

Influence of A-site Cation on Structure and Dielectric Properties in A2DyBiO6 (A = Mg, Ca, Sr, Ba) Double Perovskites
Simona Feraru A , Adrian I. Borhan A , Petrisor Samoila B , Gigel G. Nedelcu C , Alexandra R. Iordan A and Mircea N. Palamaru A DA Faculty of Chemistry, Alexandru Ioan Cuza University of Iasi, Iasi, 700506, Romania.
B Petru Poni Institute of Macromolecular Chemistry, Iasi, 700487, Romania.
C Faculty of Physics, Alexandru Ioan Cuza University of Iasi, Iasi, 700506, Romania.
D Corresponding author. Email: palamaru@uaic.ro
Australian Journal of Chemistry 67(2) 250-255 https://doi.org/10.1071/CH13300
Submitted: 14 June 2013 Accepted: 18 September 2013 Published: 17 October 2013
Abstract
Double perovskite metal oxides with formula A2DyBiO6 (A = Mg, Ca, Sr, Ba) were synthesised by a sol–gel auto-combustion method, using citric acid as the combustion agent. The effects of A-site cation on the structure, morphology, and dielectric properties were examined. The synthesis was monitored using Fourier transform infrared spectroscopy (FTIR) to indicate the absence of organic phase. X-ray diffraction (XRD) analysis showed that the compounds have three different perovskite structures. Structural characterisation of the samples was evaluated using XRD patterns. Scanning electron microscopy showed that all samples are formed by agglomerated particles. Dielectric properties were evaluated using dielectric permittivity and dielectric losses. Cole–Cole plots show a single semicircle for all materials, indicating that the double perovskites obtained are composed of well conducting grain boundaries and poorly conducting grains.
Introduction
Double perovskites, with A2BB′O6 formula, are formed by doubling the unit cell of a simple perovskite-type, ABO3. These materials have been intensively studied in recent years, especially due to their capacity to incorporate many elements such as alkaline earth (A-site), transition metal, or lanthanide cations (B, B′-site) in their structures. Therefore, the structure of these materials can change due to octahedral tilting (BO6, B′O6) to build a double perovskite-type lattice.[1] This leads to the possibility of preparing materials for specific applications. These features are very important for many magnetic materials[2,3] and electronic devices that can be operated successfully in a wide range of frequencies.[4] These double perovskites are also used in various fields such as catalysis,[5] photocatalysis,[6] and solid oxide fuel cell (SOFC) materials.[7]
Few reports have been made about double perovskites with Bi-cation on the B′-site. Byeon et al.[8] studied the synthesis and characterisation of Ba2BiVFeIIIO6. Macquart and Kennedy[9] investigated the structure of Ba2-xSrxLuBiO6 double perovskite. Harrison et al.[10] reported the synthesis, structural characterisation, and magnetic properties of Ba2MBiO6 (M = Ce to Lu) double perovskites. Lenz and Müller-Buschbaum[11] studied the structures of Ba2BiMO6, (M = Y, Dy) double perovskites and concluded that these compounds exhibit a cubic structure with a Fm-3m space group. Horyń et al.[12] studied Sr2REBiO6 (RE = La and lanthanides) double perovskites and reported that Sr2DyBiO6 has a monoclinic structure with a P21/n space group. In all of these studies, the compounds were prepared using a solid-state reaction method, except for the synthesis of Ba2BiVFeIIIO6 by Byeon et al.,[8] which was carried out under a high pressure oxygen atmosphere.
In a previous paper, we studied the compound, Ca2DyBiO6.[13] Electrical measurements revealed that this compound was the best dielectric material, displaying a dielectric constant of 40 with very low dielectric loss. Therefore, starting from Ca2DyBiO6, the A-site cation was substituted in order to improve its electrical properties.
Hence, the aim of the present work is to study, for the first time, the effect of the A-site cation on the structure and dielectric properties in A2DyBiO6 (A = Mg, Ca, Sr, Ba) double perovskite-type oxides. An important technique for double perovskite preparation is the sol–gel method because this procedure allows the control of chemical composition, solid texture, size, and shape of the particles.[14] In this study, the materials are obtained by a sol–gel auto-combustion method using citric acid as a chelating and fuel agent. Our results showed that the dielectric properties were improved with larger ionic radius cation substitution at the A site.
Experimental
Nanopowders of A2DyBiO6 (A = Mg, Ca, Sr, Ba) double perovskite-type oxides were prepared in air using a sol–gel auto-combustion method with citric acid C6H8O7 (Merck) as a chelating and fuel agent. Analytical grade precursors, Mg(NO3)2·4H2O (Merck), Ca(NO3)2·4H2O (Merck), Sr(NO3)2·4H2O (Merck), Ba(NO3)2·4H2O (Merck), Dy(NO3)3·6H2O (Merck), and Bi(NH3)2C6H7O7·H2O (Merck) were mixed in stoichiometric proportions. A solution of citric acid was mixed with metal nitrate mixtures in a 3 : 1 molar ratio of citric acid to metallic cation. The solutions obtained were brought to an alkaline pH to avoid precipitation. For A2DyBiO6 (A = Mg, Ca, Sr, Ba) double perovskite-type oxides, the following chemical equation could be written:
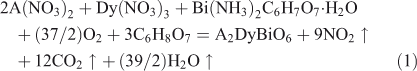
Eqn 1 is a proposed equation following a stoichiometric calculation that takes into account the redox process. The required amounts of metal salts to obtain the desired compounds have not been determined based on Eqn 1, but based on the mass reports, considering the molecular mass of general formula proposed.
The mixed solutions were heated up to 353 K under continuous stirring over a water bath in order to obtain viscous gels. The dried gels were gradually heated on a sand bath at 623 K for 1 h, when the auto-combustion was clearly observed and all the gels were converted to powders. The obtained powders were thermally treated in three steps: up to 773 K for 7 h, up to 973 K for 7 h, and up to 1173 K for 9 h.
Infrared (IR) and X-ray diffraction (XRD) analyses were recorded in powder form at different sintering temperatures. The progress of the synthesis and the disappearance of the organic phase were monitored by IR spectroscopy using a Bruker spectrophotometer TENSORTM 27-type with Fourier transform with an ATR cell at 2 cm–1 resolution in the IR range of 4000 to 400 cm–1. The double perovskite phase formation was established using powder X-ray diffraction, recorded by means of a Shimadzu LabX 6000 diffractometer equipped with graphite monochromator and CuKα (λ = 1.5406 Å) radiation. The samples fixed in the reflection mode were analysed under ambient atmosphere with a scanning rate of 0.02° s–1 over the 2θ = 20–80° range.
The morphology of the sintered powders at 1173 K was investigated by scanning electron microscopy (SEM) using a Vega Tescan-type scanning transmission electron microscope. The micrographs were made in fracture, i.e. powders heated at 1173 K were pressed into pills without other thermal treatment.
The dielectric measurements were carried out using the two-terminal direct current method. The dependence of dielectric permittivity and dielectric losses on frequency was studied using an Agilent 4292–1 device, in the range of 40 Hz to 10 MHz. The powders were thermally treated at 1173 K, pressed into disks at 400 kPa cm–2 without subsequent calcinations, and were inserted between two electrodes.
Results and Discussion
IR Results
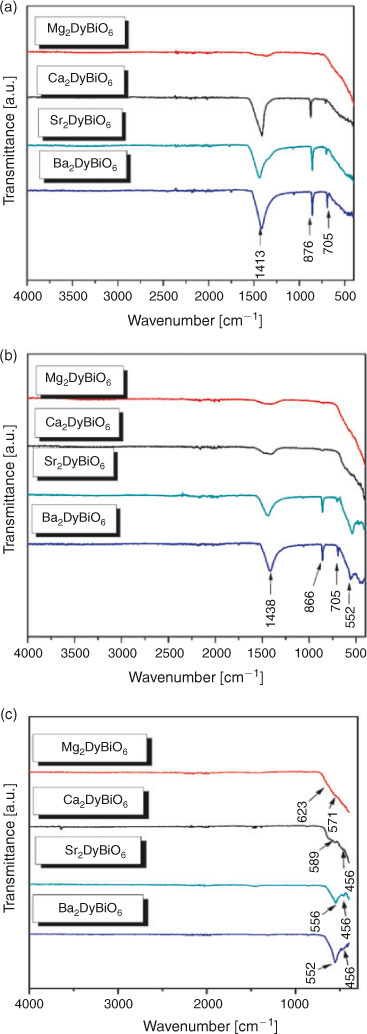
IR spectra for the A2DyBiO6 samples treated at 773, 973, and 1173 K, recorded at room temperature, are presented in Fig. 1a–c.
|
IR spectra of pure metal oxide samples are very similar, showing two bands characteristic to the symmetric (υs) and anti-symmetric (υas) octahedral BiO6 stretching vibrations. The shift of stretching vibration to lower wavelengths is due to stronger interactions between the cation and oxygen atoms (owing to the electronegativity of the metal).[15,16]
Therefore, the IR spectra at 773 (Fig. 1a) and 973 K (Fig. 1b) depict the specific symmetric (υs) and anti-symmetric (υas) bands for metal–oxygen stretching vibration in the range of 900 to 400 cm–1 and the absorption peak of nitrate groups at around 1417 cm–1.
Furthermore, the IR spectra for A2DyBiO6 samples heated at 1173 K (Fig. 1c) showed the disappearance of the nitrate groups. Note the decrease in intensity of the symmetric (υs) band with the introduction of cations with smaller ionic radii in the system. Also, the anti-symmetric υas stretching vibration (589 to 552 cm–1) of double perovskite-type oxide[15] characteristic of octahedral BiO6, and the bands at 437 cm–1characteristic of Dy–O stretching vibration,[17] become predominant with increasing temperature. This asymmetric band intensity variation leads to the idea that the Bi–O bond strength increases with increasing ionic radii of A2+cations, which means stabilisation of compound structures.
The presence of two lower bands at 623 and 571 cm–1 can also be seen in the IR spectra of Mg2DyBiO6, which can be attributed to the characteristic bands of a pyrochlore structure.[18]
XRD Results
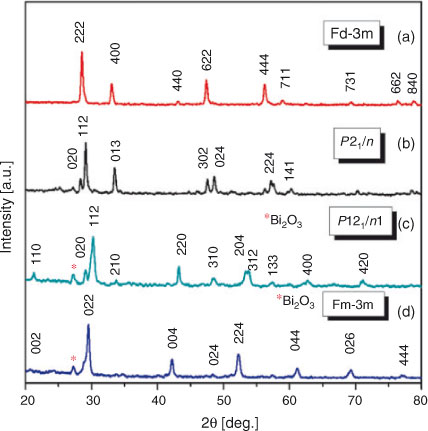
XRD patterns of A2DyBiO6 materials heated at 1173 K are presented in Fig. 2a–d. According to literature data, Ca2DyBiO6 and Sr2DyBiO6 double perovskites were found to have monoclinic structures with P21/n[19] and P121/n1[12] space groups, respectively. The strongest reflection peak at a 2θ value of around 30° identified in Ca2DyBiO6 and Sr2DyBiO6 samples are assigned to (1 1 2). Mg2DyBiO6 double perovskite is indexed as a defective cubic pyrochlore structure with a Fd-3m space group,[20] while Ba2DyBiO6 double perovskite crystallised in cubic system has a Fm-3m space group.[11] Unfortunately, the existence of a secondary phase can be observed for the Mg and Ba samples and this was identified as Bi2O3 (cubic system, COD1010313), according to the diffraction peak at 2θ = 27.3°.
|
Crystallite size (D) was calculated from XRD using the Debye–Scherrer[21] equation (Eqn 2):

where D = crystallite size, λ = wavelength of X-ray source (CuKα = 1.5405 Å), β1/2 (rad) = half maximum of diffraction plane (h k l), θ = angle of diffraction.
The pseudo-cubic pyrochlore structure of the Mg2DyBiO6 compound contains large holes that can lead to structural defects and therefore defaults to higher crystallites. A distorted structure leads to crystallite (D) with larger size, as can be seen in Table 1.

|
The best ordered structure was obtained in the case of the Ba2DyBiO6 compound, which has smaller sizes of crystallites (28 nm).
SEM Results
Fig. 3a–d shows the SEM images for all samples heated at 1173 K. All micrographs were made in fracture (powders thermally treated at 1173 K were pressed into pills without other additional thermal treatment). It can be observed that the materials are formed by agglomerated particles. Generally, the decrease in particle size is directly proportional to the increase in specific surface of these oxides. The irregular variation of SBET values for the studied samples (3 m2 g–1 for Mg2DyBiO6, 1 m2 g–1 for Ca2DyBiO6, 12 m2 g–1 for Sr2DyBiO6, and 6 m2 g–1 for Ba2DyBiO6) can be due to the different degrees of agglomeration of grains and number of fine pores. From the SEM image of the sample with Mg, it can be seen that the particles are crowded close together and almost ‘glued’ to each other. The presence of holes is most likely caused by the exhaust gases from the combustion process.

|
Dielectric Results
Fig. 4 a, b depicts the variation of real parts (ϵ′) and imaginary parts (ϵ″) of dielectric constant with frequency. The dielectric constant dependence of the frequency for all A2DyBiO6 (A = Mg, Ca, Sr, Ba) double perovskites were studied in the frequency range of 40 Hz to 10 MHz at room temperature. It can be clearly observed that all obtained materials exhibit a normal dielectric dispersion because the dielectric constants (ϵ′ and ϵ″) decrease with increasing frequency. One can observe that the dielectric constant decreases with increasing frequency, reaching a constant value at high frequencies. This variation can be observed in all of the sample cases. This phenomenon is a normal dielectric dispersion caused by electron displacement as a response to an applied field or defects and oxygen vacancies, known as Maxwell–Wagner-type interfacial polarisation.[22] By changing A in A2DyBiO6 compounds, the dielectric constant also changes. It is observed that Ba2DyBiO6 exhibits higher values of dielectric constant at all frequencies. Compared with the rest of the samples, Ba2DyBiO6 shows a much higher value of dielectric constant and the decrease in dielectric constant as a function of frequency is much sharper. It can be seen that in the case of Mg2DyBiO6, the dielectric constant reached the linear plateau at low frequency, probably caused by the defects presented in the pyrochlore structure. According to the experimental range of frequencies reported in the literature, the dielectric constant (ϵ′) of these defective pyrochlore materials is predominantly determined by the contribution of ionic polarisation.[20] In other double perovskite structure-containing samples, the dipole follows the field at low frequencies, but as the frequency increases, the dipoles start lagging behind the field and the real part (ϵ′) of the dielectric constant decreases. At high frequencies, the dielectric constant becomes independent of frequency, as reported by Laishram et al.,[23] because the dipoles can no longer follow the field.

|
The experimental data indicate that by changing A from Ca to Sr and Ba the permittivity increases, which may be related to the decrease in concentration of dipoles[20] due to the increase in unit-cell volume.
The variation of dielectric loss values with log (frequency) of all materials are shown in Fig. 4c. It can be seen that the dielectric loss values range from 0.03 to 0.68 at 10 MHz. It is clear that Mg2DyBiO6 has smaller loss values throughout the range of frequencies. In perovskites, an important contribution to high losses is attributed to oxygen vacancies, usually present in such systems. The increase in the real part of dielectric constant values and of tan δ, particularly at lower frequencies, by changing A from Ca to Sr and Ba, indicates important contributions from conductivity relaxation combined with Maxwell–Wagner phenomena[23] to the total dielectric response.
All double perovskite samples show values of permittivity ranging between one and seven, and low losses at room temperature in the whole range of frequencies, indicating good dielectric character of these materials compared with that from an earlier report by Vijayakumar et al.[24]
Fig. 5a–d shows the Cole–Cole plots for all compounds obtained by plotting the variation of imaginary part (ϵ″) versus real part (ϵ′) of the dielectric constant. It can be observed that the Cole–Cole plots for all samples are characterised by one semicircle. It can be noted that the grain boundary resistance contributed to the dielectric measurements and is directly proportional to the polarisability of the studied samples. From the variation of the dielectric constant, it can be observed that the values of grain boundary resistance increase with increasing divalent cation (A2+) ionic radius.

|
Conclusions
A2DyBiO6 (A = Mg, Ca, Sr, Ba) double perovskites were successfully prepared by a sol–gel auto-combustion method using citric acid as a combustion agent.
The IR spectra showed bands characteristic of metal–oxygen bonds in the perovskites. XRD revealed the formation of double perovskite phases at 900°C containing the minor impurity Bi2O3 in Sr2DyBiO6 and Ba2DyBiO6 double perovskites. Ca2DyBiO6 and Sr2DyBiO6 were found to have monoclinic structures, whilst Ba2DyBiO6 had a cubic structure, and Mg2DyBiO6 was found to have a defective pseudo-cubic pyrochlore structure. SEM images revealed the formation of holes in the structure of Mg2DyBiO6 double perovskite, probably caused by the gases from the combustion process. Microstructures of Ca2DyBiO6, Sr2DyBiO6, and Ba2DyBiO6 compounds showed homogenous crowded particles.
All double perovskite samples showed values of permittivity ranging between one and seven and low losses at room temperature in the whole range of frequencies, indicating good dielectric character of these materials. Cole–Cole plots show a single semicircle for all materials, which means that all polycrystalline double perovskites are composed of well-conducting grain boundaries and poorly conducting grains.
Acknowledgements
This work was supported by the European Social Fund in Romania, under the responsibility of the Managing Authority for the Sectorial Operational Programme for Human Resources Development 2007–2013 (grant POSDRU/107/1.5/S/78342), and the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 264115. STREAM is kindly acknowledged by PS.
References
[1] R. Shaheen, J. Bashir, Solid State Sci. 2010, 12, 1496.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpt1altrs%3D&md5=f8f28fac4ef90763caddd3b115d0120fCAS |
[2] C. M. Bonilla, D. A. L. Téllez, J. A. Rodríguez, E. V. López, J. Roa-Rojas, Physica B 2007, 398, 208.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvF2ltrg%3D&md5=55a7ea61470ac57784b8546dc86dac43CAS |
[3] B. García-Landa, C. Ritter, M. R. Ibarra, J. Blasco, P. A. Algarabel, R. Mahendiran, J. García, Solid State Commun. 1999, 110, 435.
| Crossref | GoogleScholarGoogle Scholar |
[4] R. V. K. Mangalam, E. Suard, A. Sundaresan, Physica B 2009, 404, 154.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFahtbbP&md5=850474fcf703966cebea651857ae2a4cCAS |
[5] R. Hu, R. Ding, J. Chen, J. Hu, Y. Zhang, Catal. Commun. 2012, 21, 38.
| Crossref | GoogleScholarGoogle Scholar |
[6] T. Hatakeyama, S. Takeda, F. Ishikawa, A. Ohmura, A. Nakayama, Y. Yamada, A. Matsushita, J. Yea, J. Ceram. Soc. Jpn. 2010, 118, 91.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXitleltLo%3D&md5=8e4dba65ed4176def82971300b213297CAS |
[7] D. Marrero-López, J. Peña-Martínez, J. C. Ruiz-Morales, M. Gabás, P. Núñez, M. A. G. Aranda, J. R. Ramos-Barrado, Solid State Ionics 2010, 180, 1672.
| Crossref | GoogleScholarGoogle Scholar |
[8] S. H. Byeon, C. G. Demazeau, H. J. Choy, C. L. Fournes, Mater. Lett. 1991, 12, 163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XltVWltw%3D%3D&md5=0cc74110af2345ba91263d47b6a56effCAS |
[9] R. B. Macquart, B. J. Kennedy, Chem. Mater. 2005, 17, 1905.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXitFegu70%3D&md5=e2d1fede68645ed7278726716e9f2343CAS |
[10] T. A. W. Harrison, P. J. K. Reis, J. A. Jacobson, F. L. Schneemeyer, V. J. Waszczak, Chem. Mater. 1995, 7, 2161.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXovFChurk%3D&md5=6a1229f39e45257a07994521c872cc7eCAS |
[11] A. Lenz, H. K. Müller-Buschbaum, J. Less-Common Met. 1990, 161, L15.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXlt1CgtLk%3D&md5=2a7753b2f791601d74a80a861d9b69c4CAS |
[12] R. Horyń, M. Wotcyrz, A. Wojakowski, A. J. Zaleski, J. Alloy. Compd. 1996, 242, 35.
| Crossref | GoogleScholarGoogle Scholar |
[13] S. Feraru, P. M. Samoila, V. Nica, A. R. Iordan, M. N. Palamaru, Chem. Pap. – Chem. Zvesti 2013, 67, 1311.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXoslWgs7Y%3D&md5=cff421395de045d4382fc22a1bcff8abCAS |
[14] S. Z. Tian, J. C. Zhao, C. D. Qiao, X. L. Ji, B. Z. Jiang, Mater. Lett. 2006, 60, 2747.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmslChu7o%3D&md5=26693de3b2c34251ead5601b8eb15cdeCAS |
[15] A. E. Lavat, J. E. Baran, Vib. Spectrosc. 2003, 32, 167.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXms1ahu7w%3D&md5=f5c540d1c0002b63897ccb53860072e3CAS |
[16] A. E. Lavat, J. E. Baran, J. Alloy. Compd. 2008, 460, 152.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmvVyrsrY%3D&md5=9473393056351fd62352ae557e8e3e7eCAS |
[17] C. Vijayakumar, H. P. Kumar, S. Solomon, J. K. Thomas, P. R. S. Warian, A. John, J. Alloy. Compd. 2009a, 480, 167.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnt1egsrY%3D&md5=2e8d7d6734971a32edf3e4f937eab154CAS |
[18] M. Mączka, A. V. Knyazev, Yu. N. Kuznetsova, M. Ptak, L. Macalik, J. Raman Spectrosc. 2011, 42, 529.
| Crossref | GoogleScholarGoogle Scholar |
[19] A. E. Lavat, J. E. Baran, Mater. Res. 2011, 14, 472.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XivVyitro%3D&md5=0aa92a7bee7acda16119643d0cf46789CAS |
[20] S. Senthil Kumar, K.V.O. Nair, J. James, J. Solid State Chem. 2004, 177, 3873.
| Crossref | GoogleScholarGoogle Scholar |
[21] C. A. Triana, D. A. Landiz Téllez, J. Arbey Rodríguez, F. Fajardo, J. Roa-Rojas, Mater. Lett. 2012, 82, 116.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpsVOhtLs%3D&md5=cad1c86ea4ccb8822211bfa2b3e4717bCAS |
[22] P. K. Jana, S. Sudipta, B. K. Chaudhuri, J. Phys. D: Appl. Phys. 2007, 40, 556.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVygt74%3D&md5=b51c69bd3f610a5f901bea3a25e9ad75CAS |
[23] R. Laishram, S. Phanjoubam, H. N. K. Sarma, C. Prakash, J. Phys. 1999, D32, 2151.
[24] C. Vijayakumar, H. Padma Kumar, V. T. Kavitha, S. Solomon, J. K. Thomas, P. R. S. Wariar, J. Koshy, J. Alloy. Compd. 2009b, 475, 778.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXktlymsLY%3D&md5=3038ccbc1d45fac9a7905032ae2e6acaCAS |