11] Herein, we present a detailed study on the use of molecular sieves as water scavengers in MAOS involving two classical model transformations.
Results and Discussion
In order to study the effect of microwave irradiation on organic transformations requiring molecular sieves as water scavengers, two classical model systems were selected, namely acetal formation (Scheme 1) and imine formation (Scheme 2). In both cases, the presence of water scavengers is generally required in order to drive the reaction to a high degree of conversion.[12] We were particularly interested to investigate these transformations both in the presence and absence of sieves and to compare these data with results obtained using microwave heating. For both reaction systems, datasets obtained from runs at different temperatures were compared using gas chromatography-flame ionisation detection (GC-FID) monitoring of the reaction progress.
|
|
Acetal Formation
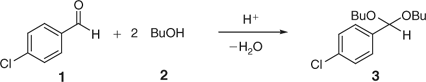
Acetal formation from an aldehyde and an alcohol component proceeds through the nucleophilic attack of alcohol on the carbonyl group. The main problem in acetal formation in acidic medium is to shift the equilibrium to the right by reducing the concentration of the formed water in the reaction system.[13] After some experimentation, we arrived at a suitable model system for a reliable kinetic analysis that involved the acetal formation from 4-chlorobenzaldehyde (1) with n-butanol (2) in a suitable non-polar solvent (Scheme 1). The use of alcohols with lower molecular weight such as ethylene glycol or i-propanol in conjunction with standard 4 Å molecular sieves was not optimal, as apparently these comparatively small alcohols can compete with the formed water for the pores in the 4 Å molecular sieves.
Our initial experiments involved 0.5 mmol of 4-chlorobenzaldehyde (1), 1.2 mmol n-butanol (2), 10 mol-% of p-toluenesulfonic acid (PTSA, monohydrate) and 150 mg of freshly activated 4 Å molecular sieves (see Experimental) in 3 mL of toluene. Although the experiment with molecular sieves at room temperature displayed a ~90% conversion after a few hours, the same experiment performed without sieves reached a maximum at ~30% conversion, clearly demonstrating the positive effect of the molecular sieves in removing water from the equilibrium. A solvent screen further established that toluene was indeed one of the best solvents to perform the acetal formation, with results using other solvents such as chloroform, 1,2-dichloroethane, 1,2-dichlorobenzene, 1,2-dimethoxyethane or THF showing inferior conversions under otherwise identical conditions. It has to be emphasized that none of the solvents used in these studies were pre-dried. The amount of water initially contained in the solvent therefore will have an influence on the equilibrium yield of product. Along similar lines, it was quickly established that 10 mol-% of PTSA proved to be optimal for obtaining high conversions.
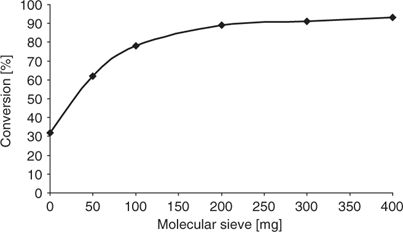
Of key importance in all transformations requiring dehydrating agents is the amount of molecular sieves added to the reaction mixture. As shown in Fig. 1, we established that a quantity of 200 mg of activated 4 Å molecular sieves provided a high degree of conversion (~90%). Increasing the amount further only slightly increased conversion to acetal 3. We therefore used an amount of 200 mg of sieves per 0.5 mmol of aldehyde building block for all further experiments.
|
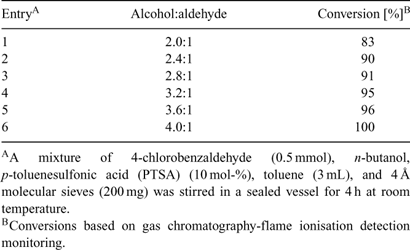
Along similar lines, the stoichiometric ratio between aldehyde and alcohol was investigated (Table 1). Although using 4 equiv of alcohol, full conversion was obtained after 4 h at room temperature, we chose to work with 2.4 equiv in all subsequent experiments in order to be able to more accurately measure any differences between microwave heating and standard processing. For preparative work, however, the use of 4 equiv of n-butanol is clearly advisable.
|
After the extensive reaction optimization described above, we finally started to investigate the influence of reaction temperature on the acetal formation shown in Scheme 1. In traditional microwave synthesis, reactions can be performed at a significantly faster rate because of the comparatively high processing temperatures employed.[1,2] The influence of reaction temperature on any process requiring molecular sieves is therefore of critical importance. We were surprised to find that for the acetal formation in Scheme 1, higher reaction temperatures using conventional heating resulted in lower conversions (Fig. 2). Although reactions run at room temperature under our carefully optimized conditions led to 90% conversion after 4 h, a significantly lower amount of acetal product 3 was formed at 50°C, 70°C, and 90°C. By monitoring the conversion over time (Fig. 2), we established that the reaction equilibrium was reached after 1–2 h, with very little changes in reaction composition occurring after that time. Only for the room temperature run, where the equilibrium is apparently reached more slowly, an increased formation of acetal 3 was observed after 4 h.
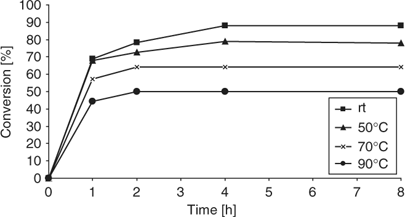
|
We ascribe the lower conversions reached at higher temperatures to the fact that water is more effectively adsorbed to molecular sieves at lower temperatures.[9] At higher temperatures, most of the water will be outside the sieves and therefore will be able to hydrolyze the acetal back to the aldehyde and alcohol components. In order to demonstrate the reversible nature of this process, we performed an experiment where the acetalization was initially performed at room temperature for 4 h, giving the expected ~90% conversion. The reaction mixture was then heated to 90°C for an additional 4 h, which led to a reduced conversion back to 50% in agreement with the data shown in Fig. 2. This experiment clearly shows the equilibrium-type behaviour of the acetalization and again demonstrated the effectiveness of molecular sieves to remove water at ambient conditions. At higher temperatures, the water-scavenging ability of molecular sieves is apparently reduced.
The results presented above are in good agreement with studies on the hydrolytic stability of acetal 3 at higher temperatures. As shown in Table 2, acetal 3 is stable towards hydrolysis in the absence of an acidic catalyst (PTSA), even in the presence of an excess of water (entries 1 and 2). By introducing 10 mol-% of the catalyst in the monohydrate form, significant hydrolysis takes place (entry 3), which cannot be fully suppressed by the molecular sieves at 90°C (entry 4). By adding an excess amount of water (~3 equiv, 27 mg, one drop), the capacity of the sieves is exceeded and almost full hydrolysis to the aldehyde is observed (entry 5).

|
Having all these data in hand, we finally investigated the influence of microwave irradiation on acetal formation in the presence of molecular sieves. In a first series of experiments, we established that molecular sieves are indeed strong microwave absorbers. In the experiments shown in Fig. 3, weighed quantities of molecular sieves were suspended in a microwave-transparent solvent (carbon tetrachloride) contained in a transparent quartz reaction vessel and exposed to microwave irradiation at constant power for 2 min.[14] As shown in Fig. 3, even small quantities of molecular sieves are able to raise the bulk temperature of the mixture significantly. It should be emphasized that in the absence of molecular sieves, no heating occurs and that it can be assumed that the temperature of the molecular sieves is significantly higher than the monitored bulk temperature of the solvent under these specific conditions.[15] This would in principle open up the possibility to use molecular sieves as passive heating elements for otherwise low-absorbing reaction mixtures.[16]
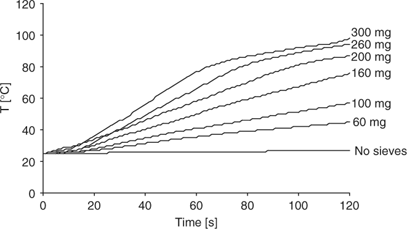
|
Based on the data obtained using conventional heating for the acetalization reaction (Fig. 2), we ultimately performed an identical experiment using controlled microwave irradiation at 70°C. A comparison of the kinetic data obtained for microwave and conventional heating revealed that, within experimental error, the same results were obtained: at higher temperature, the conversion in the acetalization is lower and the use of molecular sieves becomes ineffective, regardless if microwave or conventional heating is employed (Fig. 4). Owing to the fact that molecular sieves are strong microwave absorbers and will heat rapidly under microwave conditions (Fig. 3), the use of these materials as water scavengers in microwave-assisted reactions is probably not practical. However, it may be possible to affix the molecular sieves in the upper region of a modified 10 mL Pyrex vial, which typically remains cold during the microwave heating process, being outside the actual microwave cavity and not in contact with the microwave-heated solvent. If the reaction is performed above the boiling point of the solvent, the formed water may be (azeotropically) distilled to the upper parts of the vial where the sieves are located. Under these conditions, the use of sieves may be possible (J. T. Shaw, pers. comm.). In our hands, an acetalization experiment at 130°C in toluene (in the absence of sieves) did not provide a satisfactory result, leading to 30% conversion after 1 h. Although the formation of small water droplets at the top part of the vial could be observed, in the absence of an effective water scavenging mechanism, this method apparently is ineffective.
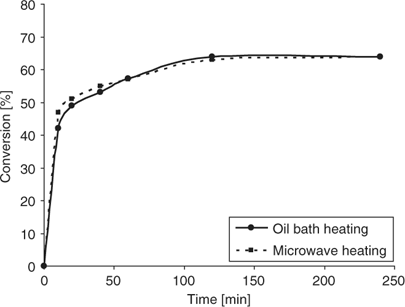
|
Imine Formation
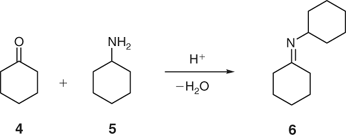
To verify the results obtained for acetalization (Scheme 1), we additionally studied the acid-catalyzed formation of imines from ketones and amines. Primary amines can add to aldehydes and ketones to produce imines, which are often sensitive to hydrolysis, in particular if derived from aliphatic aldehydes.[17] In general, ketones react more slowly than aldehydes, and higher temperatures and longer reaction times are required. In addition, the equilibrium must often be shifted, usually by removal of water, either azeotropically by distillation, or by use of a water scavenger such as TiCl4,[18] or molecular sieves.[19] For our model studies, we selected the reaction between cyclohexanone and cyclohexylamine, again employing PTSA as a catalyst (Scheme 2). Initial investigations in toluene at room temperature confirmed that the use of 4 Å molecular sieves for this reaction was required in order to obtain an acceptable degree of conversion.
Similarly to the investigations described above for acetalization, the reaction was subsequently optimized at room temperature using different solvents (Table 3), molar ratios of building blocks (Table 4), and amounts of sieves. After considerable experimentation, we arrived at conditions that employed 1,2-dichloroethane (DCE) as a solvent, 1.2 equiv of amine, 10 mol-% of PTSA catalyst and 200 mg of 4 Å molecular sieves.
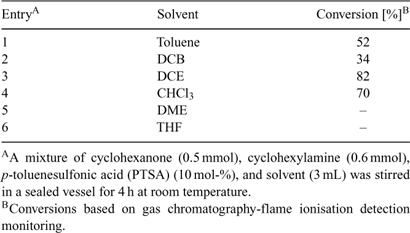
|

|
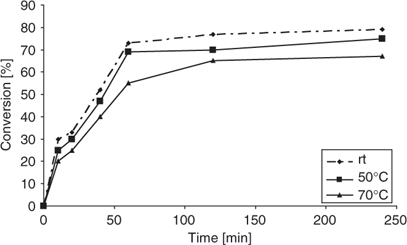
Similarly to acetal formation, the reaction of cyclohexylamine with cyclohexanone (Scheme 2) was then performed at different temperatures using oil-bath and microwave heating. In agreement with the results described above for acetal formation, we found that also in the case of imine formation, the molecular sieves worked best at room temperature, and were not really effective at higher temperatures, although the effects were not as pronounced as with acetal formation (Fig. 5). At room temperature, an 86% conversion to imine was obtained after 8 h, whereas at 90°C, the conversion was significantly lower (55%). For imine formation at higher temperatures, side products were seen after prolonged reaction times by GC-MS monitoring, which further contributed to the low product yields. As expected, the mode of heating had no significant influence on imine product formation and kinetic studies at 70°C revealed no significant difference between microwave heating and conventional heating (data not shown).
|
Conclusions
In the present publication, we have evaluated the performance of molecular sieves as water scavengers in microwave-assisted transformations. After careful kinetic analysis of two model reactions (acetal and imine formation), it appears that molecular sieves cannot be used efficiently as water scavengers under standard sealed-vessel microwave conditions. The reasons for this are not so much connected to the strong microwave absorbance of these zeolite-based materials but rather to the fact that the effectiveness of molecular sieves as water scavengers is generally highest at ambient temperatures. As shown for both acetal and imine formation, performing these reversible reactions at a higher temperature (microwave or conventional heating) in the presence of molecular sieves leads to significantly reduced conversions compared with experiments at room temperature.
Experimental
General Instrumentation
Microwave experiments (Figs 3 and 4) were recorded using a Discover Labmate single-mode microwave reactor (CEM Corp.) employing custom-made high-purity quartz vessels (capacity 10 mL).[14] For comparison experiments between oil-bath and microwave heating (Fig. 4), the temperature profiles for both sets of experiments were monitored with a fibre-optic probe inserted into the vessel protected by a sapphire immersion well.[14]
1H and 13C NMR spectra were recorded on a Bruker AMX 360 in CDCl3. Low-resolution mass spectra were obtained on an Agilent 1100 LC/MS instrument using atmospheric pressure chemical ionization (APCI) in positive or negative mode. GC-FID analysis was performed on a Trace-GC (ThermoFisher) with a flame ionization detector using an HP5 column (30 m × 0.250 mm × 0.025 μm). After 1 min at 50°C, the temperature was increased in 25°C min–1 steps up to 300°C and kept at 300°C for 4 min. The detector gas for the flame ionization was H2 and compressed air (5.0 quality).
4 Å Molecular sieves were purchased from Aldrich Chem. Co. (Cat. No. 20860–4). All solvents and chemicals were obtained from standard commercial vendors and were used without any further purification.
General Procedure for Activating 4 Å Molecular Sieves under Microwave Irradiation
A sample of 30 g of 4 Å molecular sieves (fresh bottle) was placed in a 100 mL round-bottom flask and irradiated in a domestic microwave oven with full power (800 W) for ~80 s, which caused the temperature to rise to ~250°C (temperature measurement immediately after irradiation by a standard thermometer). Immediately after switching off the microwave power, the sieves were put under vacuum (900 Pa, membrane pump) for 15 min. This cycle was repeated five times.
Kinetic Study of Acetal Formation (Scheme 1)
A 5 mL Pyrex vessel equipped with a stir bar was charged with 4-chlorobenzaldehyde (1) (70 mg, 0.5 mmol), n-butanol (2) (110 μL, 90 mg, 1.2 mmol), PTSA monohydrate (9 mg, 0.05 mmol), 3 mL of solvent, and the appropriate amount of 4 Å molecular sieves. The vessel was sealed with a septum and the reaction mixture was either exposed to microwave irradiation or immersed in a preheated oil bath at different temperatures and times as shown in Tables 1 and 2, and Figs 1–3. After being cooled to ambient temperature, the mixture was immediately quenched with triethylamine (1 mL), washed with 2 M KOH solution (5 mL), and dried over potassium sulfate before injection into a GC-FID.
Kinetic Study of Imine Formation (Scheme 2)
A 5 mL Pyrex vessel equipped with a stir bar was charged with cyclohexylamine (5) (69 μL, 59 mg, 0.6 mmol), cyclohexanone (4) (50 μL, 47 mg, 1.2 mmol), 3 mL of solvent, PTSA monohydrate (9 mg, 0.05 mmol), and the appropriate amount of 4 Å molecular sieves. The vessel was sealed with a septum and the reaction mixture was either exposed to microwave irradiation or immersed in a preheated oil bath at different temperatures and times shown in Tables 3, 4 and in Fig. 4. After being cooled to ambient temperature, the mixture was immediately quenched with triethylamine (1 mL), washed with 2 M KOH solution (5 mL), and dried over potassium sulfate before injection into a GC-FID.
1-Chloro-4-(dibutoxymethyl)benzene 3
A mixture of 4-chlorobenzaldehyde (1) (1.4 g, 10 mmol), n-butanol (2) (3.8 mL, 2.96 g, 40 mmol), PTSA monohydrate (190 mg, 1 mmol), 30 mL of toluene, and 4 g of 4 Å molecular sieves was stirred in a sealed 100 mL round-bottom flask at room temperature for 4 h. After completion of the reaction (GC-FID monitoring), the mixture was quenched with triethylamine (5 mL), washed with 2 M KOH solution (20 mL), and dried over potassium sulfate. The solvent was evaporated under reduced pressure and the pure acetal product was isolated as a colourless oil[20] in quantitative yield and >98% purity (GC-FID). δH 0.94 (6H, t, J 7.3, 2CH3), 1.42 (4H, tq, J 7.3, 7.1, 2CH2CH3), 1.61 (4H, tt, J 7.3, 7.1, 2CH2CH2), 3.51 (4H, m, 2OCH2), 5.50 (1H, s, CH), 7.34 (2H, d, J 8.4, 2ArH), 7.43 (2H, d, J 8.4, 2ArH). δC 13.7, 19.4, 31.8, 65.0, 100.8, 128.1, 128.2, 133.9, 137.7. m/z (APCI) 197 (100, [M – C4H9O•]+), 140 (65, [M – C7H16O•]+).
Synthesis of N-Cyclohexylidenecyclohexanamine 6
A mixture of cyclohexylamine (5) (2.3 mL, 1.98 g, 20 mmol), cyclohexanone (4) (1.0 mL, 0.94 g, 10 mmol), PTSA monohydrate (190 mg, 1 mmol), 30 mL of DCE, and 4 g of 4 Å molecular sieves was stirred in a sealed 100 mL round-bottom flask at room temperature for 4 h. After completion of the reaction (GC-FID monitoring), the mixture was quenched with triethylamine (5 mL), washed with 2 M KOH solution (20 mL), and dried over potassium sulfate. The solvent was evaporated under reduced pressure and the pure imine product was isolated as a colourless oil[21] in 95% yield and >98% purity (GC-FID). δH 1.20–1.38 (6H, m), 1.53–1.93 (10H, m), 2.18–2.30 (4H, m), 3.20–3.28 (1H, m). δC 24.7, 25.6, 27.5, 29.0, 34.1, 40.2, 57.9, 170.8 (C=N). m/z (APCI) 180 (100, [M + 1•]+).
Acknowledgement
M.B. thanks Shahid Beheshti University and the Ministry of Science, Research and Technology of Iran for a scholarship. We gratefully acknowledge Professor Minoo Dabiri for facilitating M.B.’s leave.
[1]
[2]
(b) A. De La Hoz,
A. Díaz-Ortiz,
A. Moreno,
Chem. Soc. Rev. 2005, 34, 164.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
|
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
| Crossref | GoogleScholarGoogle Scholar |
CAS |
