

Halogen Functionalization of Aluminium Fumarate Metal–Organic Framework via In Situ Hydrochlorination of Acetylenedicarboxylic Acid*
Tobie J. Matemb Ma Ntep A , Wei Wu A , Hergen Breitzke B , Carsten Schlüsener A , Bastian Moll A , Laura Schmolke A , Gerd Buntkowsky B and Christoph Janiak A C
B and Christoph Janiak A C
A Institut für Anorganische Chemie, Heinrich-Heine-Universität Düsseldorf, D-40204 Düsseldorf, Germany.
B Eduard-Zintl-Institut für Anorganische und Physikalische Chemie, Technische Universität Darmstadt, Alarich-Weiss-Straße. 4, D-64287 Darmstadt, Germany.
C Corresponding author. Email: janiak@uni-duesseldorf.de
Australian Journal of Chemistry 72(10) 835-841 https://doi.org/10.1071/CH19221
Submitted: 13 May 2019 Accepted: 9 August 2019 Published: 10 September 2019
Abstract
The successful chloro-functionalization of aluminium fumarate (MIL-53-Fum) was achieved by in situ hydrochlorination of acetylenedicarboxylic acid on reaction with aluminium chloride resulting in the formation of the aluminium chlorofumarate metal–organic framework (MIL-53-Fum-Cl = [Al(OH)(Fum-Cl)]) in a one-pot reaction. The chloro functional groups decorating the pores enhance gas (CO2, CH4, and H2) sorption capacities and affinity compared with the non-functionalized MIL-53-Fum. The functionalization also results in a 2-fold increase in the selective adsorption of CO2 over CH4 compared with MIL-53-Fum.
Introduction
Composed of metal units that are bridged by organic ligands into porous networks, metal–organic frameworks (MOFs) allow for various functional groups to be introduced into the material, hence tuning of the chemistry of their pore surface to meet specific properties and potential applications.[1] Such functionalization of MOFs can be done pre-synthetically, post-synthetically or in situ.[2] For the latter, linker or cluster functionalization and MOF formation occur in a one-pot reaction, which offers the advantage of reducing synthesis steps and cost when compared with post-synthetic modification, as well as yields products that might at times not be obtained by direct synthesis.[3]
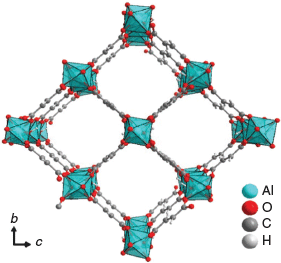
We recently reported the halogen functionalization of MOFs via in situ hydrohalogenation of linkers containing alkyne moieties.[4] More precisely, chlorofumarate-based UiO-type zirconium MOF was obtained by in situ hydrohalogenation of the triple bond of acetylenedicarboxylic acid. Aluminium-based MOFs are of great interest owing to the lightness and low toxicity of aluminium, as well as the abundance of its sources. Aluminium fumarate was one of the first commercialized Al-MOFs owing to its easy and inexpensive synthesis route at large scale.[5] Owing to its high thermal, chemical, and hydrothermal stability, aluminium fumarate holds promise for adsorption-based heat transformation applications,[6] adsorption of fluoride from water,[7] carbon dioxide capture from wet gas stream,[8] shape-based separation of hydrocarbons,[9] and as anode material for lithium ion batteries.[10] Aluminium fumarate, commercialized under the trademark Basolite A520, is also named MIL-53-Fum because it is isostructural with the aluminium terephthalate MOF MIL-53(-BDC).[11] The MIL-53 framework is built up from chains of trans-corner-sharing AlO6 octahedra, linked together by fumarate to form lozenge-shaped one-dimensional channels having a cross-section of ~5.7 × 6.0 Å2 (Fig. 1).[12]
|
The short fumarate linker in this material yields narrow pores (ultramicroporous MOF), which are beneficial with respect to the optimum adsorption of some gases and vapours like CO2 and water. For example, the Al-fumarate MOF (MIL-53-Fum) and the Zr-fumarate MOF (MOF-801) both display an S-shaped water sorption isotherm. The high hydrophilicity of these MOFs is demonstrated by a water step at a low relative pressure range of P/P0 = 0.2–0.3 (for MIL-53-Fum) and 0.05–0.1 (for MOF-801) favourable for application in thermally driven adsorption heat exchangers.[13,14] This hydrophilicity was attributed to the small pores, which dictate easy water molecule interaction with the framework and formation of hydrogen bonds between neighbouring water molecules.[15] Also, the short fumarate linker enables minimization or even cancelling of the hysteresis loop between the water adsorption and desorption branches in MIL-53-Fum and MOF-801 compared with what is observed for their respective terephthalate parents MOFs MIL-53-BDC and UiO-66.[15,16] Further, the small pore system in ultramicroporous MOFs is the basis for cost-effective selective removal of CO2 from flue gas as well as for the selective exclusion of branched paraffins.[17,18] Introducing moderately sized polar functional groups (chlorine for instance) into the pore system of MIL-53-Fum is of interest to improve its sorption capacity without compromising its gas selectivity. Surprisingly, no functionalized version of MIL-53-Fum has yet been reported. Reinsch et al. recently described the synthesis of an Al-MOF based on the short methylfumarate linker (MIL-68Mes).[19] However, grafting a CH3 group to the fumarate linker yielded an Al-MOF with the structure of MIL-68,[20] unlike the MIL-53-type structure of aluminium fumarate. Thus, synthesizing a functionalized version of MIL-53-Fum remains a non-accessed research area to explore.
Herein, we report the synthesis of the new aluminium chlorofumarate MOF (MIL-53-Fum-Cl), which is isoreticular with the well-known aluminium fumarate. As MIL-53-Fum-Cl is obtained by in situ generation of the chlorofumarate linker, the nature of the latter is examined along with the porosity and vapour and gas sorption properties of the new material.
Results and Discussion
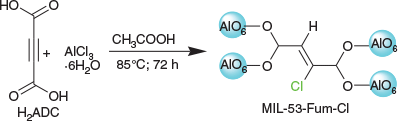
The reaction in DMF of acetylenedicarboxylic acid (H2ADC) with aluminium chloride hexahydrate in the presence of acetic acid modulator after 72 h at 85°C yielded a light brown–yellowish microcrystalline powder, designated MIL-53-Fum-Cl (Scheme 1).
|
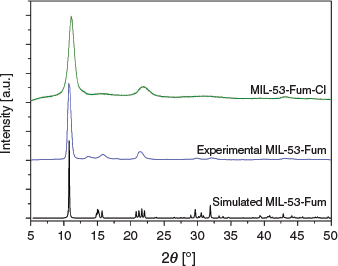
The powder X-ray diffraction pattern (PXRD) of the material obtained is similar to that of aluminium fumarate (MIL-53-Fum) (Fig. 2), thus revealing their isostructural nature.
|
Poor crystallinity is well known for MIL-53-Fum (Basolite A520),[12] and hence can also be expected for the analogue MIL-53-Fum-Cl. However, the fairly broad reflections in the PXRD pattern of MIL-53-Fum-Cl also derive from the very small particle size of the obtained material (see SEM image below). Conducting the synthesis using formic acid as modulator produced no improvement on the PXRD pattern (see PXRD pattern in Fig. S17, Supplementary Material).
In addition, a slight shift of reflections in the PXRD pattern of MIL-53-Fum-Cl can be a result of its flexibility (as in MIL-53-Fum). The position of the lowest reflection and the one at ~21° 2θ shift slightly depending on the degree of hydration and number of guests in the pores (see Fig. S12 in the Supplementary Material, where the position of these reflections varies with the solvent in the material). The PXRD patterns of MIL-53-Fum-Cl treated with different solvents show variable shifts of the strongest reflections at ~11° and 21° 2θ angles depending on the nature of the solvent in the pores of the material (Fig. S12). This supports the notion of the slight flexibility of MIL-53-Fum-Cl. Its flexibility is more noticeable than was previously observed for MIL-53-Fum.[12,13] This could mean that the chlorofunctionalization of MIL-53-Fum induces an increased flexibility in its framework. At the same time, the introduction of the chloro functional group can result in a slight change in the unit cell parameters with respect to MIL-53-Fum.[21]
It was observed that conducting the reaction without addition of acetic acid did not yield any solid product. This shows that acetic acid plays an important role in the formation of the new material. The role of monocarboxylic acids (formic, acetic, benzoic acid) has been clearly elucidated for the modulated synthesis of Zr-MOFs and Hf-MOFs,[22] whereas few Al-MOFs have been obtained by the modulated synthesis approach.[23] However, the mechanism of modulation remains to be studied in the case of Al-MOFs.
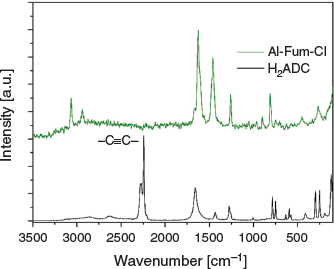
The Raman spectrum (Fig. 3) of MIL-53-Fum-Cl shows the disappearance of the strong band at 2250 cm−1 relative to the spectrum of the initial H2ADC linker. This band is characteristic of the stretching vibrations of the –C≡C– triple bond. The disappearance of this band indicates the transformation of the –C≡C– triple bond during MOF formation. The band at ~3100 cm−1 is ascribed to the =C–H vibration. In the IR spectrum (Fig. S1, Supplementary Material), the bands at 1616 and 1414 cm−1 correspond respectively to the asymmetric and symmetric stretching mode of coordinated carboxylate (–COO). Their respective wavenumber values, within the same range with those obtained for aluminium fumarate (νas 1606 cm−1 and νs 1424 cm−1), (Fig. S2, Supplementary Material) suggest the same coordination mode of linkers in both materials. The band at 748 cm−1 in the IR spectrum of MIL-53-Fum-Cl is attributable to the vibration of the C–Cl bond.[24]
|
Solid-state NMR (ssNMR) analysis gives more insight into the nature of the linker in the new MOF. The peaks at 130 and 140 ppm in the 13C spectrum (Fig. 4a) are ascribed to two non-symmetrically bonded olefinic carbon atoms (C=C). One olefinic carbon atom is bonded to a proton with a chemical shift at 6.9 ppm, as revealed by their correlation in the 1H–13C frequency switched Lee Goldburg heteronuclear correlation (FSLG HETCOR) spectrum (Fig. 4c). This chemical shift is confirmed by homonuclear decoupled 2D FSLG proton spectra, recorded with the same parameters as for the FLSG HECTOR spectra. Fig. S4 (Supplementary Material) depicts the projection on the indirect dimension of this spectrum. The peak at 170 ppm with a shoulder is ascribed to the two carboxylate carbon atoms of the linker. The ssNMR analysis also reveals the presence of the acetate modulator in the framework. The peak at 20 ppm in the 13C spectra is due to the acetate –CH3 group, while its carboxylate group –COO yields the peak at 180 ppm. This suggests the coexistence of defective sites within the material, where the acetate modulator is partly coordinated in place of the linker. This type of defect is well known for example in UiO-type MOFs obtained by monocarboxylic acid modulation.[25] The 1H ssNMR spectrum (Fig. 4b) presents three peaks at 6.9, 2.1, and 1.6 ppm attributable to the olefinic proton, the proton of the bridging µ-OH, and the methyl protons of acetate respectively. The correlation peak at ~170 ppm(13C)/2 ppm(1H) in the HETCOR spectrum recorded at 500 μs (Fig. S4) results from the long-range interaction between the proton of the bridging µ-OH and the carboxylate of the linker, as they are both connected to the same aluminium centre. If this interpretation holds true, this correlation should be absent in HETCOR spectra recorded with lower contact times, i.e. in HETCOR spectra that are only sensitive to short-range interactions. Fig. 4c depicts the HETCOR spectrum recorded with 200 µs contact time, where this correlation is indeed absent. Note that some peaks may slightly shift or split owing to different build-up times during the cross-polarization process, and also that cross-polarization spectra are not quantitative.

|
The number of defects was determined from the liquid 1H NMR analysis of a digested sample of MIL-53-Fum-Cl as ~0.4 missing Fum-Cl linker per unit formula of MIL-53-Fum-Cl ([Al(OH)(Fum-Cl)]) (see details in Supplementary Material). The number of missing linkers obtained from NMR analysis is confirmed by its estimation from thermogravimetric analysis (TGA) (see details in Supplementary Material).
From the X-ray photoelectron (XP) spectrum (Fig. S8, Supplementary Material), the presence of chlorine is observed in MIL-53-Fum-Cl. The peak at ~200 eV binding energy accounts for an organic chlorine.[26] Thus, there has been formation of a C–Cl bond during the reaction. This chlorine atom is obviously bonded to the second olefinic carbon atom that was observed in the ssNMR analysis (signal b in Fig. 4a). Peaks at 73.7 and 74.5 eV binding energy are due to Al–O and Al–OH respectively.[27] Scanning electron microscopy (SEM) energy-dispersive X-ray spectroscopy (EDX) elemental mapping (Fig. 5) further shows the presence of chlorine homogenously distributed over the whole MIL-53-Fum-Cl sample range. The molar atomic aluminium to chlorine (Al : Cl) ratio of ~1 : 0.7 (from EDX, details in Supplementary Material) is different from the expected 1:1 ratio for an ideal MIL-53-type MOF (ideal formula: [Al(OH)(linker)]). This is however in agreement with the occurrence of ~0.4 missing linker per formula unit of the synthesized MIL-53-Fum-Cl.

|
The aforementioned analysis results indicate a transformation of the reacting acetylenedicarboxylic acid molecule into a chlorofumarate linker in the newly formed MOF (Scheme 1). A trans hydrochlorination (addition of HCl) on the –C≡C– triple bond of acetylenedicarboxylic acid has taken place. HCl is produced in situ during the reaction of AlCl3 with water or with the acetic acid modulator to an intermediate aluminium hydroxide acetate Al(OH)(OOCCH3)2. The MOF is subsequently formed from the substitution of the acetate by the chlorofumate (see Scheme 2). This suggested mechanism is corroborated by the presence of missing linkers whose sites are occupied instead by coordinated acetate from incomplete substitution. Moreover, the basic aluminium acetate Al(OH)(ac)2·xH2O has previously been reported as a precursor for the synthesis of Al-MOFs.[28] An equivalent mechanism was also studied for monocarboxylic acid-modulated synthesis of Zr-MOFs and Hf-MOFs.[21]
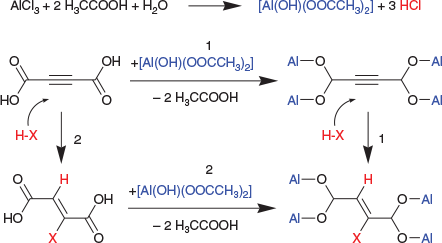
|
A similar transformation has previously been found for the formation of a chlorofumarate-based UiO-type Zr-MOF (HHU-2) from acetylenedicarboxylic acid reacting with zirconium chloride.[4]
Structural information could be obtained from 27Al ssNMR spectroscopy analysis. The 27Al ssNMR spectrum of activated MIL-53-Fum-Cl (Fig. 6) exhibits a peak at –13 ppm corresponding to an octahedral six-coordinate Al–O.[29] MIL-53-Fum-Cl is therefore constructed of AlO6 octahedra. The shoulder displayed at −13 ppm indicates the presence of two slightly different Al sites, and is due to the asymmetry of the chlorofumarate linker and to the presence of linker defective sites with coordinated acetate. Furthermore, the presence of the Al–OH group is seen in the XP spectrum (Fig S8, Supplementary Material). Analogously with the formula of MIL-53-type MOFs like aluminium fumarate, the ideal formula of the desolvated material is [Al(OH)(OOC–HC=CCl–COO)] ([Al(OH)(Fum-Cl)]). Its exact formula, taking into account the number of linker defects from liquid NMR, is [Al(OH)1.4(OOC–HC=CCl–COO)0.6(OOCCH3)0.4]. It is worth noting that the number of defects in materials obtained from a monocarboxylic acid-modulation synthesis usually depends on the relative amount of modulator used. Therefore, the linker defects ratio may vary from one synthesis batch to the other.

|
The thermal stability from TGA under air (Fig. S10, Supplementary Material) shows that MIL-53-Fum-Cl is stable to ~300°C. Its stability is lower than that of MIL-53-Fum, which is stable to ~400°C, but is still within the same range. Also, the chemical stability of MIL53-Fum-Cl is lower than that of MIL-53-Fum. The crystallinity of MIL-53-Fum-Cl was slightly altered after stirring in water or aqueous NaOH (pH 12) for 24 h at room temperature. The material completely dissolved in aqueous HCl (pH 1) to give a colourless solution. However, MIL-53-Fum-Cl was found to be stable in ethanol, methanol, DMF, and acetone with the same treatments (see Fig. S12 in Supplementary Material for details). The lower chemical stability can be attributed to the presence of a large number of linker defective sites that are occupied by coordinated acetate instead, as revealed in the ssNMR analysis. A synthesis method that considerably reduces the number of defects is expected to provide higher chemical stability. It has been demonstrated with highly chemically stable MOFs like UiO-66 that a large number of defects compromises their stability.[30]
The Brunauer–Emmett–Teller (BET) surface area of MIL-53-Fum-Cl was evaluated from nitrogen adsorption experiments as 800 m2 g−1, while micropore and total pore volumes were calculated as 0.21 and 0.48 cm3 g−1 respectively (see Fig. S13, and details in Supplementary Material). Its surface area and micropore volume are smaller than that of aluminium fumarate MIL-53-Fum (1021 m2 g−1 and 0.48 cm3 g−1). This is in agreement with pore occupancy by the chlorine atoms resulting in a decrease of surface area and micropore volume.
Sorption experiments of carbon dioxide at 273 K, methane at 273 K, and hydrogen at 77 K established the microporosity of MIL-53-Fum-Cl as they display type I isotherms (Fig. 7).[31] The CO2 adsorption capacity of MIL-53-Fum-Cl at 273 K and 1 bar (100 kPa) is 4.6 mmol g−1. This value is higher than the CO2 adsorption capacity of MIL-53-Fum (3.6 mmol g−1) which has a surface area of 984 m2 g−1. Thus, there is a 27 % increase in the CO2 adsorption capacity of MIL-53-Fum-Cl compared with the non-functionalized MIL-53-Fum.

|
The CH4 adsorption capacity of MIL-53-Fum-Cl at 273 K and 1 bar is 1.5 mmol g−1, corresponding to a 15 % increase in comparison with that of MIL-53-Fum (1.3 mmol g−1).
The H2 adsorption capacity of MIL-53-Fum-Cl is 8.40 mmol g−1 at 77 K and 1 bar and is slightly lower than that of MIL-53-Fum (8.97 mmol g−1) under the same conditions. A summary of the porosity characteristics and gas adsorption capacities of MIL-53-Fum-Cl and MIL-53-Fum is given in Table 1. This is expected as hydrogen uptake capacity at 77 K is known to strongly depend on the surface area of the adsorbent.[32] However, when normalized to the BET surface area, the H2 sorption capacity of MIL-Fum-Cl (10.5 µmol m−2) is still higher than that of MIL-53-Fum (9.1 µmol m−2). Furthermore, all CO2, CH4, and H2 adsorption isotherms display a higher initial slope and uptake compared with MIL-53-Fum, indicating a higher affinity for these gases. The affinity constants for MIL-53-Fum-Cl obtained from the Toth model fits of CO2, CH4, and H2 isotherms are 2.73, 1.34, and 2.18 bar−1 respectively (see Supplementary Material for details). These values are higher than those obtained with MIL-53-Fum (1.09, 0.41, 1.87 bar−1 for CO2, CH4, and H2 respectively). The increased gas sorption capacity and affinity in MIL-53-Fum-Cl compared with MIL-53-Fum is indicative of stronger CO2–, CH4–, or H2–MOF interactions. This is confirmed by the higher isosteric heat of CO2 adsorption (Qst) in MIL-53-Fum-Cl (average 35 kJ mol−1) compared with the Qst of 22 kJ mol−1 obtained for MIL-53-Fum (Fig. S16, Supplementary Material). The zero-coverage isosteric heat of CO2 adsorption (Qst0) in MIL-53-Fum-Cl is much higher, reaching a value of 55 kJ mol−1 (see details of calculation and plots of Qst in the Supplementary Material). These observations can rationally be attributed to the presence of polar chloro functional groups on the pore surface of MIL-53-Fum-Cl, inducing C–Cl⋯O=C=O, C–Cl⋯C=O, and C–Cl⋯H–H interactions. Halogen atoms as molecular entities are, on account of their high electronegativity, the basis of inter- and intra-molecular interactions known as halogen bonding.[33] However, the pore confinement due to steric occupation by chlorine atoms could also be contributing to the observed CO2 adsorption enhancement in MIL-53-Fum-Cl.[34]

|
The CO2 versus CH4 selectivity was calculated for a 50 : 50 (v/v) CO2/CH4 mixture at 273 K and 1 bar pressure. Applying IAST (ideal adsorbed solution theory) calculations on Toth-fitted isotherms yielded a selectivity of CO2 over CH4 (SCO2/CH4) value of 8.5 for MIL-53-Fum-Cl, approximately twice the value of 4.5 for MIL-53-Fum (calculation details in the Supplementary Material).
MIL-53-Fum-Cl shows a non-reversible S-shaped water vapour adsorption isotherm with a considerable hysteresis loop between the adsorption and desorption branches (Fig. 8). In the case of MIL-53-Fum, the S-shaped water adsorption isotherm is characterized by a steep increase in the narrow pressure range P/P0 = 0.2–0.3, with the isotherm reaching a plateau, and uptake capacity of 410 mg g−1 at P/P0 = 0.9. Unlike MIL-53-Fum, MIL-53-Fum-Cl shows a fairly gradual water uptake without reaching a plateau to a water uptake capacity of 300 mg g−1 at P/P0 = 0.9. The water adsorption behaviour of MIL-53-Fum-Cl can be explained by strong H2O–MOF interactions due to the presence of polar chlorine groups on the surface. The different shape of the desorption branch relative to the adsorption one indicates partial deterioration of the framework during water sorption. This was verified by a decrease in the surface area after water sorption from 800 to 570 m2 g−1, although the PXRD pattern post water sorption showed retention of crystallinity (see Fig. S12 in Supplementary Material).
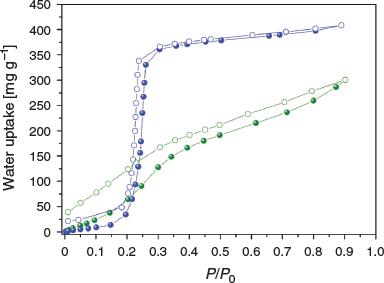
|
Conclusions
In summary, the in situ hydrohalogenation of acetylenedicarboxylic acid in the presence of AlCl3·6H2O under acetic acid modulation was shown to be a route for obtaining chloro-functionalized aluminium fumarate. The modified aluminium fumarate (MIL-53-Fum-Cl) features enhanced gas uptake capacity, affinity, and CO2 versus CH4 selectivity relative to the non-functionalized MOF, which highlights the benefit of chlorination. This synthesis of the first functionalized version of aluminium fumarate paves the way to other possible functionalization of this important adsorbent as a means to improving its properties.
Supplementary Material
Experimental details including materials used, syntheses, methods; infrared, Raman, NMR, EDX, and XP spectra; PXRD in the chemical stability test; nitrogen sorption isotherms; fitting of gas adsorption isotherms, and selectivity calculations are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
We thank the German Academic Exchange Service (DAAD) for funding to TJMMN and the German Federal Ministry of Education and Research (BMBF) for project Optimat, grant no. 03SF0492C.
References
[1] (a) H. Furukawa, K. E. Cordova, M. O’Keeffe, O. M. Yaghi, Science 2013, 341,| Crossref | GoogleScholarGoogle Scholar | 23990564PubMed |
(b) C. Janiak, J. K. Vieth, New J. Chem. 2010, 34, 2366.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) S. M. Cohen, Chem. Rev. 2012, 112, 970.
| Crossref | GoogleScholarGoogle Scholar | 21916418PubMed |
(b) K. K. Tanabe, S. M. Cohen, Chem. Soc. Rev. 2011, 40, 498.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Torrisi, R. G. Bell, C. Mellot-Draznieks, Cryst. Growth Des. 2010, 10, 2839.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Chavan, J. G. Vitillo, M. J. Uddin, F. Bonino, C. Lamberti, E. Groppo, K.-P. Lillerud, S. Bordiga, Chem. Mater. 2010, 22, 4602.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) X. Kong, T. He, Y. Zhang, X. Wu, S. Wang, M. Xu, G. Si, J. J. Li, Chem. Sci. 2019, 10, 3949.
| Crossref | GoogleScholarGoogle Scholar | 31015934PubMed |
(b) X.-M. Zhang, Coord. Chem. Rev. 2005, 249, 1201.
| Crossref | GoogleScholarGoogle Scholar |
[4] T. J. Matemb Ma Ntep, H. Reinsch, B. Moll, E. Hastürk, S. Gökpinar, H. Breitzke, C. Schlüsener, L. Schmolke, G. Buntkowsky, C. Janiak, Chem. – Eur. J. 2018, 24, 14048.
| Crossref | GoogleScholarGoogle Scholar |
[5] M. Gaab, N. Trukhan, S. Maurer, R. Gummaraju, U. Müller, Microporous Mesoporous Mater. 2012, 157, 131.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) F. Jeremias, D. Fröhlich, C. Janiak, S. K. Henninger, RSC Adv. 2014, 4, 24073.
| Crossref | GoogleScholarGoogle Scholar |
(b) H. Kummer, F. Jeremias, A. Warlo, G. Füldner, D. Fröhlich, C. Janiak, R. Gläser, S. K. Henninger, Ind. Eng. Chem. Res. 2017, 56, 8393.
| Crossref | GoogleScholarGoogle Scholar |
[7] S. Karmakar, J. Dechnik, C. Janiak, S. De, J. Hazard. Mater. 2016, 303, 10.
| Crossref | GoogleScholarGoogle Scholar | 26513559PubMed |
[8] J. A. Coelho, A. M. Ribeiro, A. F. P. Ferreira, S. M. P. Lucena, A. E. Rodrigues, D. C. S. de Azevedo, Ind. Eng. Chem. Res. 2016, 55, 2134.
| Crossref | GoogleScholarGoogle Scholar |
[9] B. Bozbiyik, J. Lannoeye, D. E. De Vos, G. V. Baron, J. F. M. Denayer, Phys. Chem. Chem. Phys. 2016, 18, 3294.
| Crossref | GoogleScholarGoogle Scholar | 26752453PubMed |
[10] Y. Wang, Q. Qu, G. Liub, V. S. Battaglia, H. Zheng, Nano Energy 2017, 39, 200.
| Crossref | GoogleScholarGoogle Scholar |
[11] T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille, G. Férey, Chem. – Eur. J. 2004, 10, 1373.
| Crossref | GoogleScholarGoogle Scholar | 15034882PubMed |
[12] E. Alvarez, N. Guillou, C. Martineau, B. Bueken, B. Van de Voorde, Cl. Le Guillouzer, P. Fabry, F. Nouar, F. Taulelle, D. de Vos, J.-S. Chang, K. H. Cho, N. Ramsahye, T. Devic, M. Daturi, G. Maurin, C. Serre, Angew. Chem. Int. Ed. 2015, 54, 3664.
| Crossref | GoogleScholarGoogle Scholar |
[13] F. Jeremias, D. Fröhlich, C. Janiak, S. K. Henninger, RSC Adv. 2014, 4, 24073.
| Crossref | GoogleScholarGoogle Scholar |
[14] M. V. Solovyeva, L. G. Gordeeva, T. A. Krieger, Y. I. Aristov, Energy Convers. Manage. 2018, 174, 356.
| Crossref | GoogleScholarGoogle Scholar |
[15] H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson, O. M. Yaghi, J. Am. Chem. Soc. 2014, 136, 4369.
| Crossref | GoogleScholarGoogle Scholar | 24588307PubMed |
[16] J. Canivet, J. Bonnefoy, C. Daniel, A. Legrand, B. Coasne, D. Farrusseng, New J. Chem. 2014, 38, 3102.
| Crossref | GoogleScholarGoogle Scholar |
[17] S. Shalini, S. Nandi, A. Justin, R. Maitya, R. Vaidhyanathan, Chem. Commun. 2018, 13472.
| Crossref | GoogleScholarGoogle Scholar |
[18] A. H. Assen, Y. Belmabkhout, K. Adil, P. M. Bhatt, D.-X. Xue, H. Jiang, M. Eddaoudi, Angew. Chem. Int. Ed. 2015, 54, 14353.
| Crossref | GoogleScholarGoogle Scholar |
[19] H. Reinsch, T. Homburg, N. Heidenreich, D. Fröhlich, S. Hennninger, M. Wark, N. Stock, Chem. – Eur. J. 2018, 24, 2173.
| Crossref | GoogleScholarGoogle Scholar | 29227561PubMed |
[20] (a) S.-H. Lo, C.-H. Chien, Y.-L. Lai, C.-C. Yang, J. J. Lee, D. S. Raja, C.-H. Lin, J. Mater. Chem. A Mater. Energy Sustain. 2013, 1, 324.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. Barthelet, J. Marrot, G. Férey, D. Riou, Chem. Commun. 2004, 520.
| Crossref | GoogleScholarGoogle Scholar |
(c) Q. Yang, S. Vaesen, M. Vishnuvarthan, F. Ragon, C. Serre, A. Vimont, M. Daturi, G. De Weireld, G. Maurin, J. Mater. Chem. 2012, 22, 10210.
| Crossref | GoogleScholarGoogle Scholar |
[21] (a) H. Reinsch, S. Waitschat, S. M. Chavan, K. P. Lillerud, N. Stock, Eur. J. Inorg. Chem. 2016, 4490.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. De Vos, N. Stock, Chem. Commun. 2015, 12578.
| Crossref | GoogleScholarGoogle Scholar |
[22] (a) G. Wißmann, A. Schaate, S. Lilienthal, I. Bremer, A. M. Schneider, P. Behrens, Microporous Mesoporous Mater. 2012, 152, 64.
| Crossref | GoogleScholarGoogle Scholar |
(b) O. V. Gutov, S. Molina, E. C. Escudero-Adán, A. Shafir, Chem. – Eur. J. 2016, 22, 13582.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke, P. Behrens, Chem. – Eur. J. 2011, 17, 6643.
| Crossref | GoogleScholarGoogle Scholar |
[23] (a) S.-H. Lo, C.-H. Chien, Y.-L. Lai, C.-C. Yang, J. J. Lee, D. S. Raja, C.-H. Lin, J. Mater. Chem. A Mater. Energy Sustain. 2013, 1, 324.
| Crossref | GoogleScholarGoogle Scholar |
(b) E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long, O. M. Yaghi, J. Am. Chem. Soc. 2010, 132, 14382.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. W. B. Teo, A. Chakraborty, S. Kayal, Microporous Mesoporous Mater. 2018, 272, 109.
| Crossref | GoogleScholarGoogle Scholar |
(d) W. Liang, L. Li, J. Hou, N. D. Shepherd, T. D. Bennett, D. M. D’Alessandro, V. Chen, Chem. Sci. 2018, 9, 3508.
| Crossref | GoogleScholarGoogle Scholar |
[24] A. De Lorenzi, S. Giorgianni, R. Bini, Mol. Phys. 1999, 96, 101.
[25] (a) G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye, K. P. Lillerud, Chem. Mater. 2016, 28, 3749.
| Crossref | GoogleScholarGoogle Scholar |
(b) G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga, K. P. Lillerud, Chem. Mater. 2014, 26, 4068.
| Crossref | GoogleScholarGoogle Scholar |
[26] E. Papirer, R. Lacroix, J.-B. Donnet, G. Nan, P. Fioux, Carbon 1995, 33, 63.
| Crossref | GoogleScholarGoogle Scholar |
[27] M. R. Alexander, G. E. Thompson, G. Beamson, Surf. Interface Anal. 2000, 29, 468.
| Crossref | GoogleScholarGoogle Scholar |
[28] N. Tannert, S.-J. Ernst, C. Jansen, H.-J. Bart, S. K. Henninger, C. Janiak, J. Mater. Chem. A Mater. Energy Sustain. 2018, 6, 17706.
| Crossref | GoogleScholarGoogle Scholar |
[29] (a) M. Haouas, F. Taulelle, C. Martineau, Prog. Nucl. Magn. Reson. Spectrosc. 2016, 94–95, 11.
| Crossref | GoogleScholarGoogle Scholar | 27247283PubMed |
(b) P. Soubayrol, G. Dana, Magn. Reson. Chem. 1996, 34, 638.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. Muller, W. Gessner, Chem. Phys. Lett. 1981, 79, 59.
| Crossref | GoogleScholarGoogle Scholar |
[30] A. W. Thornton, R. Barbarao, A. Jain, F. Trouselet, F.-X. Coudert, Dalton Trans. 2016, 4352.
| Crossref | GoogleScholarGoogle Scholar | 26733113PubMed |
[31] M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol, K. S. W. Sing, Pure Appl. Chem. 2015, 87, 1051.
| Crossref | GoogleScholarGoogle Scholar |
[32] M. P. Suh, H. J. Park, T. K. Prasad, D.-W. Lim, Chem. Rev. 2012, 112, 782.
| Crossref | GoogleScholarGoogle Scholar | 22191516PubMed |
[33] G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati, G. Terraneo, Chem. Rev. 2016, 116, 2478.
| Crossref | GoogleScholarGoogle Scholar | 26812185PubMed |
[34] (a) Q. Wang, J. Bai, Z. Lu, Y. Pan, X. You, Chem. Commun. 2016, 443.
| Crossref | GoogleScholarGoogle Scholar |
(b) Q.-G. Zhai, X. Bu, X. Zhao, D.-S. Li, P. Feng, Acc. Chem. Res. 2017, 50, 407.
| Crossref | GoogleScholarGoogle Scholar |
* Dedicated to Professor Richard Robson in commemoration of his seminal contribution to science.