

Synthesis, X-ray Crystal Studies and Metal Picrates Extraction Properties of Lipophilic Benzocrown Ethers
Ewa Kowalska A , Jaywant Phopase A and Nicholas Gathergood A BA Department of Chemical Sciences and National Institute of Cellular Biotechnology, Dublin City University, Glasnevin 9, Ireland.
B Corresponding author. Email: nick.gathergood@dcu.ie
Australian Journal of Chemistry 63(9) 1348-1357 https://doi.org/10.1071/CH10112
Submitted: 6 March 2010 Accepted: 27 May 2010 Published: 9 September 2010
Abstract
The preparation of 16 macrocycles based on 2-(2-hydroxyethoxy) phenol with both aliphatic and aromatic linkers has been achieved. Macrocycles varying in ring sizes (23–28 atoms), number of aromatic groups (2–4), and donor atoms (6–10 including oxygen and nitrogen) were synthesized. Binding affinities were also assessed by extraction studies against a series of metal picrates. X-ray crystal structures were solved for four macrocycles.
Introduction
Macrocyclic ligand complexes play a significant role in many fundamental biological systems, such as photosynthesis and the transport of oxygen in mammalian and other respiratory systems.[1]
Nature has created many elegant chiral hosts, including cyclic peptides (e.g. valinomycin), and nonactin, containing ester and ether binding sites.[2] Compounds that can bind and transport ions are referred to as ionophores. The antibacterial properties of ionophores arise from their perturbation of ion gradients across the cell.[3] The presence of heteroatoms that act as ligands is key for the complexation of inorganic cations. Complexation of alkali metal cations and their non-covalent interactions with host molecules have engaged considerable interest owing to their importance in chemical[4] and biological systems.[5] In recent years, large metal complexes (e.g. La,[6] Cu,[7] Co,[8] Zn[9]) with chiral ligands have also been recognized as efficient catalysts in asymmetric synthesis.
We are interested in designing and synthesizing new hosts (Fig. 1) with varied conformational, electronic and chemical properties for ionophoric antibiotics, metal catalysis and sensor applications. Herein, we describe our progress towards a series of benzocrown ethers, with a lipophilic exterior and hydrophilic central cavities of different sizes possessing oxygen and nitrogen atoms that can serve as ligands for metal cations and small organic molecules. A modular, short and convergent synthesis was a priority, with the scope to prepare a diverse library. The compounds reported herein are the basis for the development of a stereoselective synthesis of macrocycles incorporating the morphine alkaloid scaffold (highlighed in blue, Fig. 1). Metal binding affinity, metal templation effects and X-ray crystallographic data were obtained to assist the development of future medicinal chemistry, macrocycle synthesis (e.g. templation effects), asymmetric catalysis and analytical chemistry applications.
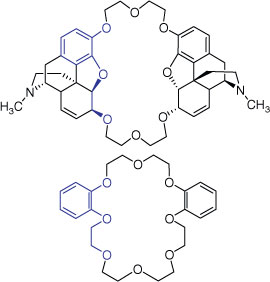
|
Results and Discussion
Synthesis of phenoxy-linked open structures 2–6 and 8 have been reported previously.[10–13] Macrocyclic precursors 2–8 were synthesized following the known literature procedure.[12] Commercially available 2-(2-hydroxyethoxy)phenol 1 was reacted with the corresponding dihalides in DMF at 120°C in the presence of 3 equiv. of K2CO3 as base to give the desired product (Scheme 1). These reactions occurred in moderate to high yields varying from 54% for 2 to 92% yield for 8. Diol macrocycle precursor 7 containing an ortho-substituted aromatic linker was synthesized in 76% yield.
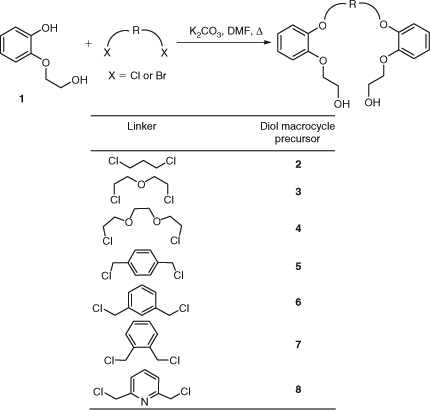
|
Alkyl dibromides were also used for the alkylation of diol 1. In all cases, yields were lower compared with the dichlorides, except for diol 2, which is prepared from an aliphatic dihalo linker without ethereal backbone (see Table 1). Diol 4 was synthesized using 1,2-bis-(2-iodoethoxy)ethane; however, a significant drop in yield (54% yield, from 81%) for 1,2-bis-(2-chloroethoxy)ethane was observed.
|
The preparation of benzocrown ethers was based on the approach that the macrocyclization process can be promoted either by suppressing the intermolecular reaction (resulting in polymerization) or facilitating the intramolecular cyclization. We chose to combine these approaches by suppressing competing reactions, via the use of moderate to high dilution conditions, while promoting cyclization via the template effect. First, synthesis of macrocycles 9 and 10 containing both ethereal and ester groups was performed by an acylation reaction on the corresponding linear precursors 5 and 6 respectively. In both cases, the reactions were carried out on a 1 mmol scale in refluxing dichloromethane in the presence of 8 equiv. of K2CO3 (Scheme 2). Macrocycles 9 and 10 were isolated by column chromatography in 44 and 48% yield respectively.
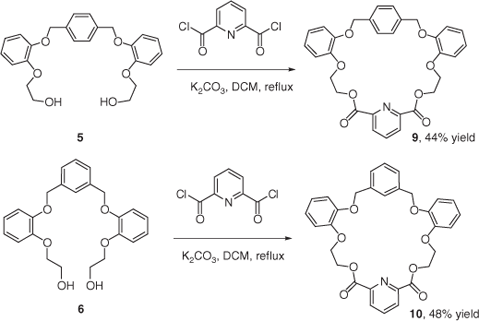
|
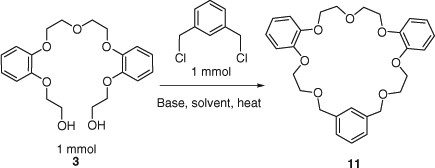
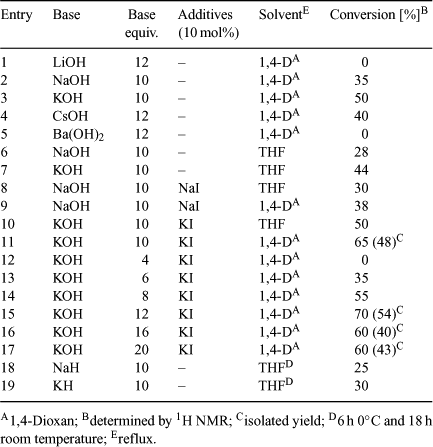
Before the synthesis of the target macrocycles 11–26, optimization of the macrocyclization reaction conditions was performed. Preparation of macrocycle 11 from diol 3 was chosen as a model reaction for the optimization of reaction conditions (Scheme 3). The ring-closure reaction was carried out on diol 3 to determine the most favourable reaction conditions, considering various factors such as equivalents of base, additives (NaI, KI), choice of solvent and concentration. Bases (e.g. NaH, NaOH, KH, KOH, CsOH and Ba(OH)2) in different solvents (e.g. THF, 1,4-dioxan) were screened (Table 2).
|
|
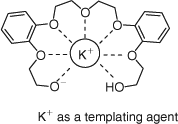
Among the metal hydroxides, potassium hydroxide proved to be more effective in promoting alkylation of the primary alcohol, with subsequent macrocyclization (entries 1–5). Experiments employing hydroxides with a small cationic radius, e.g. LiOH, and relatively large cationic radius, such as Ba(OH)2, gave poor results, with the formation of the product not observed. This is most likely due to the fact that the cations with a smaller radius than potassium cannot fit effectively to template the macrocycle for efficient ring closure.[14] Fig. 2 depicts the behavior of the macrocyclic precursor 3, in the presence of a potassium cation templating agent. This shape is the consequence of interactions between the metal cation and oxygen donor atoms. In the absence of a templating agent, 3 adopts a linear conformation and in that shape, oligomeric products are more likely to be formed.
|
Synthesis yield of the desired product was higher in the experiments carried out in 1,4-dioxan (entries 2, 3 versus 6, 7). The presence of the iodide salt improved reaction yields further by nucleophilic displacement of Cl with KI (Finkelstein reaction) via an alkyl iodide intermediate.[15] Presence of 10 mol% of KI in the ring-closure reaction to synthesize 11 improved conversion by 15% (entries 3 versus 11). From the series of experiments with varied amount of bases, it was observed that the optimal number of equivalents of base required to effect ring closure was 12 (entries 11–17), with the preferred base being potassium hydroxide. No macrocycle was formed in experiments carried out using less than 6 equiv. of base. These experiments demonstrated that in general, if THF is replaced by 1,4-dioxan, conversion of starting materials tends to increase (entries 10, 11). Synthesis of 11 occurs most efficiently using at least 10 equiv. of KOH and 0.1 equiv. of KI in refluxing 1,4-dioxan (entries 11, 15). The optimal molar ratio of base to substrate was 12 for the highest conversion, in the presence of KI. In addition, the effect of sodium and potassium hydrides on the macrocyclization was investigated. Reactions were stirred at 0°C in THF for 6 h, then at room temperature overnight; however, only low conversions were recorded (entries 18, 19).
A study to establish the optimum concentration of the diol macrocycle precursor for the macrocyclization reaction was also investigated, to determine whether it is the template effect of K+ or high dilution that benefits the reaction most. Experiments were carried out on a 1 mmol scale of 3 in 1,4-dioxan together with 12 equiv. of potassium hydroxide and 0.2 equiv. of potassium iodide.
For the concentration range 4–25 mM, conversions between 60 and 75% were observed, with a maximum at 8 mM. At 50 mM, the conversion decreased to 40% and only trace product formed at 125 mM. This suggests the importance of the template effect, which appears to play a key role in the macrocyclization step. Previous experiments (Table 2) also show that the use of bases with lithium or sodium counter cations proceeded with lower reaction conversions compared with reactions carried out in the presence of potassium bases.
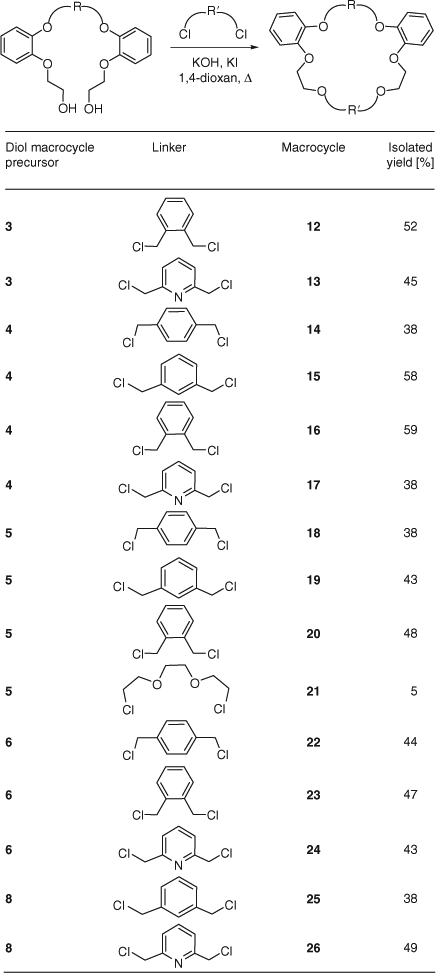
Preparation of macrocycles 12–26 from a variety of dialkyl chlorides using the optimized conditions (Table 2, entry 15) was carried out on a 1 mmol scale, employing 12 mmol of metal hydroxide under dilute conditions (8 mM) in 1,4-dioxan at reflux temperature (Scheme 4).
|
This synthetic approach gave good yields (38–59%), allowing the preparation of macrocycles with varied ring sizes (23–28 atoms), number of aromatic rings (2–4), and number of donor atoms (6–10 including oxygen and nitrogen).
In all the cases, the ring-closure reaction gave higher yields of the respective macrocycles with the shortest spacer meta-xylene (12, 16, 20, 23) compared with longer spacers (20 versus 21). The symmetry exhibited by these benzocrown macrocycles can be seen from both 1H and 13C NMR spectroscopy. Whereas structures 2–6 and 8 proved to be excellent precursors to undergo ring-closure reaction, compound 7 did not give any macrocycle. Every attempt to introduce different linkers into the structure of 7 failed.
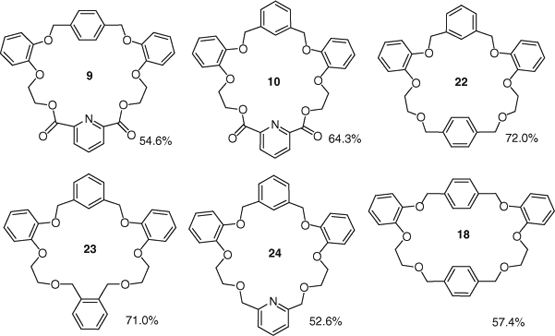
The complexation behaviour of macrocycles 9–14, 18–19 and 22–26 towards alkali, alkaline and transition metal cations was assessed by metal picrate extraction from aqueous solution into chloroform. The percentage extraction values for metal cations were calculated according to a procedure described by Pedersen.[16] The extraction data show significant binding of several macrocyclic structures towards sodium picrate (Table 3). Among the 13 macrocyclic systems, the highest Na+ extraction values were observed for 22 (72.0%), 23 (71.0%), 10 (64.3%), 18 (57.4%) and 9 (54.6%) (Fig. 3). The common feature linking these compounds is the presence of four aromatic rings in their structures arranged to form a rigid cavity surrounded by six donor oxygen atoms. According to the data in the table and from analysis of cavity sizes in the crystallographic literature, we would expect metal picrates with cation diameters larger than sodium to be bound more effectively.[16b] The most efficient cation extractions were observed with macrocycles containing four aromatic rings (e.g. 22, 23, 24) instead of three, in which the xylene linker is replaced by an ethereal chain (e.g. 12, 13, 14). This suggests that the presence of four aromatic rings in the macrocycle prevents binding to the larger cation. Macrocycle 11 exhibits relatively low extraction values for Na+, 21.1%, and even lower for K+, 6.8%, whereas for macrocycle 14, the cavity selectivity towards Na+ over K+ increased and extraction values were 41.7 and 4.0% respectively. Macrocycles containing a pyridine ring 9, 10 and 24 gave high extraction values for Na+ (64.3, 54.6 and 52.6 respectively) and proved to have significant binding affinity for La3+ (9.0, 10.1 and 7.2%) and to bind relatively weakly to K+. Macrocycle 22 showed an excellent 72% extraction value for Na+ and 4.8% for K+. The results show that the highest extraction values of Na+ are consistent with tetrabenzo 23- and 24-crown-6 polyether rings. Significant extractability of Na+ has also been observed for macrocycles containing four aromatic rings including the pyridine ring and ester groups. 10 and 9 extract Na+ with 64.3 and 54.6% extraction values respectively. Macrocycles 22 and 23 coordinate Na+ with almost identical extractability (71–72%), whereas extractability towards sodium picrate with 9, 18 and 24 was between 52.6 and 57.4%.

|
|
All macrocycles screened exhibit low to moderate extractability towards potassium and caesium picrates. Compound 23 gave the highest extraction levels (23.1%) for potassium, with an order of Na+ > K+ > Cs+ and was also the only macrocycle to exhibit binding over 3% towards caesium picrate (16%). Compared with 23 (lower part contains ortho-substituted aromatic linkers), the cavities of hosts 19 and 26 are relatively large (upper and lower part contains para or meta-substituted aromatic linkers), and their binding parameters correspond to the scaffold of 24-crown-8. Macrocycle 23 exhibits good extraction for potassium and caesium compared with compound 12, in which the upper part of the macrocycle contains an ethereal chain with extra oxygen instead of an aromatic ring. It appears that the presence of one or more extra aromatic rings may not only change or rearrange the conformation of the binding pocket but also provide an additional source of π-electrons. Therefore, it is likely that complex formation is stabilized by the cation–π interaction when the metal cation is surrounded by the four benzene rings.[9] Percentage extractabilities for all tested ligands towards alkaline earth metals Mg2+, Ca2+, Sr2+ and Ba2+ were low, <2.1%.
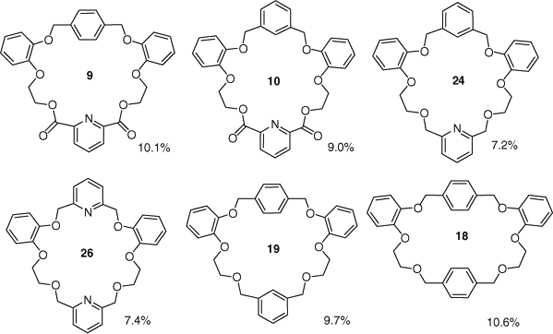
Among transition metal binding abilities investigated, 25 and 26 exhibited low extractability of up to 3.2% and up to 4.3% respectively, whereas higher extraction values (>5%) were observed in macrocycles 9, 10, 13, 18, 19, 24, 25 and 26 towards lanthanum picrate (Table 3 and Fig. 4). Compounds 9 and 18 displayed highest extraction values of 10.1 and 10.6%, whereas 10 and 19 showed >9% extractability towards La3+. The presence of aromatic rings (e.g. meta- or para-substituted benzene or pyridine) increased the extractability of La3+ compared with extraction values for macrocycles containing extra oxygen like 11 (1.3%) or 14 (4.7%). As expected, macrocycles with large cavity size showed higher binding affinity towards La3+ compared with the macrocycles with a small cavity size (9 versus 10, 18 versus 19). Ligands 12 and 13 are poor hosts for all the alkali and transition metal picrates tested. The presence of three rigid ortho-substituted aromatic rings in the macrocyclic structure of 12 might direct the two attached arms in a semiconvergent arrangement, which may influence the binding.
Apart from interactions between the cation and donor oxygen atoms, the complexation process may also be favoured by cation–π interactions with π-electrons from the aromatic rings. None of the tested ligands exhibited a high affinity for potassium and caesium, whose hydroxides played a significant role in the ring-closure step. The optimal metal for the preorganization of the acyclic precursor to facilitate ring closure does not necessary have optimal fit with the macrocyclic product. Careful selection of templates to assist synthesis versus binding metal studies is always an important compromise. The extraction studies do not take into account the stoichiometry of the complexes, and the ratio of complexing agent:metal picrate is assumed to be 1:1 for the purpose of the calculations. However, it is well known that the ratio can be more complex and formation of 1:2 or 2:3 complexes could also take place.[17,18]
Conclusions
We report a concise, two-step general methodology for the preparation of 16 new lipophilic benzocrown ether-based macrocycles with varying cavity sizes. Reaction conditions for the ring-closure step have been optimized in order to improve yields. Thirteen macrocycles have been tested for cation binding. Alkali metal picrate extraction studies from water into chloroform confirmed the binding properties of the different hosts with metal picrates. Although an ability to extract multiple metal picrates was more common, some of these products (22 > 23 > 10 > 18 > 9) exhibit significant binding towards sodium and lanthanum picrate (9 and 18). The preference exhibited by several macrocycles to bind with sodium suggests that the binding affinities of the novel crown ether analogues for cations are not only based on ionic size. Further investigations would be required in order to determine whether these macrocycles (9 and 18) with their higher affinity for La3+ might have applications as possible cocatalysts in asymmetric synthesis.
The work presented is a preface to the preparation of chiral macrocycles based on the morphine alkaloid scaffold and is ongoing. A study of the antimicrobial activity of the compounds reported herein will be published in due course.
Experimental
Methods and Materials
NMR spectra were obtained on a Bruker AC 400 NMR spectrometer operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. The 1H and 13C NMR shifts are expressed in ppm relative to tetramethylsilane and were internally referenced to the residual solvent signal. Coupling constants values (J) are in Hertz [Hz]. Chemical shift assignments for 1H and 13C spectra were assisted with correlation spectroscopy (COSY), distortionless enhancement of polarisation transfer (DEPT) and heteronuclear multiple quantum coherence (HMQC) experiments. The splitting patterns for NMR spectra are designated as follows: s (singlet), d (doublet), t (triplet), q (quartet), p (quintet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dt (doublet of triplet) and m (multiplet). Crystal data were collected using a Bruker SMART APEX CCD area detector diffractometer. A full sphere of reciprocal space was scanned by phi-omega scans. Pseudo-empirical absorption correction based on redundant reflections was performed by the program SADABS (X-1). The structures were solved by direct methods using SHELXS-97 (X-2) and refined by full matrix least-squares on F2 for all data using SHELXL-97 (X-2). Hydrogen atoms were added at calculated positions and refined using a riding model. Their isotropic temperature factors were fixed to 1.2 times (1.5 times for methyl groups) the equivalent isotropic displacement parameters of the carbon atom the H atom is attached to. Anisotropic thermal displacement parameters were used for all non-hydrogen atoms.
Melting point determinations were carried out using a Stuart SMP3 melting point apparatus. Infrared spectra were recorded on a Perkin Elmer Fourier-transform (FT)-IR system spectrum GX spectrometer and are reported in wavenumbers [cm–1]. High-resolution mass spectrometry (HRMS) was carried out on a liquid chromatography time-of-flight mass spectrometer. All chemicals were purchased from commercial suppliers, and used without further purification. THF was distilled from sodium with benzophenone as indicator under nitrogen. Dichloromethane was distilled from CaH2, DMF and 1,4-dioxan were purchased anhydrous from Sigma–Aldrich.
General Synthetic Procedure for Phenoxy-Linked Open Structures 2–8
A mixture of 2-(2-hydroxyethoxy)phenol 1 (10.0 mmol), alkyl dihalide (5.0 mmol) and potassium carbonate (15.0 mmol) in dry DMF (5.0 mL) was stirred for 7 h at 120°C. The reaction mixture was then cooled to room temperature, distilled water (30 mL) added and the precipitate filtered. The solid was washed with copious amounts of water. Crude product was recrystallized from chloroform/hexane or when necessary separated by flash chromatography.[15]
1,2-Bis[2-(2-hydroxyethoxy)phenoxy]-o-xylene 7
The general procedure was used with 2-(2-hydroxyethoxy)phenol 1 (10.0 mmol), and α,α′-dibromo-o-xylene (1.32 g, 5.0 mmol). Recrystallization from chloroform/hexane (5:1) gave the title compound as a white powder (1.56 g, 3.8 mmol, 76% yield), and 78% yield (1.60 g, 3.9 mmol) when α,α′-dichloro-o-xylene was used as a starting material. Mp 66–68°C. νmax (KBr)/cm–1 3267, 2901, 1590, 1505, 1395, 1323, 1256, 1126, 1040, 1000. δH (400 MHz, CDCl3) 7.37 (2H, dd, J 5.6 and 3.6, ArH), 7.23 (2H, dd, J 5.6 and 3.6, ArH), 6.88–6.77 (8H, m, ArH), 5.26 (4H, s, –OCH2Ar), 3.96 (4H, t, J 4.2, –OCH2CH2OH), 3.75 (4H, t, J 4.2, –OCH2CH2OH). δC (100 MHz, CDCl3) 148.27, 146.96, 134.32, 128.27, 127.33, 121.27, 120.34, 114.89, 112.83, 69.70, 69.22, 60.09. HRMS (EI, 70 eV) found [M + H]+ 411.1805; C24H27O6+ requires 411.1802.
|
General Synthetic Procedure for Benzocrown Ethers 9, 10
Diol macrocyclic precursor (1.0 mmol) was dissolved in dry dichloromethane (150 mL) under a nitrogen atmosphere, potassium carbonate (1.66 g, 12.0 mmol) was added, followed by the acyl dichloride (1.0 mmol) in one portion and the mixture was refluxed over night. The reaction mixture was then quenched with distilled water (70 mL), the organic phase separated, dried over MgSO4, filtered, and organic solvent removed via rotary evaporation. Purification of the product was carried out by column chromatography on silica gel.
14,20-Dioxo-3,10,13,21,24,31-hexaoxa-38-azapentacyclo[31.2.2.115,19.04,9.025,30]octatriaconta-1(35),4(9),5,7,15,17,19(38),25(30),26,28,33,36-dodecaen-8-yl 9
The general procedure was used with 5 (0.41 g, 1.0 mmol) and 2,6-pyridine dicarbonyl chloride (0.21 g, 1.0 mmol). Product 9 was isolated by column chromatography (silica; EtOAc elution) as a white solid (0.24 g, 0.44 mmol, 44% yield), mp 185–187°C. Rf 0.64 (EtOAc). νmax (KBr)/cm–1 2932, 1759, 1591, 1503, 1460, 1388, 1293, 1254, 1126, 1019. δH (400 MHz, [D6]DMSO) 8.18 (2H, d, J 7.6, ArH), 7.71 (1H, t, J 7.6, ArH), 7.16–6.92 (12H, m, ArH), 4.99 (4H, s, –OCH2Ar), 4.79 (4H, t, J 4.4, –OCH2CH2O–), 4.47 (4H, t, J 4.4, –OCH2CH2O–). δC (100MHz, [D6]DMSO) 163.78, 148.31, 147.90, 147.83, 139.21, 136.40, 128.19, 126.89, 121.60, 121.34, 115.20, 113.89, 69.70, 66.17, 63.58. HRMS (EI, 70 eV) found [M + H]+ 542.1810; C31H28NO8+ requires 542.1809.
14,20-Dioxo-3,10,13,21,24,31-hexaoxa-38-azapentacyclo[31.3.1.115,19.04,9.025,30]octatriaconta-1(36),4(9),5,7,15,17,19(38),25(30),26,28,33(37),34-dodecaen-8-yl 10
The general procedure was used with 6 (0.41 g, 1.0 mmol) and 2,6-pyridine dicarbonyl chloride (0.21 g, 1.0 mmol). Product 10 was isolated by column chromatography on silica gel using EtOAc as a white solid (0.26 g, 0.48 mmol, 48% yield), mp 164–166°C. Rf 0.66 (EtOAc). νmax (KBr)/cm–1 2942, 1743, 1588, 1506, 1450, 1378, 1291, 1256, 1125, 1081. δH (400 MHz, CDCl3) 7.92 (2H, d, J 7.6, ArH), 7.66 (1H, t, J 7.6, ArH), 7.44 (1H, s, ArH), 7.12–7.10 (3H, m, ArH), 6.94–6.84 (8H, m, ArH), 4.92 (4H, s, –OCH2Ar), 4.69 (4H, t, J 4.4, –OCH2CH2O–), 4.37 (4H, t, J 4.4, –OCH2CH2O–). δC (100 MHz, CDCl3) 163.17, 148.45, 148.42, 146.89, 136.71, 136.43, 128.08, 127.07, 126.67, 125.40, 121.48, 121.39, 116.32, 115.07, 71.21, 66.70, 63.39. HRMS (EI, 70 eV) found [M + H]+ 542.1791; C31H28NO8+ requires 542.1809.
General Synthetic Procedure for Benzocrown Ethers 11–26
The diol macrocyclic precursor (1.0 mmol) was dissolved in dry 1,4-dioxan (80 mL) under a nitrogen atmosphere, then potassium hydroxide (0.67 g, 12.0 mmol) was added and the mixture was heated at 60°C for 30 min. Alkyl dichloride (1.0 mmol) and potassium iodide (0.03 g, 0.2 mmol) were added in one portion and reaction mixture heated at reflux for 36 h. Solvent was removed by rotary evaporation, and distilled water (70 mL) was added to the residue. The aqueous phase was extracted with chloroform (6 × 30 mL). The organic phase was dried over MgSO4 and solvent removed via rotary evaporation. Purification of the product was carried out by column chromatography on silica gel.
3,6,13,16,19,26,29-Heptaoxatetracyclo[29.3.1.07,12.020,25]pentatriaconta-1(35),7(12),8,10,20(25),21,23,31,33‐nonaene 11
The general procedure was used with 3 (0.38 g, 1.0 mmol) and α,α′-dichloro-m-xylene (0.17 g, 1.0 mmol). Product 11 was isolated by column chromatography on silica gel using EtOAc/CHCl3 (1:1) as a white solid (0.19 g, 0.40 mmol, 40% yield), mp 90–92°C. νmax (KBr)/cm–1 2930, 1594, 1505, 1449, 1357, 1256, 1123, 1041. Rf 0.81 (EtOAc/CHCl3, 1:1). δH (400 MHz, CDCl3) 7.35 (1H, s, ArH), 7.22–7.18 (3H, m, ArH), 6.86–6.78 (8H, m, ArH), 4.61 (4H, s, –OCH2Ar), 4.11 (4H, t, J 4.6, –OCH2CH2O–), 4.08 (4H, t, J 4.8, –OCH2CH2O–), 3.82 (4H, t, J 4.6, –OCH2CH2O–), 3.80 (4H, t, J 4.8, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.40, 149.02, 138.45, 128.41, 127.43, 127.11, 121.89, 121.60, 115.87, 114.42, 73.46, 70.29, 69.72, 69.30, 68.78. HRMS (EI, 70 eV) found [M + H]+ 481.2215; C28H33O7+ requires 481.2221.
2,5,14,17,24,27,30-Heptaoxatetracyclo[29.4.0.07,12.018,23]pentatriaconta-1(31),7,9,11,18 (23),19,21,32,34-nonaen-8-yl 12
The general procedure was used with 3 (0.38 g, 1.0 mmol) and α,α′-dichloro-o-xylene (0.17 g, 1.0 mmol). Product 12 was isolated by column chromatography on silica gel using CHCl3 as a white solid (0.25 g, 0.50 mmol, 50% yield), mp 109–111°C. Rf 0.30 (CHCl3). νmax (KBr)/cm–1 2931, 1593, 1509, 1452, 1260, 1130, 1030. δH (400 MHz, CDCl3) 7.33 (2H, dd, J 5.6 and 3.2, ArH), 7.22 (2H, dd, J 5.6 and 3.2, ArH), 6.88–6.79 (8H, m, ArH), 4.71 (4H, s, CH2Ar), 4.13–4.08 (4H, m, –OCH2CH2O–), 3.95 (4H, t, J 4.4, –OCH2CH2O–), 3.77 (4H, t, J 4.6, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.40, 148.68, 136.71, 129.02, 127.78, 121.81, 121.32, 115.30, 113.59, 71.29, 70.21, 69.58, 68.90, 68.69. HRMS (EI, 70 eV) found [M + H]+ 481.2240; C28H33O7+ requires 481.2221. HRMS (EI, 70 eV) Na adduct found [M + Na]+ 503.2052; C28H32O7Na+ requires 503.2040.
3,6,13,16,19,26,29-Heptaoxa-35-azatetracyclo[29.3.1.07,12.020,25]pentatriaconta- 1(34),7(12),8,10,20(25),21,23,31(35),32-nonaen-8-yl 13
The general procedure was used with 3 (0.38 g, 1.0 mmol) and 2,6-bis(chloromethyl)pyridine (0.17 g, 1.0 mmol). Product 13 was isolated by column chromatography on silica gel using EtOAc/CHCl3 (3:1) as a white solid (0.21 g, 0.45 mmol, 45% yield), mp 150–152°C. Rf 0.25 (EtOAc/CHCl3, 3:1). νmax (KBr)/cm–1 2933, 1599, 1509, 1460, 1258, 1131, 1058. δH (400 MHz, CDCl3) 7.67 (1H, t, J 7.6, ArH), 7.35 (2H, d, J 7.6, ArH), 6.85–6.81 (8H, m, ArH), 4.82 (4H, s, CH2Ar), 4.14 (4H, t, J 4.4, –OCH2CH2O–), 4.03 (4H, t, J 4.8, –OCH2CH2O–), 3.93 (4H, t, J 4.4, –OCH2CH2O–), 3.83 (4H, t, J 4.8, –OCH2CH2O–). δC (100 MHz, CDCl3) 158.08, 149.17, 149.04, 137.41, 121.60, 121.37, 119.89, 114.87, 113.80, 74.58, 70.29, 69.63, 69.42, 69.31. HRMS (EI, 70 eV) found [M + H]+ 482.2169; C27H32NO7+ requires 482.2173.
3,6,13,16,19,22,29,32-Octaoxatetracyclo[32.2.2.07,12.023,28]octatriaconta-1(36),7,9,11,23(28),24,26,34,37-nonaen-10-yl 14
The general procedure was used with 4 (0.42 g, 1.0 mmol) and α,α′-dichloro-p-xylene (0.17 g, 1.0 mmol). Product 14 was isolated by column chromatography on silica gel using EtOAc/CHCl3 (1:1) as a white solid (0.19 g, 0.38 mmol, 38% yield), mp 104–106°C. Rf 0.63 (EtOAc/CHCl3, 1:1). νmax (KBr)/cm–1 2928, 1595, 1507, 1454, 1334, 1259, 1125, 1048. δH (400 MHz, CDCl3) 7.33 (4H, s, ArH), 6.88–6.77 (8H, m, ArH), 4.62 (4H, s, CH2Ar), 4.12 (4H, t, J 4.4, –OCH2CH2O–), 4.07 (4H, t, J 4.8, –OCH2CH2O–), 3.79 (4H, t, J 4.4, –OCH2CH2O–), 3.74 (4H, t, J 4.8, –OCH2CH2O–), 3.56 (4H, s, –OCH2CH2O–). δC (100 MHz, CDCl3) 148.98, 137.71, 127.93, 121.49, 121.46, 114.31, 114.07, 73.15, 70.88, 69.79, 69.18, 69.06, 68.41. HRMS (EI, 70 eV) found [M + H]+ 525.2486; C30H37O8+ requires 525.2483.
3,6,13,16,19,22,29,32-Octaoxatetracyclo[32.3.1.07,12.023,28]octatriaconta-1(37),7(12),8,10,23(28),24,26,34(38), 35-nonaen-8-yl 15
The general procedure was used with 4 (0.42 g, 1.0 mmol) and α,α′-dichloro-m-xylene (0.17 g, 1.0 mmol). Product 15 was isolated by column chromatography on silica gel using CHCl3/EtOAc (3:1) as a white solid (0.31 g, 0.58 mmol, 58% yield), mp 109–111°C. Rf 0.51 (CHCl3/EtOAc, 3:1). νmax (KBr)/cm–1 2928, 2872, 1594, 1511, 1447, 1348, 1259, 1126, 1047. δH (400 MHz, CDCl3) 7.28 (1H, s, ArH), 7.24–7.22 (3H, m, ArH), 6.82–6.78 (8H, m, ArH), 4.60 (4H, s, CH2Ar), 4.11 (4H, t, J 4.4, –OCH2CH2O–), 4.05 (4H, t, J 4.8, –OCH2CH2O–), 3.79 (4H, t, J 4.4, –OCH2CH2O–), 3.71 (4H, t, J 4.8, –OCH2CH2O–), 3.53 (4H, s, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.08, 148.99, 138.46, 128.56, 127.32, 127.22, 121.64, 121.51, 114.50, 73.45, 70.89, 69.80, 69.43, 69.22, 68.80. HRMS (EI, 70 eV) Na adduct found [M + Na]+ 547.2320; C30H36O8Na+ requires 547.2308.
2,5,14,17,24,27,30,33-Octaoxatetracyclo[32.4.0.07,12.018,23]octatriaconta-1(34),7(12),8,10,18(23),19,21,35,37-nonaen-19-yl 16
The general procedure was used with 4 (0.42 g, 1.0 mmol) and α,α′-dichloro-o-xylene (0.17 g, 1.0 mmol). Product 16 was isolated by column chromatography on silica gel using CHCl3/EtOAc (3:1) as a white solid (0.32 g, 0.59 mmol, 59% yield), mp 91–93°C. Rf 0.5 (CHCl3/EtOAc, 3:1). νmax (KBr)/cm–1 2922, 1594, 1511, 1454, 1261, 1127, 1055. δH (400 MHz, CDCl3) 7.35 (2H, dd, J 5.6 and 3.2, ArH), 7.20 (2H, dd, J 5.6 and 3.2, ArH), 6.81–6.76 (8H, m, ArH), 4.69 (4H, s, CH2Ar), 4.08–4.03 (8H, m, –OCH2CH2O–), 3.78–3.75 (8H, m, –OCH2CH2O–), 3.62 (4H, s, –OCH2CH2O–). δC (100 MHz, CDCl3) 147.94, 147.91, 135.58, 127.89, 126.70, 120.38, 120.31, 113.16, 113.07, 70.02, 69.90, 68.66, 68.09, 67.92, 67.80. HRMS (EI, 70 eV) Na adduct found [M + Na]+ 547.2315; C30H36O8Na+ requires 547.2308.
3,6,13,16,19,22,29,32-Octaoxa-38-azatetracyclo[32.3.1.07,12.023,28]octatriaconta-1(37),7(12),8,10,23(28),24,26,34(38),35-nonaen-8-yl 17
The general procedure was used with 4 (0.42 g, 1.0 mmol) and 2,6-bis(chloromethyl)pyridine (0.17 g, 1.0 mmol). Product 17 was isolated by column chromatography on silica gel using EtOAc/CHCl3 (3:1) as a white solid (0.20 g, 0.38 mmol, 38% yield), mp 144–146°C. Rf 0.19 (EtOAc/CHCl3, 3:1). νmax (KBr)/cm–1 2932, 2866, 1594, 1508, 1452, 1258, 1125, 1055. δH (400 MHz, CDCl3) 7.89 (1H, t, J 7.2, ArH), 7.54 (2H, d, J 7.2, ArH), 6.88–6.80 (8H, m, ArH), 4.78 (4H, s, CH2Ar), 4.15 (4H, t, J 4.2, –OCH2CH2O–), 4.07 (4H, t, J 4.8, –OCH2CH2O–), 3.90 (4H, t, J 4.2, –OCH2CH2O–), 3.76 (4H, t, J 4.8, –OCH2CH2O–), 3.56 (4H, s, –OCH2CH2O–). δC (100 MHz, CDCl3) 157.68, 148.78, 137.21, 121.40, 120.23, 113.83, 113.45, 73.61, 70.80, 69.92, 69.34, 68.71. HRMS (EI, 70 eV) found [M + H]+ 526.2432; C29H36NO8+ requires 526.2435.
3,10,13,20,23,30-Hexaoxapentacyclo[30.2.2.215,18.04,9.024,29]octatriaconta-1(34),4(9),5,7,15,17,24(29),25,27,32,35,37-dodecaene 18
The general procedure was used with 5 (0.41 g, 1.0 mmol) and α,α′-dichloro-p-xylene (0.17 g, 1.0 mmol). Product 18 was isolated by column chromatography on silica gel using CHCl3 as a white solid (0.19 g, 0.38 mmol, 38% yield), mp 135–137°C. Rf 0.32 (CHCl3). νmax (KBr)/cm–1 1591, 1506, 1451, 1257, 1216, 1124, 1097, 1049, 1015. δH (400 MHz, CDCl3) 7.41 (4H, s, ArH), 7.15 (4H, s, ArH), 6.97–6.82 (8H, m, ArH), 5.03 (4H, s, CH2Ar), 4.59 (4H, s, CH2Ar), 4.15 (4H, t, J 4.2, –OCH2CH2O–), 3.79 (4H, t, J 4.2, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.29, 148.56, 137.50, 136.78, 127.70, 127.62, 121.70, 121.08, 114.79, 113.12, 73.00, 71.02, 68.83, 68.08. HRMS (EI, 70 eV) found [M + H]+ 513.2265; C32H33O6+ requires 513.2272.
3,10,13,21,24,31-Hexaoxapentacyclo[31.2.2.115,19.04,9.025,30]octatriaconta-1(35),4(9),5,7,15,17,19(38),25(30),26, 28,33,36-dodecaen-34-yl 19
The general procedure was used with 5 (0.41 g, 1.0 mmol) and α,α′-dichloro-m-xylene (0.17 g, 1.0 mmol). Product 19 was isolated by column chromatography on silica gel using CHCl3 as a white solid (0.22 g, 0.43 mmol, 43% yield), mp 159–161°C. Rf 0.26 (CHCl3). νmax (KBr)/cm–1 2924, 1591, 1505, 1455, 1374, 1254, 1121, 1121, 1008. δH (400 MHz, CDCl3) 7.37 (4H, s, ArH), 7.24–7.12 (4H, m, ArH), 6.93–6.84 (8H, m, ArH) 4.99 (4H, s, CH2Ar), 4.61 (4H, s, CH2Ar), 4.15 (4H, t, J 4.4, –OCH2CH2O–), 3.84 (4H, t, J 4.4, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.38, 148.72, 138.47, 136.78, 128.60, 127.59, 126.90, 126.82, 121.80, 121.29, 115.03, 113.70, 73.71, 71.02, 69.42, 68.89. HRMS (EI, 70 eV) found [M + H]+ 513.2291; C32H33O6+ requires 513.2272.
3,10,13,22,25,32-Hexaoxapentacyclo[32.2.2.04,9.015,20.026,31]octatriaconta-1(36),4(9),5,7,15(20),16,18,26(31),27,29,34,37-dodecaen-16-yl 20
The general procedure was used with 5 (0.41 g, 1.0 mmol) and α,α′-dichloro-o-xylene (0.17 g, 1.0 mmol). Product 20 was isolated by column chromatography on silica gel using CH2Cl2 as a white solid (0.26 g, 0.48 mmol, 48% yield), mp 175–177°C. Rf 0.44 (CH2Cl2). νmax (KBr)/cm–1 2933, 1591, 1509, 1452, 1259, 1125, 1028. δH (400 MHz, CDCl3) 7.41 (4H, s, ArH), 7.36 (2H, dd, J 5.6 and 3.2, ArH), 7.21 (2H, dd, J 5.6 and 3.2, ArH), 6.96–6.82 (8H, m, ArH), 5.00 (4H, s, CH2Ar), 4.67 (4H, s, CH2Ar), 4.12 (4H, t, J 4.4, –OCH2CH2O–), 3.77 (4H, t, J 4.4, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.49, 149.05, 136.85, 136.44, 128.95, 127.79, 127.43, 121.83, 121.59, 115.19, 114.52, 71.21, 70.99, 69.32, 69.05. HRMS (EI, 70 eV) Na adduct found [M + Na]+ 535.2079; C32H32O6Na+ requires 535.2091.
3,10,13,16,19,22,25,32-Octaoxatetracyclo[32.2.2.04,9.026,31]octatriaconta-1(36),4,6,8,26(31),27,29,34,37-nonaen-6-yl 21
The general procedure was used with 5 (0.41 g, 1.0 mmol) and 1,2-bis-(2-chloroethoxy)ethane (0.19 g, 0.10 mL, 1.0 mmol). Product 21 was isolated by column chromatography on silica gel using EtOAc/Et2O (1:1) as a white solid (0.027 g, 0.05 mmol, 5% yield), mp 84–86°C. Rf 0.28 (EtOAc/Et2O, 1/1). νmax (KBr)/cm–1 2931, 1593, 1506, 1452, 1336, 1255, 1125, 1046. δH (400 MHz, CDCl3) 7.42 (4H, s, ArH), 6.92–6.81 (8H, m, ArH), 5.05 (4H, s, CH2Ar), 4.12 (4H, t, J 4.4, –OCH2CH2OAr), 3.84 (4H, t, J 4.4, –OCH2CH2OAr), 3.69 (4H, t, J 4.6, –OCH2CH2O–), 3.54 (4H, t, J 4.6, –OCH2CH2O–), 3.52 (4H, s, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.38, 148.61, 136.99, 127.71, 121.87, 121.32, 115.48, 113.82, 71.21, 71.06, 70.79, 70.72, 69.74, 69.09. HRMS (EI, 70 eV) found [M + H]+ 525.2477; C30H37O8+ requires 525.2483. HRMS (EI, 70 eV) Na+ adduct found [M + Na]+ 547.2284; C30H36O8Na+ requires 547.2302.
3,6,13,21,28,31-Hexaoxapentacyclo[31.2.2.115,19.07,12.022,27]octatriaconta-1(35),7(12),8,10,15,17,19(38),22(27),23,25,33,36-dodecaene 22
The general procedure was used with 6 (0.41 g, 1.0 mmol) and α,α′-dichloro-p-xylene (0.17 g, 1.0 mmol). Product 22 was isolated by column chromatography on silica gel using CHCl3 as a white solid (0.22 g, 0.44 mmol, 44% yield), mp 155–157°C. Rf 0.21 (CHCl3). νmax (KBr)/cm–1 2922, 1592, 1505, 1455, 1374, 1254, 1122, 1008. δH (400 MHz, CDCl3) 7.37 (4H, s, ArH), 7.25–7.11 (4H, m, ArH), 6.86–6.82 (8H, m, ArH), 4.99 (4H, s, CH2Ar), 4.60 (4H, s, CH2Ar), 4.14 (4H, t, J 4.2, –OCH2CH2O–), 3.83 (4H, t, J 4.2, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.37, 148.71, 138.45, 136.78, 128.71, 127.69, 126.88, 126.82, 121.81, 121.28, 115.02, 113.69, 73.70, 71.01, 69.38, 68.89. HRMS (EI, 70 eV) found [M + H]+ 513.2285; C32H33O6+ requires 513.2272.
3,10,13,22,25,32-Hexaoxapentacyclo[32.3.1.04,9.015,20.026,31]octatriaconta-1(37),4(9),5,7,15(20),16,18,26(31),27,29,34(38),35-dodecaen-8-yl 23
The general procedure was used with 6 (0.41 g, 1.0 mmol) and α,α′-dichloro-o-xylene (0.17 g, 1.0 mmol). Product 23 was isolated by column chromatography on silica gel using CHCl3 as a white solid (0.16 g, 0.29 mmol, 29% yield), mp 84–86°C. Rf 0.34 (CHCl3). νmax (KBr)/cm–1 2930, 1592, 1506, 1452, 1378, 1255, 1124, 1097, 1021. δH (400 MHz, CDCl3) 7.67 (1H, s, ArH), 7.27–7.23 (3H, m, ArH), 7.21 (2H, dd, J 5.6 and 3.2, ArH), 7.15 (2H, dd, J 5.6 and 3.2, ArH), 6.93–6.77 (8H, m, ArH), 5.02 (4H, s, CH2Ar), 4.65 (4H, s, CH2Ar), 4.08 (4H, t, J 4.4, –OCH2CH2O–), 3.72 (4H, t, J 4.4, –OCH2CH2O–). δC (100 MHz, CDCl3) 149.78, 149.01, 138.04, 136.78, 129.21, 128.29, 127.67, 127.21, 127.09, 122.20, 121.37, 116.61, 114.33, 72.19, 71.42, 69.01, 68.72. HRMS (EI, 70 eV) Na adduct found [M + Na]+ 535.2092; C32H32O6Na+ requires 535.2091.
3,10,13,21,24,31-Hexaoxa-38-azapentacyclo[31.3.1.115,19.04,9.025,30]octatriaconta-1(36),4(9),5,7,15,17,19(38),25(30),26,28,33(37),34-dodecaen-8-yl 24
The general procedure was used with 6 (0.41 g, 1.0 mmol) and 2,6-bis(chloromethyl)pyridine (0.17 g, 1.0 mmol). Product 24 was isolated by column chromatography on silica gel using EtOAc/CHCl3 (3:1) as a white solid (0.22 g, 0.43 mmol, 43% yield), mp 178–180°C. Rf 0.39 (EtOAc/CHCl3, 3:1). νmax (KBr)/cm–1 2937, 1597, 1508, 1459, 1377, 1258, 1127, 1059, 1008. δH (400 MHz, CDCl3) 7.67 (1H, t, J 7.6, ArH), 7.54–7.46 (6H, m, ArH), 7.02–6.83 (8H, m, ArH), 4.96 (4H, s, CH2Ar), 4.63 (4H, s, CH2Ar), 4.12 (4H, t, J 4.6, –OCH2CH2O–), 3.81 (4H, t, J 4.6, –OCH2CH2O–). δC (100 MHz, CDCl3) 158.38, 148.78, 148.69, 137.60, 137.14, 130.03, 129.45, 127.14, 121.22, 120.89, 119.45, 112.91, 111.90, 74.32, 70.98, 70.43, 69.83. HRMS (EI, 70 eV) found [M + H]+ 514.2224; C31H32NO6+ requires 514.2224.
3,10,13,21,24,31-Hexaoxa-37-azapentacyclo[31.3.1.115,19.04,9.025,30]octatriaconta-1(37),4(9),5,7,15(38),16,18,25(30),26,28,33,35-dodecaen-16-yl 25
The general procedure was used with 8 (0.41 g, 1.0 mmol) and α,α′-dichloro-m-xylene (0.17 g, 1.0 mmol). Product 25 was isolated by column chromatography on silica gel using EtOAc as a white solid (0.19 g, 0.38 mmol, 38% yield), mp 166–168°C. Rf 0.28 (EtOAc). νmax (KBr)/cm–1 2937, 1596, 1509, 1459, 1377, 1258, 1127, 1056. δH (400 MHz, CDCl3) δ 7.65 (t, 1H, J 7.6, ArH), 7.58 (s, 1H, ArH), 7.53–7.44 (m, 5H, ArH), 7.02–6.83 (m, 8H, ArH), 4.94 (s, 4H, CH2Ar), 4.62 (s, 4H, CH2Ar), 4.11 (t, 4H, J 4.4, –OCH2CH2O–), 3.81 (t, 4H, J 4.4, –OCH2CH2O–). δC (100 MHz, CDCl3) 157.57, 148.78, 148.71, 137.06, 129.98, 129.65, 129.51, 127.18, 121.30, 121.00, 119.52, 113.11, 112.13, 73.58, 71.12, 68.45, 67.89. HRMS (EI, 70 eV) found [M + H]+ 514.2222; C31H32NO6+ requires 514.2224.
3,10,13,21,24,31-Hexaoxa-37,38-diazapentacyclo[31.3.1.115,19.04,9.025,30]octatriaconta-1(37),4(9),5,7,15(38),16,18,25(30),26,28,33,35-dodecaen-16-yl 26
The general procedure was used with 8 (0.41 g, 1.0 mmol) and 2,6-bis(chloromethyl)pyridine (0.17 g, 1.0 mmol). Product 26 was isolated by column chromatography on silica gel using EtOAc as a white solid (0.25 g, 0.49 mmol, 49% yield), mp 145–147°C. Rf 0.18 (EtOAc). νmax (KBr)/cm–1 2920, 1594, 1507, 1456, 1362, 1259, 1125, 1053. δH (400 MHz, CDCl3) 7.48 (2H, d, J 7.8, ArH), 7.34 (3H, m, ArH), 7.02–6.87 (8H, m, ArH), 6.78 (1H, t, J 7.8, ArH), 5.07 (4H, s, CH2Ar), 4.61 (4H, s, CH2Ar), 4.17 (4H, t, J 4.4, –OCH2CH2O–), 3.87 (4H, t, J 4.4, –OCH2CH2O–). δC (100 MHz, CDCl3) 157.61, 156.47, 149.32, 148.90, 137.51, 137.20, 122.03, 121.58, 121.42, 119.40, 114.89, 113.52, 73.70, 72.51, 69.37, 68.66. HRMS (EI, 70 eV) found [M + H]+ 514.2245; C30H31N2O6+ requires 514.2230.
Accessory Publication
X-ray crystal studies and metal picrates extraction properties of lipophilic benzocrown ethers are available from the journal’s website.
Acknowledgements
The authors wish to thank Dublin City University (E.K.) and Irish Research Council for Science, Engineering & Technology (J.P.) for funding and Helge Muller-Bunz at University College Dublin, Ireland, for X-ray crystallography studies. Authors E.K. and J.P. contributed equally.
[1]
(a) H. R. Diril,
X. Chang,
S. K. Zhang,
J. A. Larsen,
C. G. Polenza,
H. J. Pierpont,
S. Schugar,
S. Isted,
D. N. Hendrickson,
J. Am. Chem. Soc. 1987, 109, 6207.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
