

Phenotyping CHST3 skeletal dysplasia from freezer-induced urine sediments
Edward S. X. Moh A , Andreas Zankl B C D and Nicolle H. Packer A *
A , Andreas Zankl B C D and Nicolle H. Packer A *
A ARC Centre of Excellence for Synthetic Biology, School of Natural Sciences, Faculty of Science and Engineering, Macquarie University, North Ryde, Sydney, NSW 2109, Australia.
B Department of Clinical Genetics, The Children’s Hospital at Westmead, Sydney, NSW 2145, Australia.
C Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia.
D Bone Biology Division and Kinghorn Centre for Clinical Genomics, Garvan Institute of Medical Research, Sydney, NSW 2010, Australia.
Australian Journal of Chemistry 76(8) 476-481 https://doi.org/10.1071/CH23041
Submitted: 24 February 2023 Accepted: 5 May 2023 Published: 31 May 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Skeletal dysplasias are a group of rare genetic disorders that affect growth and development of the skeleton, leading to physical deformities and other medical problems. High-throughput genome sequencing technologies have made it easier to genotype the disorder, but do not always reflect the phenotypic outcome. CHST3-related skeletal dysplasia is caused by the reduced function of the carbohydrate sulfotransferase that sulfates chondroitin sulfate glycosaminoglycans. We show in this pilot study that we were able to phenotype patients with CHST3-related skeletal dysplasia by profiling the glycosaminoglycans and identifying their potential protein carriers sequentially using freezer-induced patient urine sediments.
Keywords: carbohydrate sulfotransferase 3, clinical sample collection, Congenital disorders of glycosylation, freezer-induced urine sediments, glycosaminoglycans, proteoglycan, skeletal dysplasia, urine biomarker.
Introduction
Skeletal dysplasias are a group of rare genetic disorders that affect growth and development of the skeleton.[1,2] Skeletal dysplasias are characterised by abnormal growth and development of bones, cartilage and other connective tissues, resulting in bones that are short, malformed, or abnormally fragile, which leads to physical deformities and other medical problems.
CHST3-related skeletal dysplasia is caused by homozygous or compound heterozygous loss-of-function mutations in the carbohydrate sulfotransferase 3 (CHST3) gene, encoding the CHST3 enzyme that adds a sulfate group to the hydroxyl on the C6 of N-acetylgalactosamine (GalNAc) in chondroitin sulfate glycosaminoglycans (GAGs).[3] Patients born with CHST3-related skeletal dysplasia often have short stature, spine deformities and multiple joint dislocations. Although high-throughput genome sequencing technologies have made it easier to identify genetic variants in CHST3 in patients with suspected CHST3-related skeletal dysplasia, it is often difficult to determine if the identified variants have any functional effect on the CHST3 enzyme. To validate the loss of function of an identified CHST3 mutation, patient fibroblasts[4,5] or urine (protein after acetone precipitation)[5] can be obtained to detect the absence of the 6-O-sulfate on chondroitin sulfate GAGs. Recombinant expression and biochemical assay in vitro of CHST3 enzyme bearing the corresponding mutation is sometimes performed to determine the activity of the mutated CHST3.[5,6]
Urine-based protein biomarker analysis is popular owing to its non-invasive and cost-effective means of sample collection.[7] However, practical hurdles sometimes hinder the use of urine as a sample for biomarker discovery research. Variable sample dilutions, protein concentrations and stability often mean same-day batch collection from participants, same-day sample analysis or sample concentration using molecular cut-off filters, or large volumes of organic solvents that can be costly and difficult when sample numbers increase. These processes can sometimes put unnecessary burdens on hospitals or clinics that are not equipped with research or diagnostic facilities or instruments.
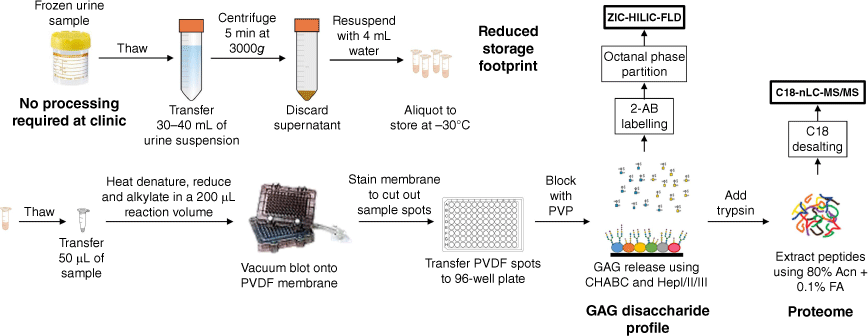
Freezer-induced urine sediments are precipitates commonly observed after a frozen urine sample thaw. They are calcium-containing crystals that entrap a significant amount of the total urinary proteins, and can be resolubilised on vigorous mixing.[8] The crystalline nature of these freezer-induced sediments presents a number of opportunities. Samples collected can be immediately frozen without pre-processing, with post-transfer to the research/diagnostic laboratory, and the sediments post thawing can be collected and stored in much smaller sample tubes suitable for biobanking (Fig. 1). In this work, we show, using freezer-induced urine sediments, that we are able to phenotype patients with CHST3-related skeletal dysplasia by analysing the protein-bound GAGs and proteome.
Workflow used in this study. Urine samples are immediately frozen, and on transfer to a research facility, thawed overnight and the sediments collected for a sequential GAG and proteome analysis. PVDF, Polyvinylidene fluoride; HILIC–FLD, hydrophilic liquid interaction chromatography–fluorescence detection; nLC, nano-liquid chromatography; 2-AB, 2-aminobenzamide; Acn, acetonitrle; FA, formic acid; PVP, polyvinylpyrrolidone
Results and discussion
Although high-throughput genomic sequencing technologies have made it routine to identify genetic variants in genes associated with skeletal dysplasias and other rare genetic disorders, determining the functional consequences of the detected variants often is labour-intensive, time-consuming and expensive. There is a need to develop simple and cost-effective methods to assess the functional impact of variants identified in patients with skeletal dysplasias.
For CHST3-related skeletal dysplasia, the functional impact of identified CHST3 variants has been assessed through the analysis of the chondroitin sulfate (CS) profile of fibroblasts and acetone-precipitated proteins of patient urine.[4,5] To simplify this process, we investigated the GAG profile of patient-derived freezer-induced urinary sediments, which only require defrosting of the frozen urine samples to analyse the GAG profile and urinary proteome.
Zwitterionic hydrophilic interaction liquid chromatography (ZIC-HILIC) enables the separation of both chondroitin/dermatan sulfate (CS) and heparan sulfate (HS) disaccharides after depolymerisation using chondroitinase and heparinase enzyme mix.[9,10] In the healthy controls, both CS and HS disaccharides can be observed, with varying overall GAG abundances (Fig. 2a, controls chromatogram). The most abundant CS disaccharide is the mono-sulfated CS-4S (sulfate on the hydroxyl of C4 of the GalNAc), with low sulfation on the hexuronic acid (2S containing) (Fig. 2b). For HS disaccharides, the non-sulfated HS is the major HS. This agrees with an unrelated study of urinary GAGs from healthy participants (n = 12), where the frozen urine samples were re-solubilised by vortexing.[11] Importantly, patient samples MQ 1 and MQ 5 had a significantly reduced CS 6-O-sulfate, observed from both the decrease in monosulfated CS-6S and disulfated CS-2S6S (Fig. 2b), and a significantly increased CS-4S relative to the healthy controls and the other two patient samples. This is consistent with the expected biochemical outcomes from patients with reduced function of the CHST3 enzyme[4,5] and consequent decrease in 6-sulfation CS. On unblinding of the identity of the samples, patients MQ 1 and MQ 5 were revealed to be the two patients who had been genotyped with CHST3-related skeletal dysplasia.
GAG disaccharide profiles from freezer-induced urine sediments, identified using a panel of common disaccharides (eight CS and eight HS and hyaluronic acid (HA)) by retention time (a), and the relative percentage abundance quantitation by fluorescence intensity (b).
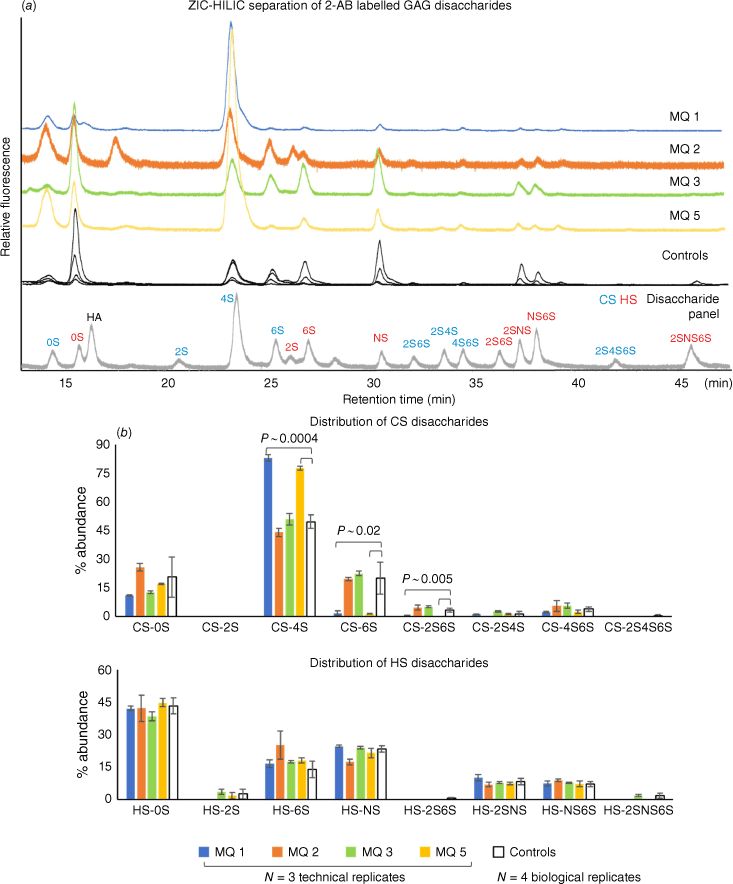
In contrast, we did not identify any significant differences in sulfation status between the diastrophic dysplasia (DTD) patients (MQ 2 and MQ 3) and the healthy controls. In these samples, both CS and HS show no significant differences in relative abundance of sulfated GAGs compared with healthy controls (Fig. 2b). DTD is a skeletal dysplasia caused by mutations in the SLC26A2 gene that encodes a sulfate anion transporter. In cultured cells, reduction in sulfate uptake due to defective SLC26A2 can be compensated through the salvage pathway from oxidation of sulfur-containing amino acids.[12] In a DTD mouse model, the reduction of CS sulfation in urine was only 5%.[13] Taken together, this suggests that phenotyping SLC26A2 mutations would not be effective based on GAGs as alternative pathways might be activated to compensate for the decreased sulfate transport.
We also used a basic bottom–up proteomics approach after the GAG depolymerisation and analysis to identify the potential carriers of these GAG chains. Immobilisation of the protein sample onto PVDF enables sequential proteomics analysis after GAG analysis, similarly to a previously published report using a filter-assisted workflow.[14] Without any depletion of abundant urinary proteins, high pH or other off-line fractionation methods,[15,16] we were able to identify 200–300 proteins per individual, and naturally, with the most abundant urinary protein, uromodulin (also known as Tamm Horsfall glycoprotein), being the top hit in all the protein searches (Supplementary Table 2). After manually curating for known GAG-carrying proteoglycans,[17] we were able to identify a list of proteoglycans, annotated with the known GAG chain, in the freezer-induced urine sediments (Table 1). These proteoglycans span both CS, HS and even keratan sulfate (KS) carriers, which was not part of the GAG profile. Sulfation levels of KS have not yet been explored in the context of SLC26A2 mutation, and KS sulfation is performed by CHST1, CHST2, CHST4 and CHST6, instead of CHST3.[18]
| Accession | Gene name | Protein name | GAG chain |
|---|---|---|---|
| P02760 | AMBP | Protein AMBP | CS |
| P98160 | HSPG2 | Basement membrane-specific heparan sulfate proteoglycan core protein | HS |
| O00468 | AGRN | Agrin | HS |
| P39060 | COL18A1 | Collagen alpha-1(XVIII) chain | HS |
| P21810 | BGN | Biglycan | CS |
| Q6UVK1 | CSPG4 | Chondroitin sulfate proteoglycan 4 | CS |
| P39059 | COL15A1 | Collagen alpha-1(XV) chain | CS and HS |
| P13727 | PRG2 | Bone marrow proteoglycan | CS |
| Q99715 | COL12A1 | Collagen alpha-1(XII) chain | CS |
| P16112 | ACAN | Aggrecan core protein | CS and KS |
| P16070 | CD44 | CD44 antigen | CS |
| P51884 | LUM | Lumican | KS |
| Q99983 | OMD | Osteomodulin | KS |
Full list of identified proteins is available in Supplementary Table S2.
Conclusion
In this pilot study, we show the potential of freezer-induced urine sediments as a working biological sample of choice for GAG phenotyping of the CHST3 mutation and bottom–up proteomics analysis. The ease of sample processing and reduced storage footprint of freezer-induced urine sediments can provide another option for sample normalisation, e.g. analyte/dry weight of sediments, instead of the commonly used creatinine content, which can be highly variable and require measurement before storage.[7] This ease of sample collection will be beneficial for patients, particularly patients with mobility issues, and reduce the logistical burden for the patients, nurses, doctors or researchers involved.
Experimental section
Urine sample collection
Urine samples from five patients with skeletal dysplasias were collected at The Children’s Hospital at Westmead (ethics approval HREC/18/SCHN/245), de-identified as samples MQ 1–MQ 5 and blinded for analysis. Among the five patients, two patients had a clinical diagnosis of CHST3-related skeletal dysplasia with confirmed homozygous or compound heterozygous mutations in the CHST3 gene. The remaining three patients had a clinical diagnosis of diastrophic dysplasia with confirmed homozygous or compound heterozygous mutations in the SLC26A2 gene. Four control urine samples (30–40 mL) were collected from healthy adult male volunteers. Urine samples were immediately frozen at −80°C.
Urine sample preparation
Frozen urine was thawed overnight at 4°C and the 30–40 mL of the urine suspension containing the freezer-induced sediments was transferred into a 50 mL Falcon tube for collection using centrifugation (5 min at 3000 g at 4°C). Only sample MQ 4 had no urine sediments observed. This was attributed to the lower volume of urine (<20 mL) collected, which was also colourless, suggesting that the patient was very well hydrated and had recently passed urine before sample collection. Hence, sample MQ 4 could not be included in the analysis. The pellet fractions from the urine samples were gently resuspended with 4 mL of deionised water to create a homogeneous suspension (final volume approx. 5 mL), and aliquots transferred into 2 mL Eppendorf tubes for storage at −30°C until analysis.
For GAG and proteome analysis, urine samples were thawed and 50 µL of homogeneous suspension was used for analysis (approx. 1% of total freezer-induced sediments). For the patient samples, four technical triplicates were analysed. Samples were reduced with 10 mM dithiothreitol, in a 200 µL reaction volume at 80°C for 30 min. Sediments were seen to have dissolved after this reduction and heating, and 50 mM iodoacetamide (final concentration) was added to alkylate the proteins at room temperature in darkness for 30 min. The protein samples were then transferred to a methanol-activated PVDF membrane using a 96-well vacuum blotter (Bio-Rad). Protein spots were visualised using 0.1% (w/v) Direct Blue in 40% (v/v) ethanol/10% (v/v) acetic acid, excised using a 4 mm hole punch, transferred into a 96-well plate and blocked with 1% (w/v) polyvinylpyrrolidone in 50% methanol as previously described.[19]
GAG analysis
The disaccharide analysis procedure was adapted from Moh et al.[9] and slightly modified. GAG disaccharides were released from the PVDF sample spots using a 20 µL enzyme mix containing 5 mU chondroitinase ABC (Sigma, Cat# C3667), 50 ng each of heparinase I/II/III (R&D Systems) in 100 mM ammonium acetate, pH 7, with 5 mM calcium chloride and incubated at 30°C overnight. Digested GAG disaccharides were collected and dried under low pressure for labelling using 2-aminobenzamide (2-AB), according to a commercially available protocol (Ludger LT-KAB-VP24-Guide-v2.0). A standard mix of eight common HS and eight common CS disaccharides (Iduron, United Kingdom), and hyaluronic acid (HA, from Streptococcus, Sigma, Cat. no. 53747, digested into disaccharides by chondroitinase ABC) were also labelled with 2-AB and used as a retention time standard. The chemical structures and shorthand notations of the disaccharides are depicted in the Supplementary information (Supplementary Fig. S1). Excess labelling reagents were removed using an octanal phase partition.[20] Cleaned samples in the aqueous layer were dried and resuspended in 32 μL 75% acetonitrile with 10 mM ammonium acetate, pH 6.8.
The labelled disaccharides were separated on a SeQuant ZIC-HILIC column (3.5 µm, 1 mm × 150 mm) at 30°C by an Agilent 1260 Infinity II LC with fluorescence detection. The mobile phases (solvent A (10 mM ammonium acetate, pH 6.8) and solvent B (90% acetonitrile in 10 mM ammonium acetate pH 6.8)) were run at a constant flowrate of 60 μL/min in microflow mode with gradient parameters as follows: 0–1 min, 100% B; 1–13 min, 93% B; 20 min, 11% B; 43 min, 25% B; 45–50 min, 60% B; 52–60 min, 100% B. Fluorescence detection was carried out with excitation and emission wavelengths set at 330 and 420 nm, respectively. Peaks were identified using the retention time standards and quantified manually from the elution curve, and significance was calculated using a two-tailed t-test.
Proteome analysis
After GAG enzyme digestion and disaccharide removal, the PVDF membrane with the remaining protein bound was cut into quarters, washed with 100 mM ammonium bicarbonate, and 0.5 μg of trypsin (Promega) added for proteolytic digestion at 37°C for 16 h. Digested peptides were extracted twice using 80% acetonitrile with 0.1% formic acid using a sonicating water bath for 5 min each. The peptides were dried under vacuum and reconstituted in 0.1% formic acid, desalted using a C18-packed tip (Empore) and dried under vacuum. The samples were reconstituted in 0.1% formic acid for injection into the nano-LC–electrospray ionisation (ESI)-MS/MS instrument.
Nano-LC–ESI-MS/MS was performed using a Dionex Ultimate 3000 RSLC system with an in-house C18 column (75 μm × 25 cm, 3 μm particles, 100 Å) coupled to a QExactive HF-X mass spectrometer. Peptides were chromatographically resolved at a flow rate of 300 nL/min with gradient parameters as follows: 0 min, 2% acetonitrile/0.1% formic acid; 6–66 min, 2–30% acetonitrile/0.1% formic acid; 76–90 min, 95% acetonitrile/0.1% formic acid; 96–110 min, 2% acetonitrile/0.1% formic acid. An acquisition window of m/z 500–2000 was used in a data-dependent acquisition mode, with the top 20 ions selected for tandem MS. The resultant mass spectra files were searched with Thermo Proteome Discoverer 2.4.1.15, using Sequest HT search engine against the UniProt Human reviewed proteome database (version as of December 2019) including the chondroitinase and heparinase enzymes used in the digest. A precursor mass tolerance of 5 ppm and fragment mass tolerance of 0.02 Da were used. Methionine oxidation (+15.995 Da) on the peptide and acetylation (+42.001 Da) on the protein N-terminus were considered as a dynamic modification while carbamidomethylation (+57.021 Da) of cysteine was considered as a static modification. Only proteins identified with three or more peptide spectrum matches were considered.
Supplementary material
Supplementary information containing structure and shorthand notations of the disaccharide panel (Supplementary Fig. S1) detailed GAG quantitation (Supplementary Table S1) and protein identification list (Supplementary Table S2) is available online.
Data availability
The fluorescence peak area intensity of the GAG profile and proteomics search output are available in the supplementary information. Proteomics raw data have been deposited to the ProteomeXchange Consortium via the PRIDE[21] partner repository with the dataset identifier PXD039081.
Acknowledgements
We acknowledge the Australian Proteomics Analysis Facility at Macquarie University for the use of the QExactive HF-X mass spectrometer.
References
2 Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A 2019; 179(12): 2393-419.
| Crossref | Google Scholar |
4 Hermanns P, Unger S, Rossi A, Perez-Aytes A, Cortina H, Bonafé L, et al. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis. Am J Hum Genet 2008; 82(6): 1368-74.
| Crossref | Google Scholar |
5 van Roij MHH, Mizumoto S, Yamada S, Morgan T, Tan-Sindhunata MB, Meijers-Heijboer H, et al. Spondyloepiphyseal dysplasia, Omani type: further definition of the phenotype. Am J Med Genet A 2008; 146A(18): 2376-84.
| Crossref | Google Scholar |
6 Tuysuz B, Mizumoto S, Sugahara K, Çelebi A, Mundlos S, Turkmen S. Omani-type spondyloepiphyseal dysplasia with cardiac involvement caused by a missense mutation in CHST3. Clin Genet 2009; 75(4): 375-83.
| Crossref | Google Scholar |
7 Sallsten G, Barregard L. Variability of urinary creatinine in healthy individuals. Int J Environ Res Public Health 2021; 18(6): 3166.
| Crossref | Google Scholar |
8 Saetun P, Semangoen T, Thongboonkerd V. Characterizations of urinary sediments precipitated after freezing and their effects on urinary protein and chemical analyses. Am J Physiol Renal Physiol 2009; 296(6): F1346-54.
| Crossref | Google Scholar |
9 Moh ESX, Nishtala K, Iqbal S, Staikopoulos V, Kapur D, Hutchinson MR, et al. Long-term intrathecal administration of morphine vs. baclofen: differences in CSF glycoconjugate profiles using multiglycomics. Glycobiology 2022; 32(1): 50-9.
| Crossref | Google Scholar |
10 Takegawa Y, Araki K, Fujitani N, Furukawa J-i, Sugiyama H, Sakai H, et al. Simultaneous analysis of heparan sulfate, chondroitin/dermatan sulfates, and hyaluronan disaccharides by gycoblotting-assisted sample preparation followed by single-step Zwitter-ionic-hydrophilic interaction chromatography. Anal Chem 2011; 83(24): 9443-9.
| Crossref | Google Scholar |
11 Han X, Sanderson P, Nesheiwat S, Lin L, Yu Y, Zhang F, et al. Structural analysis of urinary glycosaminoglycans from healthy human subjects. Glycobiology 2020; 30(3): 143-51.
| Crossref | Google Scholar |
13 Karousou E, Asimakopoulou A, Monti L, Zafeiropoulou V, Afratis N, Gartaganis P, et al. FACE analysis as a fast and reliable methodology to monitor the sulfation and total amount of chondroitin sulfate in biological samples of clinical importance. Molecules 2014; 19(6): 7959-80.
| Crossref | Google Scholar |
14 Sethi MK, Downs M, Zaia J. Serial in-solution digestion protocol for mass spectrometry-based glycomics and proteomics analysis. Mol Omics 2020; 16(4): 364-76.
| Crossref | Google Scholar |
15 Vaezzadeh AR, Briscoe AC, Steen H, Lee RS. One-step sample concentration, purification, and albumin depletion method for urinary proteomics. J Proteome Res 2010; 9(11): 6082-9.
| Crossref | Google Scholar |
16 Zhao M, Li M, Yang Y, Guo Z, Sun Y, Shao C, et al. A comprehensive analysis and annotation of human normal urinary proteome. Sci Rep 2017; 7(1): 3024.
| Crossref | Google Scholar |
17 Iozzo RV, Schaefer L. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol 2015; 42: 11-55.
| Crossref | Google Scholar |
18 Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 2023; 51(D1): D587-92.
| Crossref | Google Scholar |
19 Jensen PH, Karlsson NG, Kolarich D, Packer NH. Structural analysis of N- and O-glycans released from glycoproteins. Nat Protoc 2012; 7(7): 1299-310.
| Crossref | Google Scholar |
20 Chu A-HA, Saati AE, Scarcelli JJ, Cornell RJ, Porter TJ. Reactivity-driven cleanup of 2-aminobenzamide-derivatized oligosaccharides. Anal Biochem 2018; 546: 23-7.
| Crossref | Google Scholar |
21 Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019; 47(D1): D442-50.
| Crossref | Google Scholar |