

A Series of Imidazolyl-Containing Bisphosphonates with Abundant Hydrogen-Bonding Interactions: Syntheses, Structures, and Bone-Binding Affinity
Ling Qiu A , Jianguo Lin A B , Liqing Wang A , Wen Cheng A , Yang Cao A , Xuewen Liu A and Shineng Luo AA Key Laboratory of Nuclear Medicine, Ministry of Health, Jiangsu Key Laboratory of Molecular Nuclear Medicine, Jiangsu Institute of Nuclear Medicine, Wuxi 214063, China.
B Corresponding author. Email: linjianguo@jsinm.org
Australian Journal of Chemistry 67(2) 192-205 https://doi.org/10.1071/CH13194
Submitted: 22 April 2013 Accepted: 3 September 2013 Published: 2 October 2013
Abstract
A series of novel bisphosphonates (BPs) were designed and synthesised as longer-chain analogues of the clinically widely used BP–zoledronate (ZL). They were characterised by mass spectrometry, infrared spectroscopy, NMR spectroscopy, and single-crystal X-ray diffraction. All the crystals are zwitterions with one of the phosphonate oxygen atoms deprotonated and the hydrogen atom transferred to the nitrogen of the imidazole ring. A lot of strong hydrogen bonds are observed among the phosphonate oxygens, hydroxy groups, and protonated nitrogen atoms. An accurate, precise, and robust method was developed to determine the bone binding affinities of BPs based on high performance liquid chromatography. The results show that these five BPs have a strong affinity for hydroxyapatite and the binding capacity decreases when the substituted alkyl groups increase in size.
Introduction
Bisphosphonates (BPs), synthetic analogues of pyrophosphate (PP), contain a stable backbone structure of P–C–P which is resistant to enzymatic hydrolysis. They exhibit a high affinity for calcified matrices, such as hydroxyapatite (HA) in bone.[1] Therefore, they are commonly used to inhibit the mineralization of soft tissues and also as a therapy for bone-formation and resorption disorders, e.g. in osteoporosis, Paget’s disease, hypercalcemia of malignancy, and bone metastases of several cancers.[2] The BPs adsorb on the bone mineral by forming metal complexes with calcium, and their complexation properties are an important aspect for gaining an understanding of their mechanism of action. On the other hand, the high binding affinities of BPs for bone are of crucial biological significance since they can influence several important activities of these compounds, including uptake and retention by the skeleton, distribution and diffusion of the drug within bone, as well as the release and potential recycling of the drug and cellular functions within bone.[3] The mineral-binding affinity may also affect the appropriate dosing interval and the persistence of effect after discontinuation of medication when BPs are used clinically to treat disorders of bone resorption.[4] Therefore, it is of great importance to design and screen novel BPs with superior mineral-binding affinity.
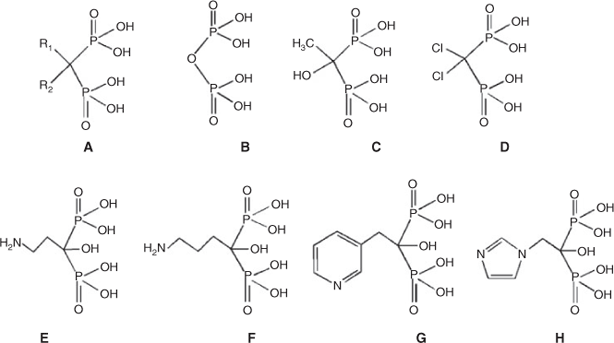
The structure activities and bone affinities of BPs are directly associated with the substituents (R1 and R2) connected both to the middle carbon and to the phosphorus atoms (see Scheme 1).[5,6] In particular, the number of donor atoms in the ligand and their connections with the metal atoms affect the bone affinity significantly. For example, the bone affinity of risedronate (G) and etidronate (C) is much greater than that of clodronate (D) owing to their tridentate adsorption versus the bidentate adsorption of clodronate on the bone mineral surface.[7] That is, a hydroxy group at the R1 position confers high affinity binding to the mineral. Varying the R2 substituent can result in differences in the antiresorptive potency of several orders of magnitude, since it is the essential determinant of antiresorptive potency.[8] The most potent antiresorptive BPs include those containing a heteroaromatic moiety with at least one nitrogen atom, such as risedronate (G) and zoledronate (ZL, H). The enhancement in the antiresorptive potency resulting from different R2 groups also appears to be linked to the bone-binding ability and biochemical activity of these drugs.[7–9] For example, ZL, containing two nitrogen atoms within a heteroaromatic ring, is at least 100 times more potent than pamidronate (E) and at least 1000 times more potent than etidronate (C).[10] The structure of the carbon chain attached to the geminal BPs also influences their biological activities. For example, from pamidronate (E) to alendronate (F) with the carbon chain increasing from 2 to 3, the potency increases 10 times.[11] In addition, two phosphonate groups in the BPs of clinical interest also have a dual function, which are required both for binding to bone mineral and for cell-mediated antiresorptive activity. Modifications to one or both phosphonate groups can dramatically reduce the affinity for bone mineral[7] and the biological potency of the BP.[6]
|
To date, a large number of BPs have been prepared and investigated.[12] We have also been involved for several years in the synthesis and biological evaluation of imidazolyl-containing BPs to develop novel agents for the diagnosis and therapy of bone diseases.[13] In a continuing effort to find novel BPs with better mineral-binding affinity, in the present work a series of imidazolyl-containing BPs were designed and synthesised (Scheme 2). These compounds can be viewed as longer-chain analogues of ZL, where the imidazolyl with different alkyl groups substituted at the 2-position is linked to the geminal BP unit by a hexylidene group. The synthesis, characterisation, and crystal structures of 1-hydroxy-6-(1H-imidazol-1-yl)hexane-1,1-bisphosphonic acid (IHDP, 3a), 1-hydroxy-6-(2-methyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic acid (MIHDP, 3b), 1-hydroxy-6-(2-ethyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic acid (EIHDP, 3c), 1-hydroxy-6-(2-propyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic acid (PIHDP, 3d), and 1-hydroxy-6-(2-butyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic acid (BIHDP, 3e) are reported. In addition, the in-vitro bone-binding affinities of these BPs are measured to understand the effect of the carbon chain length of the alkyl groups on the bone-targeting ability. The structure–activity data elucidated for these compounds may be instructive for the design and synthesis of novel BPs.

|
Experimental
Materials and Measurements
Reagents and solvents as well as HA were commercially available and used as received. Melting points were measured on a Yanaco MP-500 melting point apparatus (Shimadzu, Japan). Electrospray ionisation mass spectrometry (ESI-MS) was performed using a Waters Platform ZMD4000 LC/MS (Waters, U.S.A). 1H NMR spectra were obtained on a Bruker DRX-500 spectrometer (Bruker, Germany), and the chemical shift values were referenced to the internal tetramethylsilane (TMS). Infrared spectra were recorded on a Tensor 27 IR Fourier Transform spectrophotometer using KBr discs (Bruker, Germany).
A high performance liquid chromatography (HPLC) system was equipped with a Waters 1525 binary HPLC pump and a Waters 2487 dual wavelength absorbance detector (Waters, U.S.A). A reverse phase C18 (RP-C18) column (4.6 mm × 250 mm; 10 μm particle size) was used for HPLC analysis (Elite Analytical Instrument Co., Dalian, China). The flow rate was 1.0 mL min–1, and the mobile phase was isocratic with 80 % solvent A (4.0 mmol L–1 sodium dihydrogen phosphate and 5.0 mmol L–1 tetrabutylammonium bromide aqueous solution) and 20 % solvent B (acetonitrile). These conditions are fit for the purity determination, standard working curve, and HA-binding assay experiments.
Synthesis of BPs
Five new imidazolyl-containing BPs (3a–e) were synthesised according to the procedure outlined in Scheme 2.[12a]
General Procedure for Preparing Compound 2
The raw material 1 (0.10 mol) was dissolved in CH2Cl2 (100 mL), followed by adding KOH (8.4 g, 0.15 mol), K2CO3 (13.8 g, 0.10 mol), and (Bu)4NBr (0.7 g, 0.002 mol). A solution of Br(CH2)5COOC2H5 (0.1 mol) was added and the reaction mixture was then refluxed for 20 h. To remove the inorganic salt, the solution was filtered and the filtrate was washed with brine solution. The organic phase was dried and evaporated to give the corresponding ester, which was used without further purification in the subsequent reaction. Aqueous HCl (1.2 mol L–1, 100 mL) was added to the crude ester and the mixture was refluxed for 8 h. The solution was concentrated, and the residue was recrystallised from isopropyl alcohol to give the product 2.
6-(1H-Imidazole-1-yl)hexanoic Acid (2a)
The pale white solid was washed with acetone and recrystallised from isopropyl alcohol to obtain 12.6 g of pure white crystals. Yield: 69.3 %, mp 124–126°C. δH (400 MHz, D2O) 8.65 (s, 1H, IMZ-H), 7.44 (s, 1H, IMZ-H), 7.39 (s, 1H, IMZ-H), 4.18 (t, J 7.2, 2H, N–CH2), 2.30 (t, J 7.6, 2H, CH2COOH), 1.81–1.88 (m, 2H, CH2), 1.52–1.60 (m, 2H, CH2), 1.22–1.30 (m, 2H, CH2). m/z (ESI) 181 (compound 2a, [M – H]).
6-(2-Methyl-1H-imidazole-1-yl)hexanoic Acid (2b)
The reddish brown sticky liquid was crystallised at –5°C and washed with acetone to afford a pale white powder. Recrystallisation of the pale white powder from isopropyl alcohol gave 16.5 g of fine white crystals. Yield: 78.2 %, mp 131–133°C. δH (400 MHz, D2O) 7.34 (d, J 2.0, 1H, IMZ-H), 7.29 (d, J 2.4, 1H, IMZ-H), 4.10 (t, J 7.2, 2H, N–CH2), 2.60 (s, 3H, CH3), 2.38 (t, J 7.2, 2H, CH2COOH), 1.81–1.89 (m, 2H, CH2), 1.59–1.67 (m, 2H,CH2), 1.31–1.39 (m, 2H, CH2). m/z (ESI) 195 (compound 2b, [M – H]).
6-(2-Ethyl-1H-imidazole-1-yl)hexanoic Acid (2c)
The faint yellow sticky liquid was crystallised at 0°C and washed with acetone to afford 13.1 g of a white powder. Yield: 62.5 %, mp 88–90°C. δH (400 MHz, D2O) 7.35 (d, J 2.0, 1H, IMZ-H), 7.32 (d, J 2.4, 1H, IMZ-H), 4.12 (t, J 7.2, 2H, N–CH2), 2.95–3.01 (q, 2H, ring-CH2CH3), 2.38 (t, J 7.2, 2H, CH2COOH), 1.82–1.90 (m, 2H, CH2), 1.60–1.67 (m, 2H, CH2), 1.32–1.39 (m, 5H, CH2, CH3). m/z (ESI) 209 (compound 2c, [M – H]).
6-(2-Propyl-1H-imidazole-1-yl)hexanoic Acid (2d)
The faint yellow sticky liquid was stirred with acetone at –5°C to afford a white powder. Recrystallisation of the white powder from a mixed solvent of isopropyl alcohol/acetone (1 : 2, v/v) afforded 15.6 g of colourless crystals. Yield: 71.2 %, mp 70–72°C. δH (400 MHz, D2O) 7.36 (d, J 2.0, 1H, IMZ-H), 7.32 (d, J 2.0, 1H, IMZ-H), 4.12 (t, J 7.2, 2H, N–CH2), 2.94 (t, J 7.6, 2H, ring-CH2CH2CH3), 2.38 (t, J 7.2, 2H, CH2COOH), 1.82–1.90 (m, 2H), 1.73–1.82 (m, 2H, CH2), 1.60–1.67 (m, 2H, CH2), 1.34–1.39 (m, 2H, CH2), 0.97 (t, J 7.2, 3H, CH3). m/z (ESI) 223 (compound 2d, [M – H]).
6-(2-Butyl-1H-imidazole-1-yl)hexanoic Acid (2e)
The yellowish-brown sticky liquid was triturated with acetone at 0°C, and the mixture precipitated at –20°C. The precipitate was collected through filtration and washed with ethyl acetate. The crude product was then recrystallised from a mixed solvent of isopropyl alcohol/acetone (1 : 2, v/v) to obtain 17.9 g of colourless crystals. Yield: 75.1 %, mp 51–53°C. δH (400 MHz, D2O) 7.36 (d, J 2.0, 1H, IMZ-H), 7.32 (d, J 2.0, 1H, IMZ-H), 4.13 (t, J 7.2, 2H, N–CH2), 2.97 (t, J 7.6, 2H, ring-CH2CH2CH2CH3), 2.38 (t, J 7.2, 2H, CH2COOH), 1.83–1.90 (m, 2H, CH2), 1.69–1.77 (m, 2H, CH2), 1.60–1.67 (m, 2H, CH2), 1.32–1.43 (m, 4H, 2 × CH2). m/z (ESI) 237 (compound 2e, [M – H]).
General Procedure for Preparing Compound 3
Compound 2 (0.1 mol) was dissolved in chlorobenzene (60 mL) and heated to 110°C for 30 min. After cooling to room temperature, phosphoric acid (85 %, 15.7 mL) was added slowly, followed by phosphorus trichloride (27.3 mL). The reaction mixture was kept at 120°C for 16 h. The chlorobenzene was decanted, and the yellow residue was redissolved in HCl (9 mol L–1, 50 mL). The mixture was refluxed for 10 h. After cooling, charcoal was added to de-colour the solution. After filtration, the solvent was removed. Finally, the crude product was recrystallised from ethanol to give the white powder product 3. Recrystallisation of the white powder from water afforded white crystals.
1-Hydroxy-6-(1H-imidazol-1-yl)hexane-1,1-bisphosphonic Acid (3a)
Yield 20.6 g (62.8 %), white crystals. Mp 217–220°C. Anal. Calc. for C9H18N2O7P2: C 32.94, H 5.53, N 8.54. Found: C 33.12, H 3.69, N 8.31 %. δH (400 MHz, D2O) 8.66 (s, 1H, IMZ-H), 7.42 (s, 1H, IMZ-H), 7.37 (s, 1H, IMZ-H), 4.18 (t, J 6.8, 2H, N–CH2), 1.81–1.93 (m, 4H, N–CH2CH2, CH2–C–OH), 1.52–1.60 (m, 2H, CH2), 1.22–1.30 (m, 2H, CH2). δC (100 MHz, D2O) 140.32 (s, N=C–N), 129.70 (s, C-ring), 122.57 (s, C-ring), 78.80 (t, P–C–P), 49.20 (s, N–CH2), 38.25 (s, N–CH2CH2), 32.42 (s, CH2–C–OH), 29.18 (s, CH2), 25.90 (s, CH2). νmax (KBr)/cm–1 3396(s, br), 3162(s), 3126(s), 2951(s), 2860(s), 2389(w, br),1580(m), 1547(w), 1464(m), 1403(w), 1248(m), 1151(s), 1073(v), 989(w), 949(s), 911(m), 767(m), 471(m). m/z (ESI) 327 (compound 3a, [M – H]).
1-Hydroxy-6-(2-methyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic Acid (3b)
Yield 14.6 g (42.7 %), white crystals. Mp 214–216°C. Anal. Calc. for C10H20N2O7P2: C 35.10, H 5.89, N 8.19. Found: C 34.87, H 6.05, N 8.08 %. δH (400 MHz, D2O) 7.28 (s, 1H, IMZ-H), 7.20 (s, 1H, IMZ-H), 4.03 (t, J 7.2, 2H, N–CH2), 2.53 (s, 3H, CH3), 1.86–1.94 (m, 2H, CH2), 1.76–1.83 (m, 2H, CH2), 1.52–1.60 (m, 2H, CH2), 1.24–1.32 (m, 2H, CH2). δC (100 MHz, D2O) 148.05 (s, N=C–N), 127.12 (s, C-ring), 122.14 (s, C-ring), 78.42 (t, P–C–P), 47.81 (s, N–CH2), 37.81 (s, CH3), 31.56 (s, N–CH2CH2), 28.75 (s, CH2–C–OH), 25.59 (s, CH2), 13.45 (s, CH2). νmax (KBr)/cm–1 3279(s, br), 3163(s), 2928(s), 2387(w, br), 2290(w, br), 1609(m), 1530(w), 1450(w), 1392(w), 1279(w), 1130(m), 1106(m), 1051(s), 1019(s), 970(s), 947(m), 918(m), 759(m), 519(m). m/z (ESI) 341 (compound 3b, [M – H]).
1-Hydroxy-6-(2-ethyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic Acid (3c)
Yeild 14.0 g (39.9 %), white crystals. Mp 203–205°C. Anal. Calc. for C11H22N2O7P2: C 37.09, H 6.22, N 7.86. Found: C 36.91, H 6.50, N 7.75 %. δH (400 MHz, D2O) 7.29 (d, 1H, IMZ-H), 7.23 (d, 1H, IMZ-H), 4.04 (t, J 7.2, 2H, N–CH2), 2.91 (q, 2H, CH2CH3), 1.85–1.94 (m, 2H, CH2), 1.76–1.84 (m, 2H, CH2), 1.52–1.60 (m, 2H, CH2), 1.25–1.32 (m, 5H, –CH2CH2CH2–C–OH, CH3). δC (100 MHz, D2O) 153.32 (s, N=C–N), 128.03 (s, C-ring), 123.02 (s, C-ring), 79.30 (t, P–C–P), 48.36 (s, N–CH2), 38.62 (s, ring-CH2), 32.75 (s, N–CH2CH2), 29.95 (s, CH2–C–OH), 26.53 (s, CH2), 21.74 (s, CH2), 13.93(s, CH3). νmax (KBr)/cm–1 3387(s, br), 3138(s), 2941(s), 2394(w, br), 1601(m), 1523(w), 1467(w), 1403(w), 1227(w), 1168(m), 1122(s), 993(s), 921(m), 771(w), 459(m). m/z (ESI) 355 (compound 3c, [M – H]).
1-Hydroxy-6-(2-propyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic Acid (3d)
Yield 22.4 g (40.5 %), white crystals. Mp 206–208°C. Anal. Calc. for C12H24N2O7P2: C 38.92, H 6.53, N 7.57. Found: C 38.67, H 6.74, N 7.36 %. δH (400 MHz, D2O) 7.38 (d, J 2.0, 1H, IMZ-H), 7.32 (d, J 2.0, 1H, IMZ-H), 4.13 (t, J 7.2, 2H, N–CH2), 2.95 (t, J 7.6, 2H, CH2CH2CH3), 1.94–2.02 (m, 2H, CH2), 1.84–1.92 (m, 2H, CH2), 1.73–1.83 (m, 2H, CH2), 1.60–1.68 (m, 2H, CH2), 1.33–1.40 (m, 2H, CH2), 0.97 (t, J 7.2, 3H, CH3). δC (100 MHz, D2O) 152.16 (s, N=C–N), 128.16 (s, C-ring), 123.15 (s, C-ring), 79.44 (t, P–C–P), 48.49 (s, N–CH2), 38.84 (s, ring-CH2), 33.20 (s, N–CH2CH2), 30.40 (s, CH2–C–OH), 29.93 (s, CH2), 26.63 (s, CH2), 23.49(s, CH2), 15.73(s, CH3). νmax (KBr)/cm–1 3397(s, br), 3120(s), 2960(s), 2939(s), 2396(w, br), 2289(w, br), 1602(m), 1523(w), 1469(w), 1404(w), 1234(m), 1126(s), 1106(s), 1062(s), 989(s), 924(s), 900(m), 772(w), 477(m). m/z (ESI) 369 (compound 3d, [M – H]).
1-Hydroxy-6-(2-butyl-1H-imidazol-1-yl)hexane-1,1-bisphosphonic Acid (3e)
Yields 16.5 g (43.0 %), white powder. Mp 191–194°C. Anal. Calc. for C13H26N2O7P2: C 40.63, H 6.82, N 7.29. Found: C 40.37, H 7.06, N 7.15 %. δH (400 MHz, D2O) 7.30 (d, J 2.0, 1H, IMZ-H), 7.23 (d, J 2.0, 1H, IMZ-H), 4.06 (t, J 7.2, 2H, N–CH2), 2.90 (t, J 8.0, 2H, CH2CH2CH2CH3), 1.86–1.94 (m, 2H, CH2), 1.78–1.83 (m, 2H, CH2), 1.62–1.70 (m, 2H, CH2), 1.53–1.60 (m, 2H, CH2), 1.25–1.36 (m, 4H, –CH2CH2CH2–C–OH, –CH2CH3), 0.85 (t, J 7.6, 3H, CH3). δC (100 MHz, D2O) 152.39 (s, N=C–N), 128.64 (s, C-ring), 123.04 (s, C-ring), 79.64 (t, P–C–P), 48.72 (s, N–CH2), 38.86 (s, ring-CH2), 32.78 (s, N–CH2CH2), 31.99 (s, CH2–C–OH), 29.80 (s, CH2), 27.90 (s, CH2), 26.54 (s, CH2), 24.23 (s, CH2), 15.62 (s, CH3). νmax (KBr)/cm–1 3411(s, br), 3175(s), 3133(s), 2960(s), 2936(s), 2388(w, br), 2289(w, br), 1609(m), 1519(w), 1468(w), 1405(w), 1275(w), 1178(s), 1094(s), 1051(s), 992(s), 942(s), 784(w), 530(m). m/z (ESI) 383 (compound 3e, [M – H]).
X-Ray Crystallographic Study
Single crystals of compounds 3a–e in appropriate dimensions were used for structural determinations on a Bruker SMART APEX CCD diffractometer using graphite-monochromatised MoKα radiation (λ = 0.71073 Å) at room temperature using the ω-scan technique. Data reductions and absorption corrections were performed with the SAINT and SADABS software packages, respectively. The structures of all the crystals were solved by direct methods using the SHELXS-97 program[14] and were further refined by the full-matrix least-squares technique using the SHELXL-97 program.[15] Anisotropic displacement parameters were refined for all the non-hydrogen atoms except for the disordered atoms. The hydrogen atoms were generated theoretically, allowed to ride on their respective parent atoms, and included in structure factor calculations with assigned isotropic thermal parameters. In the case of compound 3d, the propyl in the imidazole ring appeared to be disordered over two positions with occupancy factors 0.782 and 0.218, respectively. The crystallographic data and other pertinent information for 3a–e are summarised in Table 1, while the selected bond lengths and angles are listed in Table 2. More details on the crystallographic studies as well as atom displacement parameters are presented in the .cif files (CCDC 892394–892398), and can be obtained free of charge at www.ccdc.cam.ac.uk/data_request/cif.

|

|
HA-Binding Assay
To study the bone targeting efficiency of these BPs, an in vitro HA-binding assay was set up using a HPLC method with the most usual detection system and the simplest chromatography elution conditions. First, all the BPs 3a–e with different concentrations (0.4–6.0 μmol L–1) were determined by HPLC and a standard curve was plotted for each compound from the area integral as a function of concentration (see Fig. S1). The disposition pattern of the standard curve can be described by the following linear equation: S = kC + b, where S is the average integral area under HPLC peak of an aliquot of the BP solution, C is the standard concentration of BPs, and k and b are the slope and intercept of the standard curve, respectively.
The adsorption experiments were performed at 37°C in an aqueous medium and incubated for 48 h which is sufficient for reaching the equilibrium of binding. The adsorbed amount of each BP was determined by the difference between the initial compound quantity and that contained in the supernatant solution. The adsorption profile then was obtained as a function of the BP concentration. In detail, aliquots (2 mL) of the BP solution with different concentrations were added to 25 mg of HA in a 10 mL penicillin bottle. After 15 s of treatment in a vortex apparatus, the suspension was maintained in a bascule bath at 37°C. After 48 h of incubation, the mixture was centrifuged at 5000 rpm for 5 min to ensure complete separation of the supernatant from the solid phase. The supernatant was then filtered through a microporous filter (0.22 μm pore size) and an aliquot (10 μL) of the supernatant was removed for the quantification of the BP remaining in the solution by using HPLC analysis. Based on the standard working curves obtained in advance, the amount of BP adsorbed onto HA can be determined by the following formula and expressed as the amount of BP per gram of HA (μmol g–1):
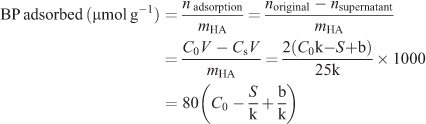
where V is the volume of the solution (2 mL), C0 is the initial concentration of the bisphosphate solution (μmol mL–1), CS is the concentration of the supernatant (μmol mL–1), and S is the average area integral under the HPLC peak of an aliquot of the supernatant (10 μL).
Results and Discussion
Synthesis of BPs
Five novel ZL analogues were synthesised by four steps according to a previous method.[12a] In the first step, carboxylic acids 2a–e were prepared through the N-alkylation reaction of 2-alkyl-1H-imidazole and Br(CH2)5COOC2H5, which is a heterogeneous reaction. To accelerate the reaction rate and achieve a satisfactory yield, tetrabutyl ammonium bromide (TBAB) was added to the reaction system as a phase transfer catalyst. Compound 2 was then obtained through the hydrolysis with concentrated hydrochloric acid. In the next step, compounds 3a–e were obtained through the phosphine acidification reaction of the corresponding carboxylic acid 2. The speed of adding PCl3 had a critical effect on the yield of the reaction. If the addition is fast, the reaction may be hard to control, and the yield is low. Since adding PCl3 led to a large amount of precipitate forming immediately, it became more and more difficult to stir. This will result in incomplete conversion from the carboxylic acid to the bisphosphonic acid. Otherwise, the reaction time may be too long. So, a moderate speed of addition (~0.4 mL min–1) was selected to control the reaction temperature and increase the yield.
On the other hand, crystallisation of the obtained faint yellow or colourless oil was also important but very tedious, as previously described by others.[12a–d] To address this problem, many changes have been made by us and now it is easy to get pure compounds. Under continuous stirring, the residual oil was added slowly to a large amount of ethanol and the crude product precipitated immediately. After 24 h, the suspension was filtered to give a fine white powder, which was very stable at room temperature. In this way, the separation and purification became easier and more efficient. The yield was moderate, even rather low (typically 35–40 %). However, there are a few advantages of this reaction including cheap and easily available reagents as well as simple operation. More importantly, the residual carboxylic acid and chlorobenzene as well as phosphoric acid can be easily separated by dissolving them in the ethanol.
Spectroscopic Characterisation
Compounds 3a–e were fully characterised by melting point, MS, and FT-IR and 1H NMR spectroscopy. From 3a to 3e with the substituent at the 2-position of the imidazole ring changing from hydrogen to butyl, the melting point decreases from 220 to 191°C. This may be attributed to the stereo-hindrance effect of the side substituent R, which leads to a more and more loose structure. The molecular ions excited by the ESI method confirmed the proposed molecular formula of all BPs.
The IR spectra of five BPs are similar to each other. All of them show that the typical strong absorption bands of the C=O stretching vibrations in the carboxylic acids 2 disappear and three new characteristic absorption bands occur. The bands in the region of 1126–1279 cm–1 are associated with the typical symmetric and asymmetric stretching vibrations of PO2 from PO3H– groups. The absorption bands at 940–1000 cm–1 and around 1080 cm–1 are attributed to the vibration modes of νs(P–OH) and δ(P–OH), respectively, which are due to the deprotonated POH groups in these compounds.[16] In addition, the absorption bands at ~3100 cm–1 correspond to the stretching vibrations of the protonated N2.
The 1H NMR spectrum of compound 3a shows three single peaks in the region of 7.37–8.66 ppm, corresponding to the protons in the aromatic imidazole ring, while those of compounds 3b–e show two doublets (3J = 2 Hz) in the region of 7.20–7.38 ppm. Similar 1H NMR spectra are also observed for the compounds 2a–e. They agree well with the expected chemical structures of all the bisphosphonic acids and carboxylic acids.
In addition, the purity of compounds 3a–e was also checked by HPLC (see Fig. 1). For each compound, only one single peak was observed. This clearly showed the high purity of the compounds. The retention times of compounds 3a–e were 5.95, 6.07, 6.50, 7.40, and 11.05 min, respectively. It is evident that the substituent at the 2-position of the imidazole ring affects the retention behaviour of these BPs. Under the same mobile phase conditions, the retention time was extended as the number of carbon atoms in the substituent increased.[17] Especially for compound 3e, the extension is the most remarkable corresponding to the butyl group which has the maximum number of C atoms within the compounds under investigation (from 5.95 to 11.05 min). This is due to the fact that the bulky lipophilic alkyl group has a significant influence on the polarity of the compound, which changes the retention time accordingly.
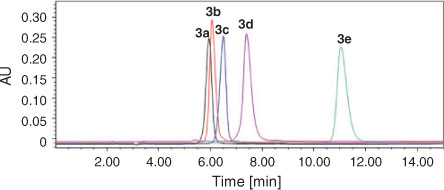
|
Crystal Structures
Single crystals of compounds 3a–e were obtained with a quality good enough for X-ray crystallography analysis. The structures of five compounds have been solved and refined (see Table 1). The single crystal X-ray diffraction studies reveal that compounds 3a–d all crystallise in the monoclinic space group P21/n and contain four irreducible molecules per unit cell, while 3e crystallises in the orthorhombic space group Pbca with eight irreducible molecules in the unit cell which is different from other compounds. In addition, although all the crystals are recrystallised from water, only compound 3b has a water solvent molecule in the crystal structure. In the asymmetric unit of 3b, there is one lattice water molecule per BP molecule. Compared with other compounds, the additional lattice water molecule in 3b may increase the donor and acceptor property towards hydrogen bonds and build a more complicated hydrogen-bonded system. In the asymmetric unit of 3d, there is disorder at the propyl group and only the propyl attached to the imidazole ring with a high occupancy factor (0.782) has been shown. The phenomenon of the propyl side chain is disordered over two sites with different occupancies were also observed in other reported structures.[18]
As anticipated, all the crystals 3a–e are zwitterions, in which one of the phosphonate oxygen atoms is ionised and the hydrogen atom is transferred to the nitrogen atom (N2) of the imidazole ring (see Fig. 2). This behaviour is not an uncommon feature for the nitrogen-containing bisphosphonates (NBPs), as has been observed previously for ZL[19] and other related compounds.[12a-d,20] According to the previous investigation,[12d] the protonated side chains of the NBPs are important for their binding to the farnesyl diphosphate synthase target and they are useful for treating various bone resorption diseases.

|
Inspection of the bond distances in the protonated imidazole ring shows that there are two classes of C–N bonds in the ring. As illustrated in the diagram for 3a, the longer bonds (C1–N1 1.374(2) Å, C2–N2 1.363(3) Å) represent essentially single-bond character at C1–N1 and C2–N2 (1.340–1.358Å),[21] while the short ones (C3–N1 1.334(3) Å, C3–N2 1.313(3) Å) imply partial double-bond character between atoms C3 and N1/N2 (1.314 Å).[21] The other double bond in the ring is found at C1–C2. This structure shows an approximately equal charge distribution on the atoms N1 and N2. As the hydrogen at the 2-position of the imidazole ring was substituted by methyl, ethyl, propyl, and butyl groups, i.e. from 3a to 3e, a slight increase was observed for all the bond lengths in the imidazole ring, particularly for the bonds N2–C3 and N2–C2 (see Table 2). This mainly stems from the repulsive force between the hydrogen atom on the protonated N2 and the alkyl group introduced at the neighbouring position.
In compounds 3a–e, the relative positions of the two phosphonate groups are nearly eclipsed when viewed along the P–C–P axis, and the chain of atoms O3–P1–C9–P2–O6 form an approximately planar ‘W’ configuration with a torsion angle of ~2.7°. On two sides of this plane, two pairs of phosphonate oxygen atoms (O1 and O4, O2 and O5), hydroxyl, and methylene groups are equally distributed. Such an arrangement provides a minimum of repulsion among the substituent R1, R2, and the phosphonate oxygens.
The geometries of atoms P1 and P2 are all approximately tetrahedral with small deviations induced by different environments of the oxygen atoms O1/O2/O3 and O4/O5/O6. This can be indicated by the bond angles O–P–O and C–P–O given in Table 2. The bond angles of O–P1–O range from 104.22° to 115.38°, while those of O–P2–O lie between 107.42° and 115.31°. The bond angles of C9–P1–O range from 104.67° to 113.51°, while those of C9–P2–O lie between 104.72° and 111.85°. As a whole, the bond angle O4–P2–O5 with a mean value of 114.31° is larger than other O–P–O bond angles with mean value of 110.06°, which is probably attributed to the repulsive interaction between two unprotonated atoms O4 and O5. Similarly, the C–P–O angles involving unprotonated oxygen atoms (O3, O4, and O5) are obviously larger than those involving protonated oxygen atoms (O1, O2, and O6). In addition, the angle between two phosphonate groups, i.e. P–C–P, decreases slightly on the whole due to the repulsive force between the side chains increasing from 3a to 3e. However, they are still close to the observed P–C–P conformation in other BPs.[12d,20]
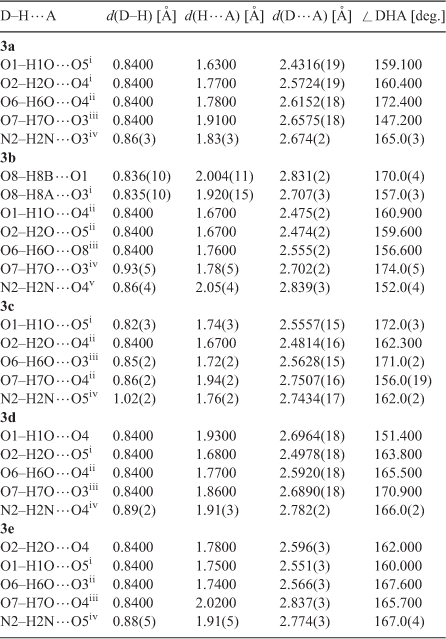
The P–O bonds in all compounds fall into two distinct classes. One contains the unprotonated oxygen atoms, i.e. P=O or P–O–, which ranges from 1.48 to 1.53 Å in length, suggesting a resonant partial double bond character (1.45–1.50 Å),[21] the other contains the protonated oxygen atoms, i.e. P–OH, which ranges from 1.53 to 1.58 Å in length, representing a single bond character (1.56–1.68 Å).[21] This is in good agreement with previous studies.[12d,20] No significant discrepancy was observed between the double bond (P=O) and single bond (P–O), indicating the importance of resonance effects in these systems. Moreover, the P–O bonds having oxygen atoms involved in strong hydrogen bonds are considerably shorter than those having the oxygen atom involved in weak hydrogen bonds. For example, in 3a the bond length of P1–O1 is 1.5277(13) Å, while those of P1–O2 and P2–O6 are 1.5622(13) and 1.5703(13) Å, respectively (see Table 2). It changes in the order of P1–O1 < P1–O2 < P2–O6, consistent with the variation of the corresponding hydrogen bond length O1–H1O⋯O5i < O2–H2O⋯O4i < O6–H6O⋯O4ii (see Table 3, which has listed the hydrogen bond details). The same is true for compounds 3b–e.
|
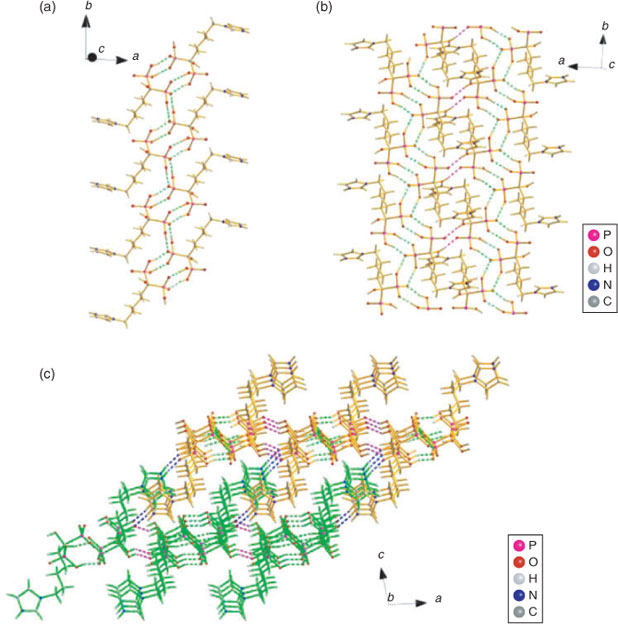
Molecules in the crystals 3a–d pack as tetramers while those in 3e as octamers, which are formed mainly due to the intermolecular hydrogen bonding interactions. The hydrogen bonding scheme for 3a–e consists of O–H⋯O and N–H⋯O interactions (Table 3) where the O⋯O and N⋯O distances are far less than the sum of the van der Waals radii of 3.04 and 3.07 Å, respectively,[22] which play an important role in stabilising the layered networks of all the crystals. The phosphonate oxygen atoms (O1, O2, and O6) and the hydroxy oxygen atom (O7) as well as the protonated nitrogen atom (N2) of the imidazole ring all serve as the hydrogen bond donors, while the phosphonyl oxygen atoms (O3, O4, and O5) in the neighbouring molecule act as the hydrogen bond receptors. In compound 3a, dimers are first generated by the symmetrical hydrogen bonds O1–H1⋯O5 and O2–H2⋯O4 between two phosphonate groups of two independent molecules, which act as a cyclic motif 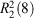 determining the formation of two eight-membered rings O4⋯H2–O2–P1–O1–H1⋯O5–P2. The dimers are then linked together and continue into a tetramer by another hydrogen bond O6–H6⋯O4 as a cyclic motif , leading to the formation of a new kind of eight-membered ring O6–H6⋯O4–P2–O6–H6⋯O4–P2. This generates a one-dimensional (1D) chain of dimers running along the b direction (Fig. 3a). The cohesion between chains of dimers is further developed by O7 and N2 by cooperative hydrogen bonds with O3, O7–H7O⋯O3iii and N2–H2N⋯O3iv, which determines the formation of two-dimensional (2D) layered network and three-dimensional (3D) supramolecular framework, respectively (see Figs 3b and 3c).
determining the formation of two eight-membered rings O4⋯H2–O2–P1–O1–H1⋯O5–P2. The dimers are then linked together and continue into a tetramer by another hydrogen bond O6–H6⋯O4 as a cyclic motif , leading to the formation of a new kind of eight-membered ring O6–H6⋯O4–P2–O6–H6⋯O4–P2. This generates a one-dimensional (1D) chain of dimers running along the b direction (Fig. 3a). The cohesion between chains of dimers is further developed by O7 and N2 by cooperative hydrogen bonds with O3, O7–H7O⋯O3iii and N2–H2N⋯O3iv, which determines the formation of two-dimensional (2D) layered network and three-dimensional (3D) supramolecular framework, respectively (see Figs 3b and 3c).
|
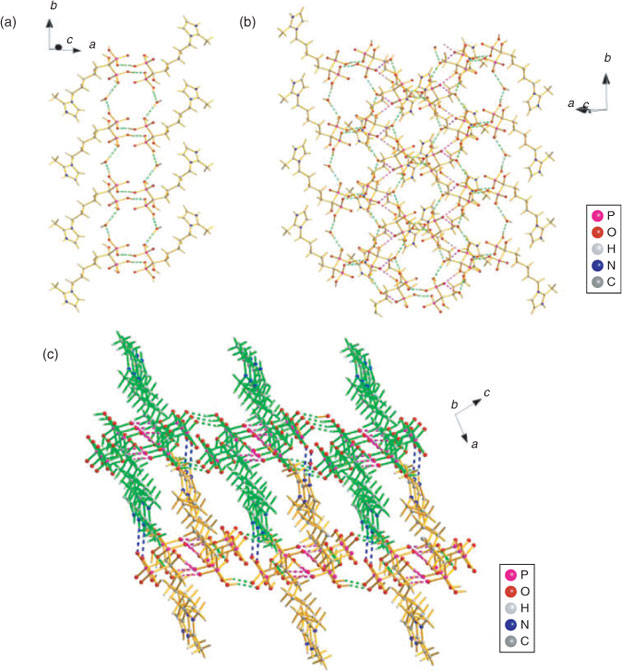
In 3b, extensive hydrogen bonding interactions are also observed among the phosphonate groups, hydroxy groups, and protonated nitrogen atoms. In addition, the lattice water molecule also provides hydrogen bonds between molecules in the tetramer, which not only serves as the hydrogen bond donor but also acts as the hydrogen bond receptor (see Fig. 4). First, the O1–H1O⋯O4ii and O2–H2O⋯O5ii hydrogen bonds between neighbouring phosphonate oxygen atoms act as a motif 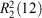 and connects the zwitterions of 3b into dimers. The dimers are then linked together by cooperative hydrogen bonds with the lattice water oxygen atom (O8) through one donor contact O8–H8B⋯O1 and a receptor contact O6–H6⋯O8, forming a chain of dimers along the b axis (Fig. 4a). The hydrogen bonds from adjacent chains, O8–H8A⋯O3i and O7–H7O⋯O3iv, lead to the formation of a 2D layered network, where the lattice water molecule gives further cohesion to this entity (Fig. 4b). Finally, the 2D layers are further stabilised by the hydrogen bonds N2–H2N⋯O4v and assembled into a 3D supramolecular network (Fig. 4c).
and connects the zwitterions of 3b into dimers. The dimers are then linked together by cooperative hydrogen bonds with the lattice water oxygen atom (O8) through one donor contact O8–H8B⋯O1 and a receptor contact O6–H6⋯O8, forming a chain of dimers along the b axis (Fig. 4a). The hydrogen bonds from adjacent chains, O8–H8A⋯O3i and O7–H7O⋯O3iv, lead to the formation of a 2D layered network, where the lattice water molecule gives further cohesion to this entity (Fig. 4b). Finally, the 2D layers are further stabilised by the hydrogen bonds N2–H2N⋯O4v and assembled into a 3D supramolecular network (Fig. 4c).
|
In 3c, the symmetrical hydrogen bonds O1–H1O⋯O5i between neighbouring phosphonate groups of two adjacent molecules form a cyclic motif and connect the zwitterions of 3c into dimers. The dimers are then linked together and continue into a 1D zigzag chain along the b axis by the hydrogen bonds O6–H6O⋯O3iii between another two phosphonate oxygens, which also act as a cyclic motif (Fig. 5a). The cohesion between chains of dimers is further developed by O2 and O7 by cooperative hydrogen bonds with O4, O2–H2O⋯O4ii and O7–H7O⋯O4ii, determining the formation of a 2D layered network (Fig. 5b). Finally, a 3D supramolecular network is further assembled by the strong hydrogen bonds between the protonated N2 atoms and phosphonate oxygen atoms (N2–H2N⋯O5iv, 2.7434(17) Å) (Fig. 5c).
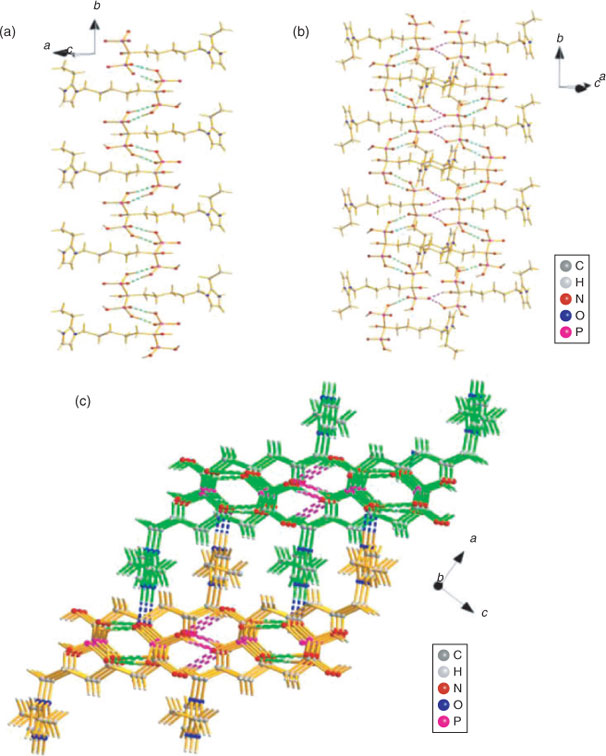
|
The hydrogen bonding observed in the crystals of 3d and 3e are quite similar to each other. The symmetrical hydrogen bonds O2–H2O⋯O5i (3d) and O6–H6O⋯O3ii (3e) between neighbouring phosphonate groups of two adjacent molecules produce a cyclic motif and connect the zwitterions of 3d/3e into dimers respectively. The dimers in 3d are then further assembled into a 1D zigzag chain along the c axis by O6–H6O⋯O4ii hydrogen bonds which act as a cyclic motif (Fig. 6a), while those in 3e are connected by the hydrogen bonds O1–H1O⋯O5i and also form a 1D zig-zag chain of dimers along the b axis (Fig. 7a). Both the 2D layered networks of 3d and 3e are formed by the interchain hydrogen bonds between the side hydroxy and the phosphonyl oxygens (3d: O7–H7O⋯O3iii, 2.6890(18) Å; 3e: O7–H7O⋯O4iii, 2.837(3) Å), see Figs 6b and 7b. As listed in Table 3, there are also strong N–H⋯O hydrogen bonding interactions between the protonated N2 atoms and phosphonate oxygen atoms in 3d and 3e (N2–H2N⋯O4iv, 2.782(2) Å and N2–H2N⋯O5iv, 2.774(3) Å), which further extend the 2D layers into the 3D supramolecular frameworks (Figs 6c and 7c).
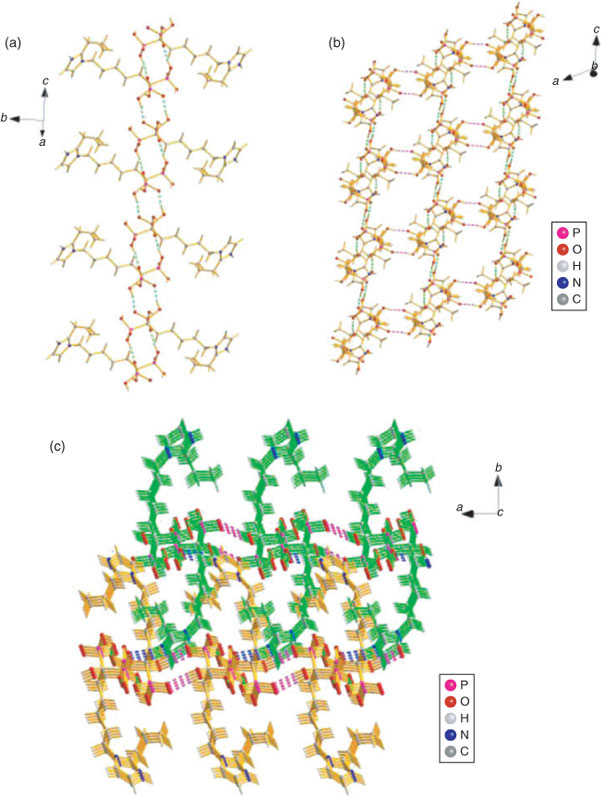
|
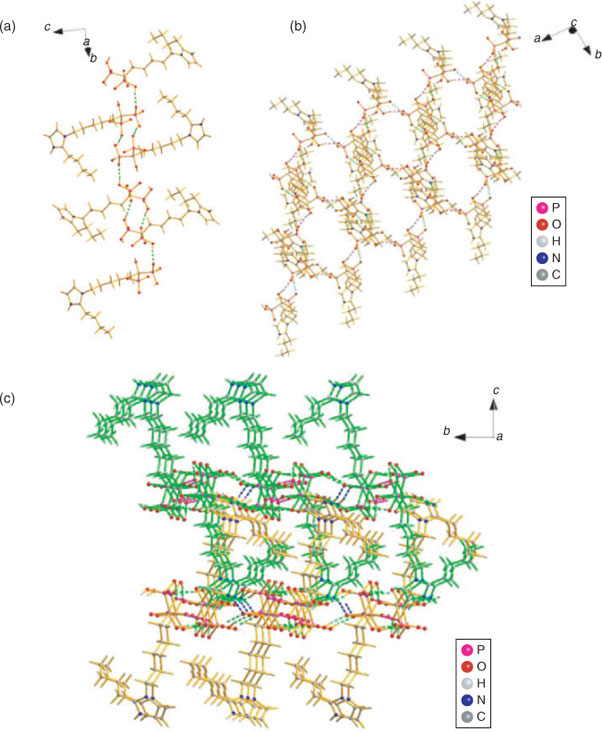
|
The structural parameters deduced from X-ray diffraction show correlations with the chemical and physical data measured for the compounds, such as melting point. For example, the strongest hydrogen bond occurs in 3a, O1–H1O⋯O5i (2.4316(19) Å), corresponding to the highest melting point of 217–220°C. It is followed by 3b with stronger hydrogen bonds (O1–H1O⋯O4ii, 2.475(2) Å and O2–H2O⋯O5ii, 2.474(2) Å), corresponding to a higher melting point of 214–216°C. The weakest hydrogen bonding occurs in 3e, O7–H7O⋯O4iii (2.837(3) Å), corresponding to the lowest melting point of 191–194°C, while 3c and 3d show an intermediate melting point (203–205 and 206–208°C) due to the intermediate hydrogen bonding (O2–H2O⋯O4ii, 2.4814(16) Å and O2–H2O⋯O5i, 2.4978(18) Å). Here, it should be pointed out that although the weakest hydrogen bond N2–H2N⋯O4v (2.839(3) Å) in 3b is slightly longer than the weakest hydrogen bond (O7–H7O⋯O4iii, 2.837(3) Å) in 3e, 3b has a higher melting point than 3e. This is because 3b has more and shorter intermolecular hydrogen bonds than 3e, which partly contribute to the additional cohesion caused by the lattice water molecules in 3b. Therefore, the hydrogen bonding in 3b is more compact than 3e. Clearly, the melting point dropping from 220 to 191°C indicates the hydrogen-bonded system varying from tight to loose for compounds 3a–e. This mainly originates from the differences in the alkyl group substituted at the neighbouring position of the protonated nitrogen atom, which may cause different stereo-hindrance effects.
HA-Binding Assay
From the above single-crystal XRD studies, it is known that extensive hydrogen bonding interactions exist among the phosphonate oxygens, hydroxy groups, and protonated nitrogen atoms as well as the lattice water molecules in compounds 3a–e. The existence of such abundant hydrogen bond donors and receptors leads to the concept that these novel BPs can interact strongly with the HA or bone mineral. So, the binding affinity of 3a–e for HA was investigated.
In the past, several methods have been developed for in vitro assay to measure binding affinities of small molecules to HA, such as X-ray photoelectron spectroscopy (XPS),[23] NMR spectroscopy,[24] fluorescent labelling,[25] and constant composition potentiostatic methods.[7] Also, a method based on fast performance liquid chromatography (FPLC) uses columns of HA, to which BPs and other phosphate-containing compounds can adsorb and be eluted.[26] Even though these methods can qualify binding affinities, a more accurate, precise, and economic method should be developed. In the present work, to quantify the bone targeting efficiency of the BPs 3a–e, a new method was set up using HPLC which employs the most common detection system and the simplest chromatography elution conditions found in experimental and clinical laboratories. In fact, it provides an accurate, precise, and robust method for quantitative analysis of these bisphosphates using UV detection and isocratic conditions.
Fig. 8 shows the adsorption profiles of 3a–e on HA as function of the concentration of BPs. The plots show that the amount of BP bound to HA increases up to a saturation level with the initial amount of each BP in solution increasing, and the binding characteristic follows the classical Langmuir adsorption isotherm. The results also clearly show that all the BPs studied in the present work have a strong affinity for HA. In their acid forms (pH 2.85), the maximum adsorption amounts of 3a–e are ~158.10, 111.10, 97.33, 91.35, and 83.59 μmol per gram of HA, respectively. So, the binding capacity appears to decrease in the order of 3a > 3b > 3c > 3d > 3e. The maximal amount of 3a bound to HA is much higher (nearly 1.5 to 1.9 fold) than those of 3b–e because of only a small difference in the substituent R2. As shown in the previous study,[26] when the risedronate was attached to an extra methyl group in the R2 side chain, the two BPs had different retention times (16.16 and 14.41 respectively) indicating the decrease of HA binding affinity. As detected by a constant composition potentiostatic method, Nancollas et al. demonstrated significant differences in the binding affinity of similar BPs to HA and carbonated apatite and established a rank order of ibandronate < alendronate < pamidronate.[7,27,28] So, the results further demonstrate that the nature of the R2 side chain has a significant influence on the binding affinity although they share a common P–C–P structure and OH at the R1 side chain. On the other hand, it is known that small differences in the R2 group lead to substantial changes in the 3D shapes and atomic orientations of the BPs according to the above single-crystal XRD studies. Therefore, it is postulated that the 3D configuration of the imidazolyl-containing BPs and the orientation of the imidazolyl group may play an important role in the mineral binding affinity.[8]
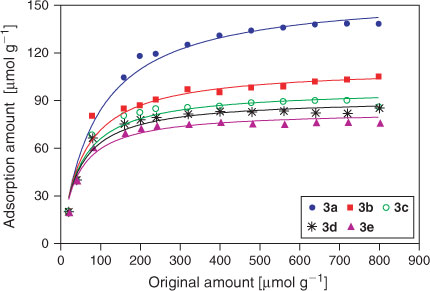
|
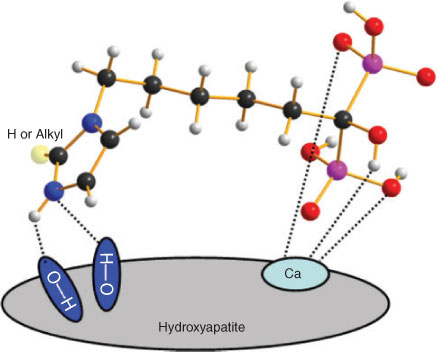
Based on the established interaction model of simpler BPs on HA where the tridentate coordination of the P–C–P ‘bone hook’ and R1 (–OH) domains interact with the calcium site,[8] there is the potential of additional interactions between the imidazolyl moiety in these more complex BPs with HA. As illustrated in Scheme 3, the protonated imidazole can interact with HA through the hydrogen bond N–H⋯O or N⋯H–O, which can optimally allow a bifurcated dual bonding arrangement. For optimal binding to HA, these hydrogen bonds require a N–H–O angle of larger than 125° and a N⋯O distance of approximately 3 Å.[8] This has been demonstrated by the optimal distance and angle of the N–H–O bond that can be formed with the alendronate –NH2 and zoledronate distal heteroaromatic nitrogen, which can account for their higher binding affinity compared with that observed for other BPs. Therefore, it is inferred that imidazolyl-containing BPs 3a–e, which can form any of these bonds, will have a higher affinity for bone mineral. However, although these BPs can theoretically orient properly for hydrogen bond formation, steric hindrance of different alkyl groups on the neighbouring position (N2) may interfere with these potential interactions, which may explain why 3b–e have a lower binding affinity than 3a. Thus, the difference in the 3D configuration allowing the BPs to form different N–H–O bonds can explain the differences observed in the HA binding affinity, which may in turn have an influence over the pharmacological potency and clinical effects of BPs.[8] Since weak binding on HA may translate to lower potency that necessitates a higher dose, we postulate that a higher affinity of BPs 3a–e for binding to HA may compensate for their low potency.
|
Conclusions
A series of novel imidazolyl-containing BPs were synthesised and identified by MS, IR spectroscopy, 1H NMR spectroscopy, XRD, and HPLC. The synthetic route was attractive for its advantages including readily available reagents and high purity of final products under easy operating conditions. There exists many strong hydrogen bonds in the crystal structures among the phosphonate oxygens, hydroxy groups, and protonated nitrogen atoms as well as the lattice water molecules to form 3D supramolecular frameworks. The HPLC method was developed to determine the HA-binding affinity of BPs and it provides an accurate, precise, and robust method for quantitative analysis. Compound 3a has the greatest affinity for HA with a maximal binding amount of 158.10 μmol g–1, which is nearly 1.5–1.9 fold that of 3b–e. These results provide a new example of how small differences in the BP structure can lead to significant changes in the bone binding affinity and capacity.
Supplementary Material
Standard concentration-area curves of BPs (Fig. 1S) and selected bond lengths and angles (Table S1) for compounds 3a–e can be found on the Journal’s website.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21001055 and 20801024), Natural Science Foundation of Jiangsu Province (BK2009077), and Key Medical Talent Project of Jiangsu Province (RC2011097).
References
[1] A. Jung, S. Bisaz, H. Fleisch, Calcif. Tissue Res. 1973, 11, 269.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3sXhsFGksLk%3D&md5=8f9dff8a3f92777b12f42a38a7ff1e45CAS | 4350498PubMed |
[2] G. A. Rodan, T. J. Martin, Science 2000, 289, 1508.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmt1Kgsb4%3D&md5=e5d2683b88b1fff31cf8e259d498a9b3CAS | 10968781PubMed |
[3] (a) S. Vasireddy, A. Talwarkar, H. Miller, Clin. Rheumatol. 2003, 22, 376.
| Crossref | GoogleScholarGoogle Scholar | 14677009PubMed |
(b) R. E. Coleman, E. V. McCloskey, Bone 2011, 49, 71.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Lipton, ExpertOpin. Pharmaco 2011, 12, 749.
[4] (a) E. Oldfield, Acc. Chem. Res. 2010, 43, 1216.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXns1Cnu7k%3D&md5=eaa324f7992400839a34337e22ba9280CAS | 20560544PubMed |
(b) R. G. Russell, Z. Xia, J. E. Dunford, U. Oppermann, A. Kwaasi, P. A. Hulley, K. L. Kavanagh, J. T. Triffitt, M. W. Lundy, R. J. Phipps, B. L. Barnett, F. P. Coxon, M. J. Rogers, N. B. Watts, F. H. Ebetino, N. Y. Ann. Acad. Sci. 2007, 1117, 209.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) D. M. Black, P. D. Delmas, R. Eastell, I. R. Reid, S. Boonen, J. A. Cauley, F. Cosman, P. Lakatos, P. C. Leung, Z. Man, C. Mautalen, P. Mesenbrink, H. Hu, J. Caminis, K. Tong, T. Rosario-Jansen, J. Krasnow, T. F. Hue, D. Sellmeyer, E.F. Eriksen, S. R. N. Cummings, Engl. J. Med. 2007, 356, 1809.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXltVSktLc%3D&md5=f9b7e5690a1152041f375a22bf376fa0CAS |
(b) G. Owens, R. Jackson, E. M. Lewiecki, Am. J. Manag. Care. 2007, 13, S290.
(c) R. C. Mühlbauer, F. Bauss, R. Schenk, M. Janner, E. Bosies, K. Strein, H. J. Fleisch, Bone. Miner. Res. 1991, 6, 1003.
| Crossref | GoogleScholarGoogle Scholar |
[6] O. L. M. Bijvoet, H. A. Fleisch, R. E. Canfield, R. G. G. Russell, Biophosphonate on Bones 1995 (Elsevier: Amsterdam).
[7] G. H. Nancollas, R. Tang, R. J. Phipps, Z. Henneman, S. Gulde, W. Wu, A. Mangood, R. G. G. Russell, F. Hebetino, Bone 2006, 38, 617.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjvFSjtr4%3D&md5=b1aa1fb1b636c78cf2cdc8b880961568CAS | 16046206PubMed |
[8] R. G. G. Russell, N. B. Watts, F. H. Ebetino, M. J. Rogers, Osteoporos. Int. 2008, 19, 733.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltFyjurw%3D&md5=ae744f39a33d6166189f17d10c24b30bCAS |
[9] J. E. Dunford, K. Thompson, F. P. Coxon, S. P. Luckman, F. M. Hahn, C. D. Poulter, F. H. Ebetino, M. J. J. Rogers, Pharmacol. Exp. Ther. 2001, 296, 235.
| 1:CAS:528:DC%2BD3MXoslaluw%3D%3D&md5=675109cd895372e72623bbbb3472df00CAS |
[10] M. R. Smith, Urol. Oncol: Semin. Orig. Investig. 2008, 26, 420.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXnvVyjurY%3D&md5=7988074847a4aef16d47aa34f2b309a6CAS |
[11] J. A. Kanis, B. J. Gertz, F. Singer, S. Ortolani, Osteoporos. Int. 1995, 5, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK2M3hvVelsg%3D%3D&md5=e6a1625b75aba40cee134596e2c5d259CAS | 7703618PubMed |
[12] (a) L. Widler, K. A. Jaeggi, M. Glatt, K. Müller, R. Bachmann, M. Bisping, A. R. Born, R. Cortesi, G. Guiglia, H. H. Jeker, R. Klein, U. Ramseier, J. Schmid, G. Schreiber, Y. Seltenmeyer, J. R. Green, J. Med. Chem. 2002, 45, 3721.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xltl2jtLw%3D&md5=58d1fdd1d646f924bee0a1c4132122c3CAS | 12166945PubMed |
(b) C. M. Szabo, M. B. Martin, E. Oldfield, J. Med. Chem. 2002, 45, 2894.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Zhang, A. Leon, Y. Song, D. Studer, C. Haase, L. A. Koscielski, E. Oldfield, J. Med. Chem. 2006, 49, 5804.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. H. Mao, S. Mukherjee, Y. Zhang, R. Cao, M. S. Ohn, Y. C. Song, Y. H Zhang, A. M. Gary, Y. G. Gao, D. Mukkamala, P. H. Michael, E. Oldfield, J. Am. Chem. Soc. 2006, 128, 14485.
| Crossref | GoogleScholarGoogle Scholar |
(e) R. T. M. Rosales, C. Finucane, S. J. Mather, P. J. Blower, Chem. Commun. 2009, 4847.
| Crossref | GoogleScholarGoogle Scholar |
(f) F. H. Ebetino, A. M. Hogan, S. Sun, M. K. Tsoumpra, X. Duan, J. T. Triffitt, A. A. Kwaasi, J. E. Dunford, B. L. Barnett, U. Oppermann, M. W. Lundy, A. Boyde, B. A. Kashemirov, C. E. McKenna, R. G. Russell, Bone 2011, 49, 20.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) X. H. Guo, S. N. Luo, H. Y. Wang, L. Zhou, M. H. Xie, W. Z. Ye, M. Yang, Y. Wang, Nucl. Sci. Technol. 2006, 17, 285.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtlelsL7P&md5=0502d0e96a2a94320f009ed03313a50eCAS |
(b) J. G. Lin, S. N. Luo, C. Q. Chen, L. Qiu, Y. Wang, W. Cheng, W. Z. Ye, Y. M. Xia, Appl. Radiat. Isot. 2010, 68, 1616.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. G. Lin, L. Qiu, W. Cheng, S. N. Luo, W. Z. Ye, Nucl. Med. Biol. 2011, 38, 619.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. Lin, L. Qiu, W. Cheng, S. N. Luo, L. Xue, S. Zhang, Appl. Radiat. Isot. 2012, 70, 848.
| Crossref | GoogleScholarGoogle Scholar |
(e) L. Qiu, J. G. Lin, S. N. Luo, Y. Wang, W. Cheng, S. Zhang, Radiochim. Acta 2012, 100, 463.
| Crossref | GoogleScholarGoogle Scholar |
[14] G. M. Sheldrick, SHELXS-97, Program for X-Ray Crystal Structure Solution 1997 (University of Götingen: Götingen, Germany).
[15] G. M. Sheldrick, SHELXL-97, Program for X-Ray Crystal Structure Refinement 1997 (University of Götingen: Götingen, Germany).
[16] (a) A. Cabeza, X. Ouyang, C. V. K. Sharma, M. A. G. Aranda, S. Bruque, A. Clearfield, Inorg. Chem. 2002, 41, 2325.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xit1Whu78%3D&md5=c8aef85e33c68169fcae2437e6be0501CAS | 11978094PubMed |
(b) H. S. Martinez-Tapia, A. Cabeza, S. Bruque, P. Pertierra, S. Garcia-Granda, M. A. G. Aranda, J. Solid State Chem. 2000, 151, 122.
| Crossref | GoogleScholarGoogle Scholar |
[17] B. Hemmateenejad, M. Shamsipur, A. Safavi, H. Sharghi, A. A. Amiri, Talanta 2008, 77, 351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFCit7zK&md5=f039ce9e30746e90415cf348642764c0CAS | 18804645PubMed |
[18] (a) K. Ravikumar, B. Sridhar, Acta Crystallogr. C 2010, 66, o97.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlemur0%3D&md5=b1660366775972c7329399d697f690a7CAS | 20124689PubMed |
(b) S. J. Li, W. D. Song, S. H. Li, J. J. Dong, J. B. Yan, Acta Crystallogr. 2010, E66, m1094.
[19] R. Ruscica, M. Bianchi, M. Quintero, A. Martinez, D. R. Vega, J. Pharm. Sci. 2010, 99, 4962.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlKjsbfN&md5=e46e9b20d8b9bbe3e2457968f0316368CAS | 20533554PubMed |
[20] (a) D. R. Vega, D. Fernández, J. A. Ellena, Acta Crystallogr. C 2002, 58, m77.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. Fernández, D. R. Vega, Acta Crystallogr. C 2003, 59, o228.
| Crossref | GoogleScholarGoogle Scholar |
(c) W. L. Gossman, S. R. Wilson, E. Oldfield, Acta Crystallogr. C. 2003, 59, m33.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Wu, S. Chen, M. Li, J. Xiang, Y. Xiao, L. Yuan, CrystEngComm 2007, 9, 907.
| Crossref | GoogleScholarGoogle Scholar |
[21] F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen, R. Taylor, J. Chem. Soc. Perkin Trans. 2 1987, II, S1.
[22] (a) A. Bondi, J. Phys. Chem. 1964, 68, 441.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2cXls1Cgsg%3D%3D&md5=b78ccfb7bc6298500d4f04a6db47751aCAS |
(b) Y. Wang, T. Okamura, W. Y. Sun, N. Ueyama, Cryst. Growth Des. 2008, 8, 802.
| Crossref | GoogleScholarGoogle Scholar |
[23] K. McLeod, G. I. Anderson, N. K. Dutta, R. S. C. Smart, N. H. Voelcker, R. Sekel, S. Kumar, J. Biomed. Mater. Res. A 2006, 79A, 271.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFarsrfO&md5=cc65180aaf458ff8c0cc058fe7801694CAS |
[24] W. Jahnke, C. Henry, ChemMedChem 2010, 5, 770.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVOltLs%3D&md5=e89796bd8c1551692e56d044176ca7f7CAS | 20209564PubMed |
[25] V. Hengst, C. Oussoren, T. Kissel, G. Storm, Int. J. Pharm. 2007, 331, 224.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsFCltr4%3D&md5=21e13de3616062a344edabeebae45321CAS | 17150316PubMed |
[26] M. A. Lawson, Z. Xia, B. L. Barnett, J. T. Triffitt, R. J. Phipps, J. E. Dunford, R. M. Locklin, F. H. Ebetino, R. G. G. Russell, J. Biomed. Mater. Res. B. 2010, 92, 149.
| 1:STN:280:DC%2BD1MfhvVGltA%3D%3D&md5=25ad7e907dd505252f1b58f7af7bed10CAS |
[27] F. H. Ebetino, P. Emmerling, B. Barnett, G. H. Nancollas, J. Bone Miner. Res. 2004, 19, S157.
[28] Z. J. Henneman, G. H. Nancollas, F. H. Ebetino, R. J. Phipps, R. G. G. Russell, J. Biomed. Mater. Res. A 2008, 85A, 993.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmsVGqsrY%3D&md5=e026886abc04f8fd82734ad7f43c0c7aCAS |