

Australian chemistry and drug discovery towards the development of antimalarials†
Brad E. Sleebs A B *
A B *
A The Walter and Eliza Hall Institute of Medical Research, Parkville, 3052, Australia.
B Department of Medical Biology, The University of Melbourne, Parkville, 3010, Australia.

Dr Sleebs is a medicinal chemist with extensive experience in early-stage Drug Discovery and Chemical Biology. Dr Sleebs received his PhD from La Trobe University and joined The Walter and Eliza Hall Institute as a Research Officer in 2005. In 2018 he was appointed a Laboratory Head in the Chemical Biology Division at the Walter and Eliza Hall Institute. His past research includes the development of anxiolytics and agents that target the BH3 family of proteins for the treatment of blood cancers. His current research focuses on developing small molecule probes to better understand biological processes that are essential to the survival of the malaria parasite and in collaboration with industry partners the development of novel antimalarial agents. |
Australian Journal of Chemistry 75(11) 849-864 https://doi.org/10.1071/CH22141
Submitted: 18 June 2022 Accepted: 18 July 2022 Published: 2 September 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Malaria, a disease caused by the Plasmodium parasite, accounts for more than 450 000 deaths annually. The devastating impact of this disease is compounded by the emergence or risk of widespread resistance to current antimalarial drugs, underscoring the need to develop new therapies. Australian scientists are at the forefront of fundamental, clinical and surveillance research, and have made significant contributions to advancing the field of malaria research. A significant component of this research has been directed toward the development of new antimalarial therapies. This perspective summarises the recent endeavours by Australian researchers in chemistry and drug discovery sciences in the identification and development of new antimalarial therapies in the global challenge to treat and eliminate malaria.
Keywords: antimalarial, Australian chemistry, Australian drug discovery, drug development, high throughput screening, malaria, parasitology, pharmacology, Plasmodium.
Introduction
Half of the world’s population is at risk of malaria infection. In humans, malaria is a disease caused by five species of Plasmodium parasite. P. falciparum is the deadliest and most prevalent in sub-Saharan Africa, while P. vivax and P. knowlesi are endemic to Southeast Asia and the Americas. P. malariae and P. ovale are geographically widespread but have significantly lower prevalence and result in mild clinical manifestations. Approximately 241 million people are infected by Plasmodium with over 627,000 deaths worldwide in 2020.[1] Malaria continues to represent a major global health challenge.
The Plasmodium parasite is transmitted to the human host by way of a bite from an infected Anopheles mosquito. Sporozoites are injected into the circulatory system until they traverse and invade a liver hepatocyte. In the liver stage or pre-erythrocytic stage, the parasite develops into a schizont releasing many merozoites into systemic circulation where they invade red blood cells initiating the asexual stage. In the asexual blood stage, multiple rounds of self-replication occur leading to erythrocyte and the symptomatic signs of disease, such as lethargy and ague. On occasions, a sexual form of the parasite known as a gametocyte develops in a host erythrocyte over 14 days. Mature gametocytes are then ingested by a mosquito upon a blood meal and immediately mature into male and female gametes inside the mosquito midgut. The fertilised gametes then develop into ookinetes that invade the midgut wall and form an oocyst on the exterior of the midgut. The mature oocyst then produces ookinetes that traverse to the salivary glands of the mosquito for transmission to another human host. The multiplex lifecycle of the malaria parasite makes studying and developing new antimalarial therapies a challenging undertaking. It also opens opportunities to target different or multiple stages of the lifecycle to develop preventative, curative and transmission blocking therapies.
Current efforts to combat malaria have concentrated on mosquito control measures, the deployment of vaccines and combination drug therapies. Preventative measures are aimed at controlling transmission of the disease by the mosquito. The implementation of insecticide-treated bed nets has limited the prevalence of infection; however, this has not completely curbed the spread of Malaria. Recently the first malaria vaccine RTS, S (Mosquirix) was approved. While this is a major achievement, this vaccine only offers modest protection.[2] Chemo-prophylactic treatments such as doxycycline and atovaquone/proguanil (Malarone) are important in controlling the disease in malaria-endemic regions and preventing the disease in travelers visiting these countries.
Current therapies for treating malaria consist of combinations of quinolines or endoperoxides. The World Health Organisation no-longer recommends the use of the non-artemisinin combination therapies, consisting of sulfadoxine-pyrimethamine, chloroquine, mefloquine and amodiaquine due to widespread resistance to these therapies. Artemisinin combination therapies (ACTs) are now the frontline therapy to treat malaria. Concerningly, there are recent reports of emerging resistance to ACTs in South-East Asia[3] and more recently in sub-Saharan Africa.[4] Reports of malaria parasites developing resistance to combination drug treatments stresses the need to identify novel therapies to combat malaria infection. Therefore, there is an urgent need to develop drug candidates that have novel chemotypes and mechanisms of action and that target multiple stages of the parasite lifecycle aligned with treatment, prophylaxis or elimination target candidate profiles.[5]
Only recently has significant benevolent funding been available to develop new treatments for malaria. These initiatives have allowed industry and academia to contribute significant in-kind infrastructure and research support that has enabled the identification, mainly through phenotypic high throughput screening, and the development of novel antimalarial chemotypes to populate pre-clinical and clinical pipelines. The most notable of the new chemical chemotypes now in clinical trials are OZ439 1, an endoperoxide,[6] DSM-265 2 a dihydroorotate dehydrogenase (DHODH) inhibitor, KAF156 3 likely a SEC61 inhibitor, MMV048 4 a phosphatidylinositol 4-kinase (PI4K) inhibitor and KAE609 5 a PfATP4 inhibitor (Fig. 1).[7] Concerningly, resistance has been observed with several candidates in human clinical trials, and with the natural attrition rate of candidates when progressing through the clinic, there is a continued need to populate the drug discovery pipeline with novel chemotypes for the treatment and elimination of malaria.

|
Australian biomedical research has been central to the fundamental understanding of biological mechanisms and pathways important for parasite development across the lifecycle. A recent review by Doolan gave a historical perspective and highlighted the groundbreaking scientific breakthroughs by Australian scientists in malaria research.[8] The advances in parasitology, pathology and epidemiology of the malaria parasite have influenced the decisions by international governing organisations on the implementation and changes in policies, and agendas in malaria control, treatment and eradication. Furthermore, the research by Australian scientists has led to advances in new technologies that have enabled the study and biological evaluation of antimalarials across the entire parasite lifecycle and has catalysed small molecule and biologic drug discovery initiatives in Australia and across the globe. This appraisal seeks to highlight chemistry and drug discovery endeavours by Australian scientists over the last 15 years that have contributed to the identification and development of new antimalarials.
Biological evaluation
Screening for antimalarials
A call to arms at the start of the twentieth centenary brought together industry and academia to enable phenotypic screening of large compound libraries against the malaria parasite. This undertaking has led to the mass screening of natural product, vendor and industry small molecule libraries sampling a large diversity of chemical space.
The Avery group at Griffith University are world leaders in antimalarial high throughput screening techniques and has established multiple platforms to enable the screening of large compound libraries against two different stages of the malaria parasite lifecycle. One assay allows the evaluation of compounds against the asexual stage parasite and drug resistant forms of the asexual stage parasite.[9] This assay platform has been used to screen multiple compound libraries and has uncovered several new chemotypes for further optimisation.[10–13] Another assay technology using an imaging-based platform was developed to evaluate compounds against both early and late sexual gametocytes.[14,15] This technology also culminated in a new method to produce sufficient numbers of gametocytes to screen large libraries of compounds[16] and is now universally applied in laboratories worldwide. Both assays have been used to support the screening of boutique compound libraries, such as Medicines for Malaria Venture (MMV) ‘Box’ sets, which include the ‘Malaria Box’ and ‘Pathogen Box’[17–20] and to support medicinal chemistry efforts in numerous antimalarial development campaigns (many cited throughout this review). More recently, the Screening Facility at the Walter and Eliza Hall Institute (WEHI) led by Dr Lowes has adapted an existing P. falciparum lactate dehydrogenase assay format to screen compound libraries and to support medicinal chemistry efforts in industry and academic partnerships (also cited throughout this review).
Understanding the mechanism by which antimalarials kill Plasmodium parasites is important for their development as it allows an improved understanding of potential resistance mechanisms, of the pharmacodynamic and pharmacokinetic relationship, and in turn safety, and, in some cases, enables structure-based optimisation. Target identification is usually undertaken by forward genetic studies, but several mechanistic-based screening assays have also been developed to identify pathway specific compounds. Typically, these are secondary or low throughput assays used to evaluate and characterise small boutique compound sets with known antimalarial or antiparasitic activities but have an unknown mechanism of action.
The Gilson/Crabb group at the Burnet Institute developed a luciferase-reporter assay to assess the ability of compounds to block the export of parasite proteins to the host red blood cell.[21] Assays were also established to identify compounds that inhibit asexual parasite invasion and egress from the host red blood cell,[22] and to uncover compounds that inhibit the new permeability pathway[23] – indispensable for nutrient exchange in the host red blood cell. Several compounds from MMV ‘Box’ sets were independently identified that block these pathways and will be important tools for unravelling new molecular targets central to parasite survival and that can become starting points for antimalarial optimisation.
The Kirk/Lehane groups at the Australian National University developed an assay to screen for compounds that lead to an increase in Na+ levels and pH in the parasite cytosol as a characteristic marker for PfATP4 inhibition.[24] PfATP4 is a plasma membrane P-type ATPase essential for controlling parasite cytosolic levels of Na+ and in turn pH. The assay was important for characterising the PfATP4 activity in the development of clinical candidates KAE609 5 and SJ733 6 (Fig. 1).[24–26] The assay was also used to screen the MMV Malaria Box and uncovered 28 compounds with diverse scaffolds that inhibited PfATP4,[27,28] including derivatives of the clinical candidates KAE609 5 and SJ733 6. This assay was also used to uncover several new chemotypes in the MMV Pathogen Box that inhibit PfATP4.[29] The Kirk/Lehane groups also developed an assay and screened the MMV Malaria Box to find compounds that affect transporters responsible for a decrease in cytosolic pH.[30] The screen found 15 compounds that affected parasite cytosolic pH, and subsequently determined by forward genetic studies that two compounds exert their effects through inhibition of P. falciparum formate nitrite transporter (PfFNT). This research led to the characterisation of the PfFNT transporter as a druggable antimalarial target.[31]
The Andrews/Poulson groups at Griffith University developed an assay where parasites were cultured under hypoxic or reduced bicarbonate conditions as an indicator of P. falciparum carbonic anhydrase activity. This assay was applied in a screen of the MMV Malaria Box and identified a set of quinoline-like compounds.[19] It was hypothesised that although the activity of these quinoline compounds was affected by both hypoxic and reduced bicarbonate conditions, the mechanism of action was likely independent of carbonic anhydrase. The McFadden group have a longstanding interest in dissecting pathways of apicoplast targeted antimalarials. They (and others[32]) have been able to show that isopentenyl pyrophosphate (IPP) biosynthesis is the sole function of the apicoplast. As part of this research, an IPP supplementation assay was employed to characterise the on-target apicoplast activity of many slow-acting antimalarials.[33]
Target-based approaches have also been applied to interrogate the on-target activities of antimalarials and library compounds against proteins important for parasite survival. The McGowan group and colleagues at Monash University deployed biochemical assays to measure compound inhibition of P. falciparum metallo-aminopeptidases M1, M17 and M18. Two compounds in the Malaria Box were found to moderately inhibit both PfA-M1 and PfA-M17,[34] although it was suspected that these peptidases are not the primary targets responsible for parasite activity of these compounds. The Wilks/Lucet group recombinantly expressed thirteen P. falciparum kinases, PfCDPK1, PfPK6, PfCK2, PfGSK3, PfCLK1, Pfmap2, PfPK7, PfNek2, PfNek4, FIKK4.2, FIKK10.2, PfCK1 and PfPK5, considered important for parasite development.[35] A thermal shift assay and ADP-Glo luciferase assay was used to assess compounds from the Malaria Box against each of these kinases. The screen found three compounds that showed modest inhibition of PfGSK3, PfPK6 and PfPK5,[19] that did not correlate with parasite activity implying these kinases were not the primary targets of these compounds. The Cowman/Sleebs groups at WEHI established a high throughput screening assay to identify binders of the ligand reticulocyte binding protein homolog 5 (Rh5) that is indispensable for parasite invasion of the host red blood cell.[36] The assay was used to screen a known drug library and the Malaria Box and found that the leukotriene inhibitor pranlukast inhibited the interaction with Rh5 with its host cell ligand basigin. Further characterisation of pranlukast and pranlukast analogues revealed Rh5 inhibitory activity did not robustly correlate with blocking parasite invasion or killing the parasite.
The Quinn group at the Eskitis Institute implemented a target-based approach using native mass spectrometry to screen a library of natural product-based fragments to find binders of high priority antimalarial drug targets. Remarkedly, 69 parasite proteins were recombinantly expressed in soluble forms amenable to native mass spectrometry detection.[37] Overall, 96 fragments were found to differentially bind to 32 of the parasite proteins, of which 79 fragments displayed modest asexual parasite activity. Native mass spectrometry was also applied to P. falciparum dUTPase to screen the same fragment library.[38] The screen found several securinine natural product derivatives that inhibited the catalytic activity of PfdUTPase and showed modest activity against stage V gametocytes.
Collectively, the screen of boutique compound sets using target-based and pathway-based assays have uncovered several starting points for further investigation. Even though the target is implied in a target-based strategy, further characterisation and target deconvolution of compounds is generally required to ensure the parasite activity observed is on target. Overall, the target-based screening assays developed provide a launch pad to screen larger compound libraries to uncover new chemotypes to feed the antimalarial drug discovery pipeline.
Antimalarial target identification
Forward genetic and chemo-proteomic methods are typically employed in the target deconvolution of antimalarials (examples provided in the chemistry section below). Metabolomic methods have also been useful in revealing details of pathway aberrations by antimalarials. The McConville, Creek and Ralph groups independently and collaboratively have established metabolomic methods[39] and determined metabolomic signatures of frontline antimalarials.[40–44] The contributions by these groups are numerous, many of which are integrated into studies mentioned in this review. One notable collaborative example was the metabolomic analysis of 96 compounds from the Malaria Box.[45] This study unveiled that a significant proportion of the 96 compounds had metabolome fingerprints consistent with that of artemisinin, quinolines, PfATP4, DHODH or isoprenoid biosynthesis inhibition, providing key information on the likely mechanism of action of these antimalarial starting points.
ADME and pharmacokinetic evaluation of antimalarials
A key component of antimalarial development is the evaluation of ADME and pharmacokinetic properties. The Charman group at Monash University has been instrumental in providing expertise and key data for many antimalarial programs across the globe, and in the development of antimalarial property guidelines.[46] The exceptional contribution to antimalarial research by the Charman group is too expansive to completely cover in this review (many examples are referenced in the chemistry section). One notable example was a program focused on the development of a synthetic ozonide scaffold to overcome the short half-life and supply demands of the artemisinin derivatives. Several iterations around the ozonide scaffold led to the lead compound OZ277 7 (Fig. 1) which was characterised by an improved pharmacokinetic profile compared to artesunate.[47] Further critical feedback on ADME and PK culminated in the development of OZ439 1 with a significantly prolonged half-life in pre-clinical models[6,48] enabling human dose modelling for development in clinical trials.[49] Other notable contributions were to the pre-clinical development of the DHODH inhibitors DSM265 2[50] and DSM421 8,[51] the PI4K inhibitors MMV048 4[52] and UCT943 9[53] and the PfATP4 inhibitor SJ733 6 (Fig. 1).[25]
Clinical evaluation of antimalarials
Clinical development of antimalarials in Australia has largely been driven by Edstein and colleagues at the Australian Defence Force Malaria and Infectious Disease Institute since the mid-1980s. Additionally, they have provided key in vitro antimalarial data for many early-stage programs many of which are mentioned in the chemistry section. The extensive works of Edstein and colleagues are too numerous to comprehensively cover in this appraisal but have contributed to pharmacokinetic/pharmacodynamic analyses in the pre-clinical and clinical development of many antimalarial therapies that are used in the field today. McCarthy and colleagues at QIMR Berghofer Medical Research Institute (now at the Doherty Institute and WEHI) have also been instrumental in developing and conducting controlled blood-stage and gametocyte challenge models in human clinical trials. Most notably these have been used to evaluate antimalarials recently developed, including artefenomel (OZ439) 1,[54,55] cipargamin (KAE609) 5,[56] DSM265 2,[57,58] MMV048 4,[59,60] SJ733 6,[61] Actelion-451840 10[62] and ZY-19489 11[63] (Fig. 1).
Chemistry
Natural product antimalarials
Historically, natural products have been an important component of antimalarial research. Key examples are the discovery of quinine, the impetus for quinoline drugs, and artemisinin. Several natural product scaffolds originating from Australian research have provided novel starting points for antimalarial development.
The Quinn and Davis groups at Griffith University have been at the forefront of curating and screening a library of natural isolates and products against the malaria parasite. This has led to the discovery of natural products with interesting antimalarial properties. A bispyrroloiminoquinone alkaloid, tsitsikammamine C 12 (Fig. 2), isolated from an Australian marine sponge showed potent activity against the P. falciparum parasite (EC50 13 nM) and efficacy in a P. berghei mouse model of malaria.[64] In another study, a screen of the Eskitis Institute Nature Bank library against the P. falciparum parasite uncovered the natural products alstonine 13 and himbeline 14 (Fig. 2) with EC50s of 0.17 and 0.58 μM, respectively.[65] Another example was a serrulatane diterpenoid 15 (Fig. 2) isolated from the Australian desert plant Eremophila microtheca. Synthetic amide derivatives of this diterpenoid scaffold were shown to have modest antimalarial activity (EC50s 1.25–5.65 µM).[66] Further screening of the natural product isolate library identified the thiazine-derived alkaloids, thiaplakortones A–D, from the Australian marine sponge Plakortislita.[67] Thiaplakortone A 16 (Fig. 2) was the most potent (EC50 51 nM) against P. falciparum. Subsequent synthesis of thiaplakortone A and its derivatisation led to analogues with improved metabolic stability and efficacy in a P. berghei mouse model.[68,69]

|
The Baell group at Monash University synthesised truncated derivatives (18) of the natural alkaloid latrunculin B 17 (Fig. 2) isolated from Red Sea marine sponge and showed they have modest activity against the malaria parasite (EC50 7 μM).[70] The latrunculin scaffold was shown to reduce parasite motility through disruption of actin dynamics. The Payne group at the University of Sydney completed the total synthesis and stereochemical assignment of gallinamide A 19 (Fig. 2),[71,72] a depsipeptide isolated from the cyanobacterium Schizothrix. Gallinamide A 19 and synthetic analogues were found to potently inhibit parasite cysteine proteases, falcipain 2 and 3, which are critical for the processing of haemoglobin in the parasite digestive vacuole providing sustenance for the developing asexual parasite.[73] Accordingly, gallinamide A 19 was shown to cause swelling of the food vacuole in the parasite, a characteristic of falcipain inhibition, and potently killed the P. falciparum parasite (EC50 50 nM). Overall, these natural products provide unique scaffolds as tools to investigate new malaria biology or starting points for antimalarial development.
Antimalarials discovered via phenotypic screening
Commonly, starting points for antimalarials are discovered by phenotypic screens of small molecule libraries on P. falciparum. The Baell group identified MIPS-0004373 20 (Fig. 3) with a bis-3-alkylthio-1,2,4-triazine scaffold originating from a screen of an internal library, that showed fast-acting and potent activity against P. falciparum asexual stage parasites (EC50 8 nM).[74] Subsequently, MIPS-0004373 20 was shown to have potent activity against sexual stage gametocytes and gametes and to potently inhibited liver stage development.[75] MIPS-0004373 20 was characterised by low metabolic stability and a short half-life in mice, but remarkably showed high efficacy (ED50 1.5 mg kg–1) by oral administration in a P. berghei mouse model.[76] The metabolic stability of the bis-triazine series was improved by replacing the liable thioether groups. The lead compound 21 was shown to maintain parasite potency in vitro (EC50 31 nM) and efficacy in a P. falciparum humanised SCID mouse model.[77] MIPS-0004373 20 has a high barrier to resistance and further investigation is ongoing to pinpoint the mechanism of action.

|
The Todd group at the University of Sydney (now located at University College London) initiated an Open Source Drug Discovery program whereby any laboratory from across the globe could collaboratively contribute chemistry and biological resources and expertise towards the optimisation of selected antimalarial scaffolds. One starting point with a pyrrole scaffold (22) (Fig. 3) that originated from a GSK Tres Cantos antimalarial screen showed potent asexual activity and gametocyte activity.[78] A concerted and creative effort by the consortium replaced certain structural motifs on the scaffold that were viewed as chemical liabilities. It was found that these changes were largely detrimental to parasite activity and eventually the series was ‘parked’. Other compound scaffolds were also investigated as part of the Open Source consortium, including the triazolopyridine series that targets PfATP4.[79] Optimised compounds from this series such as 23 (Fig. 3) have potent asexual parasite activity (EC50 0.14 nM) and oral efficacy in a P. falciparum humanised SCID mouse model. Laboratories from across the globe are continuing to work collaboratively on the optimisation of several other scaffolds.
The Sleebs/Gilson groups also initiated a program that started with a hit scaffold from the GSK Tres Cantos screen. Optimisation of the potency and ADME properties of the 2,4-amino quinazoline scaffold to produce the frontrunner compound WEB-485 24 (Fig. 3) that has potent asexual antimalarial activity (EC50 0.027 μM) and modest efficacy in a P. berghei and P. falciparum mouse models.[80,81] The future challenge of this series is to overcome the dose-limiting in vivo toxicity.[82] Target deconvolution studies on this series are ongoing to assist in mitigating toxicity. The Sleebs/Gilson groups continue to work on scaffolds identified from parasite red blood cell invasion and export screens on boutique small molecule libraries (mentioned in the Screening section).[21,22] One example was the phenylsulfonyl piperazine scaffold that was optimised to give frontrunner compound S-38 25 (Fig. 3) with an IC50 of 0.11 μM. However, the restrictive SAR negated improvement in the in vitro metabolism.[83] Therefore, in the future, this series will be used as a tool to investigate the mechanism responsible for the invasion phenotype. The Sleebs/Cowman groups at WEHI screened the Janssen Jumpstarter small molecule library and identified several hits with novel chemotypes, including the triazolopyrimidine hit scaffold.[84] The optimisation of this scaffold led to the triazolopyrimidine 26 (Fig. 3) with potent slow-acting antimalarial activity (EC50 0.07 μM). Future work will focus on correcting the metabolic stability of this scaffold which remains a challenge.
The Abbott group at La Trobe University was able to repurpose a human protein kinase A scaffold as a starting point to target kinases in P. falciparum.[85] Optimisation of the 4-cyano-3-methylisoquinoline class produced the frontrunner compound MB14 27 with modest parasite activity (EC50 1.5 μM) (Fig. 3).[86] It was subsequently deduced from forward genetic and chemo-proteomic studies that the optimised scaffold killed the malaria parasite by targeting PfATP4.[87] The Scammells/Norton group at Monash University also used a similar strategy whereby they started with a benzimidazole scaffold identified from a fragment screen against the important parasite invasion ligand AMA1.[88] Optimisation of the benzimidazole scaffold against the P. falciparum parasite gave rise to the lead compound 3r 28 (Fig. 3) with potent activity (EC50 0.006 μM) and good physicochemical properties. It was found that this compound series acts through a mechanism independent from AMA1 and is the focus of future research.[89]
Target based antimalarials
Phenotypic approaches are the most common avenue to develop antimalarials, but target-based screening strategies have also produced starting points for antimalarial development, including the clinical candidate DSM265 2.[50] The target-based approach is heavily reliant on using reverse genetics to determine the essentiality of the target for parasite survival and in turn its validity for antimalarial drug discovery. The Guddat group at the University of Queensland has a longstanding program targeting the hypoxanthine-guanine-[xanthine]-phosphoribosyltransferase (HG[X]PRT), protein essential for the function of the parasite nucleoside salvage pathway. This group has used substrate mimetics at the core of their program which has been facilitated by multiple X-ray structures of HGXPRT enabling structure-based optimisation.[90,91,92–94] This program has generated several mimetic scaffolds such as 29 (Fig. 4) with potent biochemical inhibition of both PfHGXPRT and PvHGPRT (Kis 0.15–72 μM).[95] The challenge with these mimetics is the requirement for a polar phosphate (or acidic) group limiting membrane permeability. To overcome this challenge, the team has employed several prodrugs (30) to enhance membrane permeability and achieve modest parasite activity (EC50s 2.5–12.1 μM).
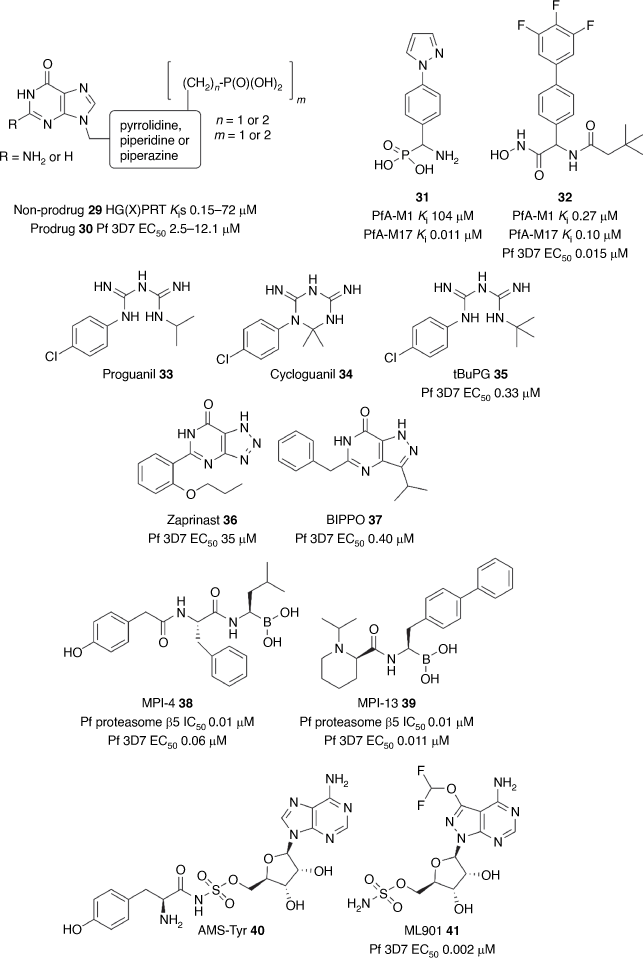
|
A target-based approach was implemented to discover inhibitors of the metallo-aminopeptidases, PfA-M1 and PfA-M17.[96] McGowan and colleagues at Monash University initiated this program by obtaining biological data to show the importance of both proteases for parasite development.[97–99] X-Ray structural data of both proteases in complex with known peptidomimetic inhibitors of aminopeptidases initiated a structure-guided design program.[100,101] The Scammells/McGowan groups started with a phosphonic arginine mimetic 31 (Fig. 4) that has potent and selective inhibition of PfA-M17 (Ki 0.011 μM).[102] The phosphonic acid was then replaced with a hydroxamic acid group and optimisation of the P′1 and P1 positions led to a potent dual inhibitor 32 (Fig. 4) of PfA-M1 and PfA-M17 (Ki 0.27 and 0.10 μM) and potent P. falciparum activity (EC50 0.015 μM).[103,104] Furthermore, the lead compound 32 has selectivity against human metalloproteases and robust metabolic stability, stimulating the next phase of development.
The Andrews/Ryan groups, while investigating a strategy to block the metabolism of proguanil 33 to cycloguanil 34, a DHFR inhibitor, designed the derivative tBuPG 35 (Fig. 4). Unlike proguanil and cycloguanil, the potent slow-acting parasite activity of tBuPG 35 (72 h IC50 0.33 μM; 96 h IC50 0.05 μM) was independent of folate and isoprenoid biosynthesis.[105] It was found that the parasite activity of tBuPG 35 in combination with cytochrome bc1 inhibitors atovaquone and ELQ300 was synergistic and as a result tBuPG 35 is under investigation as a potential replacement of proguanil as the partner agent with atovaquone in the antimalarial therapy Malarone.
The Gilson/Tonkin groups uncovered the role of cyclic-AMP and cyclic-GMP in activating protein kinase A (PKA) and protein kinase G (PKG) in parasite invasion and egress of the host RBC. 3′-5′-Cyclic nucleotide phosphodiesterases (PDEs) are key regulators of this process by hydrolysing cyclic-AMP and cyclic-GMP and stalling invasion and egress. The Thompson group used structural modelling of human and P. falciparum PDE to repurpose and fine-tune the parasite potency of the known human PDE inhibitor, zaprinast 36 (Fig. 4).[106] This strategy produced the compound, BIPPO 37 (Fig. 4), which was 90-fold more potent than zaprinast against the P. falciparum parasite (EC50 0.40 μM). In a mechanism-dependent manner, BIPPO was shown to affect the premature egress of merozoites from the asexual schizont and to prevent the capacity of the immature merozoites to invade the host erythrocyte. BIPPO serves as an important tool in the investigation of processes that underpin parasite invasion and egress.
The Tilley group’s interest in targeting the parasite proteasome was sparked by their discovery that artemisinin resistance is overcome by inhibiting the proteolytic function of the proteasome.[107] To unearth novel inhibitors of the proteasome, the Takeda library of peptide boronates was screened against P. falciparum. The hit compounds were further triaged by screening against the Pf20S and human 20S proteasome subunits. Among the hit compounds, MPI-4 38 (Fig. 4) was shown to have potent inhibition of the β1, β2 and β5 subunits of the proteasome (IC50 0.6, 0.06 and 0.01 μM, respectively) but equally potent inhibition of the human constitutive proteasome and immunoproteasome.[108] MPI-4 38 potently killed the malaria parasite (EC50 0.06 μM), but also reduced mammalian cell viability with similar potency, underlining the challenge of selectively targeting the parasite proteasome. CryoEM structures were then obtained of the P. falciparum proteasome β5 subunit in complex with peptide boronates that provided insight into optimising selectivity. Optimisation produced the frontrunner compound MPI-13 39 (Fig. 4) with 19-fold selectivity against the P. falciparum versus human proteasome β5 subunit (IC50 0.012 versus 0.23 μM), and 84-fold selectivity against the parasite versus mammalian cells (IC50 0.011 versus 0.93 μM).[109] MPI-13 39 showed robust efficacy in a P. falciparum humanised SCID mouse model and activity against male gamete and liver schizont development demonstrating that the proteasome is a multi-stage antimalarial drug target.
More recently Tilley and colleagues initially explored the application of adenosine 5′-sulfamate (AMS) as a substrate mimetic of adenosine 5′-monophosphate and as a potential modality to block protein synthesis by way of targeting parasite aminoacyl tRNA synthetases (aaRS).[110] Proteomic studies of AMS treated parasites primarily detected the AMS-Tyr adduct 40 (Fig. 4) signifying that AMS commandeered the mechanistic function and subsequently selective inhibition of TyrRS. To discover compounds with improved parasite specificity a screen of the Takeda library of nucleoside sulfamates identified ML901 41 (Fig. 4) with potent antimalarial activity (EC50 0.002 μM) and 5000-fold selectivity against mammalian cells. Forward genetic, chemo-proteomic and structural biology methods were then employed to show that ML901 41 was a selective inhibitor of TyrRS. ML901 41 was shown to potently inhibit both gamete and liver stage development and has efficacy in a P. falciparum humanised SCID mouse model, demonstrating that TyrRS is an attractive multi-stage antimalarial drug target.
The Cowman/Boddey groups researching protein trafficking by the malaria parasite within the host erythrocyte discovered that the aspartyl protease plasmepsin V is necessary for processing of the Plasmodium export element (PEXEL) on the N-terminus of parasite proteins exported to host RBC.[111] This fundamental finding led Cowman/Boddey/Sleebs to develop peptidomimetic inhibitors that mimic the PEXEL motif and inhibit plasmepsin V. The peptidomimetics initially designed including WEHI-916 42 (Fig. 5) helped define the substrate specificity of plasmepsin V, show the requirement of plasmepsin V in protein export and pharmacologically validate plasmepsin V as an antimalarial drug target.[112,113] X-Ray structures of these peptidomimetics in complex with plasmepsin V engendered an in-depth understanding of substrate recognition and inhibition of the protease, enabling the design of the peptidomimetics WEHI-842 43 and WEHI-601 44 (Fig. 5) with improved biochemical inhibition of plasmepsin V (IC50 0.019 and 0.005 μM, respectively) and parasite activity (EC50 0.43 and 0.09 μM, respectively).[114–117] These peptidomimetics were then employed to demonstrate the importance of plasmepsin V and protein export in gametocyte maturation establishing plasmepsin V as a multi-stage antimalarial target.[118]

|
In search for novel drug-like plasmepsin inhibitors, Cowman screened the MSD (Merck and Co.) library of aspartyl protease inhibitors and identified the imino pyrimidinone hit WM5 45 (Fig. 5) with potent antimalarial activity (EC50 0.01 μM).[119] Forward genetic and chemo-proteomic studies identified plasmepsin X as the molecular target. Optimisation of the imino pyrimidinone scaffold by large teams at both MSD, Wuxi and WEHI with the support of Wellcome led to the compound WM382 46 (Fig. 5) that had potent asexual stage activity (EC50 0.5 nM). Further phenotypic and chemo-proteomic experiments showed that WM382 46 potently inhibited both plasmepsin IX and X (IC50 0.51 and 0.035 nM).[119,120] WM382 46 was shown to block transmission of the parasite to the mosquito and was orally efficacious in asexual and liver stage mouse models, demonstrating that dual inhibition of plasmepsin IX and X is an attractive strategy to develop antimalarials.
Conclusion
Malaria was declared eradicated from Australia in 1981, however, there are still approximately 700–800 cases of infection each year, mostly arising from overseas-acquired malaria. Despite the low incidence of malaria, Australia has an important responsibility to support malaria elimination programs in endemic countries in the neighbouring region such as Papua New Guinea, the Solomon Islands and Vanuatu, and more broadly South-East Asia. In addition to nearby regions, the fundamental malaria research and antimalarial development undertaken in Australia and by Australian researchers overseas has contributed considerably to the worldwide effort to treat and eliminate the disease in malaria-endemic regions across the globe. The research outlined in this review further highlights that Australia’s chemistry and drug discovery sector is world-class and that continued collaborative efforts and new partnerships with Australian organisations will add significant value to antimalarial development and support the global effort to treat and eliminate malaria.
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
Conflicts of interest
The author declares no conflicts of interest.
Declaration of funding
B. E. S. was supported by the National Health and Medical Research Council of Australia (Development Grant 2014427 to B. E. S.) and the Corin Centenary Fellowship.
Acknowledgements
I acknowledge the tireless efforts of Australian researchers in the field of malaria research, of which I have attempted to capture the work of those toward antimalarial research in this review. I apologise to those researchers for the research I may have overlooked, largely due to the enormity of malaria research undertaken by the Australian scientific community. I also acknowledge that many Australian researchers are actively involved with numerous antimalarial drug discovery programs, and I am hopeful this research will be reported in future communications to further highlight the significant contribution by Australian scientists toward developing new therapies and eliminating malaria. Thank you to Prof Guillaume Lessene from the Walter and Eliza Hall Institute for helpful advice.
References
[1] WHO. World Malaria Report. Geneva: World Health Organization: Geneva, Switzerland; 2020. Available at https://www.who.int/publications/i/item/9789240015791[2] Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial Lancet 2015, 386, 31.
| Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trialCrossref | GoogleScholarGoogle Scholar |
[3] EA Ashley, M Dhorda, RM Fairhurst, C Amaratunga, P Lim, S Suon, S Sreng, JM Anderson, S Mao, B Sam, C Sopha, CM Chuor, C Nguon, S Sovannaroth, S Pukrittayakamee, P Jittamala, K Chotivanich, K Chutasmit, C Suchatsoonthorn, R Runcharoen, TT Hien, NT Thuy-Nhien, NV Thanh, NH Phu, Y Htut, KT Han, KH Aye, OA Mokuolu, RR Olaosebikan, OO Folaranmi, M Mayxay, M Khanthavong, B Hongvanthong, PN Newton, MA Onyamboko, CI Fanello, AK Tshefu, N Mishra, N Valecha, AP Phyo, F Nosten, P Yi, R Tripura, S Borrmann, M Bashraheil, J Peshu, MA Faiz, A Ghose, MA Hossain, R Samad, MR Rahman, MM Hasan, A Islam, O Miotto, R Amato, B MacInnis, J Stalker, DP Kwiatkowski, Z Bozdech, A Jeeyapant, PY Cheah, T Sakulthaew, J Chalk, B Intharabut, K Silamut, SJ Lee, B Vihokhern, C Kunasol, M Imwong, J Tarning, WJ Taylor, S Yeung, CJ Woodrow, JA Flegg, D Das, J Smith, M Venkatesan, CV Plowe, K Stepniewska, PJ Guerin, AM Dondorp, NP Day, NJ White, Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014, 371, 411.
| Spread of artemisinin resistance in Plasmodium falciparum malaria.Crossref | GoogleScholarGoogle Scholar |
[4] A Uwimana, E Legrand, BH Stokes, J-LM Ndikumana, M Warsame, N Umulisa, D Ngamije, T Munyaneza, J-B Mazarati, K Munguti, P Campagne, A Criscuolo, F Ariey, M Murindahabi, P Ringwald, DA Fidock, A Mbituyumuremyi, D Menard, Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med 2020, 26, 1602.
| Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda.Crossref | GoogleScholarGoogle Scholar |
[5] JN Burrows, S Duparc, WE Gutteridge, R Hooft van Huijsduijnen, W Kaszubska, F Macintyre, S Mazzuri, JJ Möhrle, TNC Wells, New developments in anti-malarial target candidate and product profiles. Malar J 2017, 16, 26.
| New developments in anti-malarial target candidate and product profiles.Crossref | GoogleScholarGoogle Scholar |
[6] SA Charman, S Arbe-Barnes, IC Bathurst, R Brun, M Campbell, WN Charman, FCK Chiu, J Chollet, JC Craft, DJ Creek, Y Dong, H Matile, M Maurer, J Morizzi, T Nguyen, P Papastogiannidis, C Scheurer, DM Shackleford, K Sriraghavan, L Stingelin, Y Tang, H Urwyler, X Wang, KL White, S Wittlin, L Zhou, JL Vennerstrom, Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A 2011, 108, 4400.
| Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria.Crossref | GoogleScholarGoogle Scholar |
[7] TD Ashton, SM Devine, JJ Möhrle, B Laleu, JN Burrows, SA Charman, DJ Creek, BE Sleebs, The development process for discovery and clinical advancement of modern antimalarials. J Med Chem 2019, 62, 10526.
| The development process for discovery and clinical advancement of modern antimalarials.Crossref | GoogleScholarGoogle Scholar |
[8] DL Doolan, Malaria research in Australia: looking through the lens of the past towards the future. Int J Parasitol 2021, 51, 1255.
| Malaria research in Australia: looking through the lens of the past towards the future.Crossref | GoogleScholarGoogle Scholar |
[9] S Duffy, VM Avery, Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am J Trop Med Hyg 2012, 86, 84.
| Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening.Crossref | GoogleScholarGoogle Scholar |
[10] WA Guiguemde, AA Shelat, D Bouck, S Duffy, GJ Crowther, PH Davis, DC Smithson, M Connelly, J Clark, F Zhu, MB Jiménez-Díaz, MS Martinez, EB Wilson, AK Tripathi, J Gut, ER Sharlow, I Bathurst, F El Mazouni, JW Fowble, I Forquer, PL McGinley, S Castro, I Angulo-Barturen, S Ferrer, PJ Rosenthal, JL Derisi, DJ Sullivan, JS Lazo, DS Roos, MK Riscoe, MA Phillips, PK Rathod, WC Van Voorhis, VM Avery, RK Guy, Chemical genetics of Plasmodium falciparum. Nature 2010, 465, 311.
| Chemical genetics of Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[11] VM Avery, S Bashyam, JN Burrows, S Duffy, G Papadatos, S Puthukkuti, Y Sambandan, S Singh, T Spangenberg, D Waterson, P Willis, Screening and hit evaluation of a chemical library against blood-stage Plasmodium falciparum. Malar J 2014, 13, 190.
| Screening and hit evaluation of a chemical library against blood-stage Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[12] C Le Manach, D Gonzàlez Cabrera, F Douelle, AT Nchinda, Y Younis, D Taylor, L Wiesner, KL White, E Ryan, C March, S Duffy, VM Avery, D Waterson, MJ Witty, S Wittlin, SA Charman, LJ Street, K Chibale, Medicinal chemistry optimization of antiplasmodial imidazopyridazine hits from high throughput screening of a SoftFocus kinase library: part 1. J Med Chem 2014, 57, 2789.
| Medicinal chemistry optimization of antiplasmodial imidazopyridazine hits from high throughput screening of a SoftFocus kinase library: part 1.Crossref | GoogleScholarGoogle Scholar |
[13] CD Edlin, G Morgans, S Winks, S Duffy, VM Avery, S Wittlin, D Waterson, J Burrows, J Bryans, Identification and in-vitro ADME assessment of a series of novel anti-malarial agents suitable for hit-to-lead chemistry. ACS Med Chem Lett 2012, 3, 570.
| Identification and in-vitro ADME assessment of a series of novel anti-malarial agents suitable for hit-to-lead chemistry.Crossref | GoogleScholarGoogle Scholar |
[14] S Duffy, VM Avery, Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J 2013, 12, 408.
| Identification of inhibitors of Plasmodium falciparum gametocyte development.Crossref | GoogleScholarGoogle Scholar |
[15] L Lucantoni, DA Fidock, VM Avery, Luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob Agents Chemother 2016, 60, 2097.
| Luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes.Crossref | GoogleScholarGoogle Scholar |
[16] S Duffy, S Loganathan, JP Holleran, VM Avery, Large-scale production of Plasmodium falciparum gametocytes for malaria drug discovery. Nat Protoc 2016, 11, 976.
| Large-scale production of Plasmodium falciparum gametocytes for malaria drug discovery.Crossref | GoogleScholarGoogle Scholar |
[17] S Duffy, ML Sykes, AJ Jones, TB Shelper, M Simpson, R Lang, SA Poulsen, BE Sleebs, VM Avery, Screening the medicines for malaria venture pathogen box across multiple pathogens reclassifies starting points for open-source drug discovery. Antimicrob Agents Chemother 2017, 61, e00379-17.
| Screening the medicines for malaria venture pathogen box across multiple pathogens reclassifies starting points for open-source drug discovery.Crossref | GoogleScholarGoogle Scholar |
[18] L Lucantoni, F Silvestrini, M Signore, G Siciliano, M Eldering, KJ Dechering, VM Avery, P Alano, A simple and predictive phenotypic high content imaging assay for Plasmodium falciparum mature gametocytes to identify malaria transmission blocking compounds. Sci Rep 2015, 5, 16414.
| A simple and predictive phenotypic high content imaging assay for Plasmodium falciparum mature gametocytes to identify malaria transmission blocking compounds.Crossref | GoogleScholarGoogle Scholar |
[19] WC Van Voorhis, JH Adams, R Adelfio, V Ahyong, MH Akabas, P Alano, A Alday, Y Alemán Resto, A Alsibaee, A Alzualde, KT Andrews, SV Avery, VM Avery, L Ayong, M Baker, S Baker, C Ben Mamoun, S Bhatia, Q Bickle, L Bounaadja, T Bowling, J Bosch, LE Boucher, FF Boyom, J Brea, M Brennan, A Burton, CR Caffrey, G Camarda, M Carrasquilla, D Carter, M Belen Cassera, K Chih-Chien Cheng, W Chindaudomsate, A Chubb, BL Colon, DD Colón-López, Y Corbett, GJ Crowther, N Cowan, S D’Alessandro, N Le Dang, M Delves, JL DeRisi, AY Du, S Duffy, S Abd El-Salam El-Sayed, MT Ferdig, JA Fernández Robledo, DA Fidock, I Florent, PV Fokou, A Galstian, FJ Gamo, S Gokool, B Gold, T Golub, GM Goldgof, R Guha, WA Guiguemde, N Gural, RK Guy, MA Hansen, KK Hanson, A Hemphill, R Hooft van Huijsduijnen, T Horii, P Horrocks, TB Hughes, C Huston, I Igarashi, K Ingram-Sieber, MA Itoe, A Jadhav, A Naranuntarat Jensen, LT Jensen, RH Jiang, A Kaiser, J Keiser, T Ketas, S Kicka, S Kim, K Kirk, VP Kumar, DE Kyle, MJ Lafuente, S Landfear, N Lee, S Lee, AM Lehane, F Li, D Little, L Liu, M Llinás, MI Loza, A Lubar, L Lucantoni, I Lucet, L Maes, D Mancama, NR Mansour, S March, S McGowan, I Medina Vera, S Meister, L Mercer, J Mestres, AN Mfopa, RN Misra, S Moon, JP Moore, F Morais Rodrigues da Costa, J Müller, A Muriana, S Nakazawa Hewitt, B Nare, C Nathan, N Narraidoo, S Nawaratna, KK Ojo, D Ortiz, G Panic, G Papadatos, S Parapini, K Patra, N Pham, S Prats, DM Plouffe, SA Poulsen, A Pradhan, C Quevedo, RJ Quinn, CA Rice, M Abdo Rizk, A Ruecker St, R Onge, R Salgado Ferreira, J Samra, NG Robinett, U Schlecht, M Schmitt, F Silva Villela, F Silvestrini, R Sinden, DA Smith, T Soldati, A Spitzmüller, SM Stamm, DJ Sullivan, W Sullivan, S Suresh, BM Suzuki, Y Suzuki, SJ Swamidass, D Taramelli, LR Tchokouaha, A Theron, D Thomas, KF Tonissen, S Townson, AK Tripathi, V Trofimov, KO Udenze, I Ullah, C Vallieres, E Vigil, JM Vinetz, P Voong Vinh, H Vu, NA Watanabe, K Weatherby, PM White, AF Wilks, EA Winzeler, E Wojcik, M Wree, W Wu, N Yokoyama, PH Zollo, N Abla, B Blasco, J Burrows, B Laleu, D Leroy, T Spangenberg, T Wells, PA Willis, Open source drug discovery with the malaria box compound collection for neglected diseases and beyond. PLoS Pathog 2016, 12, e1005763.
| Open source drug discovery with the malaria box compound collection for neglected diseases and beyond.Crossref | GoogleScholarGoogle Scholar |
[20] L Lucantoni, S Duffy, SH Adjalley, DA Fidock, VM Avery, Identification of MMV malaria box inhibitors of Plasmodium falciparum early-stage gametocytes using a luciferase-based high-throughput assay. Antimicrob Agents Chemother 2013, 57, 6050.
| Identification of MMV malaria box inhibitors of Plasmodium falciparum early-stage gametocytes using a luciferase-based high-throughput assay.Crossref | GoogleScholarGoogle Scholar |
[21] O Looker, MG Dans, HE Bullen, BE Sleebs, BS Crabb, PR Gilson, The medicines for malaria venture malaria box contains inhibitors of protein secretion in Plasmodium falciparum blood stage parasites. Traffic 2022,
| The medicines for malaria venture malaria box contains inhibitors of protein secretion in Plasmodium falciparum blood stage parasites.Crossref | GoogleScholarGoogle Scholar |
[22] MG Dans, GE Weiss, DW Wilson, BE Sleebs, BS Crabb, TF de Koning-Ward, PR Gilson, Screening the medicines for malaria venture pathogen box for invasion and egress inhibitors of the blood stage of Plasmodium falciparum reveals several inhibitory compounds. Int J Parasitol 2020, 50, 235.
| Screening the medicines for malaria venture pathogen box for invasion and egress inhibitors of the blood stage of Plasmodium falciparum reveals several inhibitory compounds.Crossref | GoogleScholarGoogle Scholar |
[23] BK Dickerman, B Elsworth, SA Cobbold, CQ Nie, MJ McConville, BS Crabb, PR Gilson, Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum. Sci Rep 2016, 6, 37502.
| Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[24] NJ Spillman, RJW Allen, CW McNamara, BKS Yeung, EA Winzeler, TT Diagana, K Kirk, Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 2013, 13, 227.
| Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials.Crossref | GoogleScholarGoogle Scholar |
[25] MB Jiménez-Díaz, D Ebert, Y Salinas, A Pradhan, AM Lehane, ME Myrand-Lapierre, KG O’Loughlin, DM Shackleford, M Justino de Almeida, AK Carrillo, JA Clark, AS Dennis, J Diep, X Deng, S Duffy, AN Endsley, G Fedewa, WA Guiguemde, MG Gómez, G Holbrook, J Horst, CC Kim, J Liu, MC Lee, A Matheny, MS Martínez, G Miller, A Rodríguez-Alejandre, L Sanz, M Sigal, NJ Spillman, PD Stein, Z Wang, F Zhu, D Waterson, S Knapp, A Shelat, VM Avery, DA Fidock, FJ Gamo, SA Charman, JC Mirsalis, H Ma, S Ferrer, K Kirk, I Angulo-Barturen, DE Kyle, JL DeRisi, DM Floyd, RK Guy, (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci U S A 2014, 111, E5455.
| (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium.Crossref | GoogleScholarGoogle Scholar |
[26] NJ Spillman, RJW Allen, K Kirk, Na+ extrusion imposes an acid load on the intraerythrocytic malaria parasite. Mol Biochem Parasitol 2013, 189, 1.
| Na+ extrusion imposes an acid load on the intraerythrocytic malaria parasite.Crossref | GoogleScholarGoogle Scholar |
[27] AM Lehane, MC Ridgway, E Baker, K Kirk, Diverse chemotypes disrupt ion homeostasis in the malaria parasite. Mol Microbiol 2014, 94, 327.
| Diverse chemotypes disrupt ion homeostasis in the malaria parasite.Crossref | GoogleScholarGoogle Scholar |
[28] JEO Rosling, MC Ridgway, RL Summers, K Kirk, AM Lehane, Biochemical characterization and chemical inhibition of PfATP4-associated Na+-ATPase activity in Plasmodium falciparum membranes. J Biol Chem 2018, 293, 13327.
| Biochemical characterization and chemical inhibition of PfATP4-associated Na+-ATPase activity in Plasmodium falciparum membranes.Crossref | GoogleScholarGoogle Scholar |
[29] ASM Dennis, JEO Rosling, AM Lehane, K Kirk, Diverse antimalarials from whole-cell phenotypic screens disrupt malaria parasite ion and volume homeostasis. Sci Rep 2018, 8, 8795.
| Diverse antimalarials from whole-cell phenotypic screens disrupt malaria parasite ion and volume homeostasis.Crossref | GoogleScholarGoogle Scholar |
[30] SV Hapuarachchi, SA Cobbold, SH Shafik, AS Dennis, MJ McConville, RE Martin, K Kirk, AM Lehane, The malaria parasite’s lactate transporter PfFNT is the target of antiplasmodial compounds identified in whole cell phenotypic screens. PLoS Pathog 2017, 13, e1006180.
| The malaria parasite’s lactate transporter PfFNT is the target of antiplasmodial compounds identified in whole cell phenotypic screens.Crossref | GoogleScholarGoogle Scholar |
[31] RV Marchetti, AM Lehane, SH Shafik, M Winterberg, RE Martin, K Kirk, A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum. Nat Commun 2015, 6, 6721.
| A lactate and formate transporter in the intraerythrocytic malaria parasite, Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[32] E Yeh, JL DeRisi, Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol 2011, 9, e1001138.
| Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[33] T Uddin, GI McFadden, CD Goodman, Validation of putative apicoplast-targeting drugs using a chemical supplementation assay in cultured human malaria parasites. Antimicrob Agents Chemother 2018, 62, e01161-17.
| Validation of putative apicoplast-targeting drugs using a chemical supplementation assay in cultured human malaria parasites.Crossref | GoogleScholarGoogle Scholar |
[34] A Paiardini, RS Bamert, K Kannan-Sivaraman, N Drinkwater, SN Mistry, PJ Scammells, S McGowan, Screening the medicines for malaria venture "malaria box" against the Plasmodium falciparum aminopeptidases, M1, M17 and M18. PLoS One 2015, 10, e0115859.
| Screening the medicines for malaria venture "malaria box" against the Plasmodium falciparum aminopeptidases, M1, M17 and M18.Crossref | Plasmodium falciparum aminopeptidases, M1, M17 and M18.&journal=PLoS One&volume=10&pages=e0115859-&publication_year=2015&author=A%20Paiardini&hl=en&doi=10.1371/journal.pone.0115859" target="_blank" rel="nofollow noopener noreferrer" class="reftools">GoogleScholarGoogle Scholar |
[35] IS Lucet, A Tobin, D Drewry, AF Wilks, C Doerig, Plasmodium kinases as targets for new-generation antimalarials. Future Med Chem 2012, 4, 2295.
| Plasmodium kinases as targets for new-generation antimalarials.Crossref | GoogleScholarGoogle Scholar |
[36] BE Sleebs, KE Jarman, S Frolich, W Wong, J Healer, W Dai, IS Lucet, DW Wilson, AF Cowman, Development and application of a high-throughput screening assay for identification of small molecule inhibitors of the P. falciparum reticulocyte binding-like homologue 5 protein. Int J Parasitol Drugs Drug Resist 2020, 14, 188.
| Development and application of a high-throughput screening assay for identification of small molecule inhibitors of the P. falciparum reticulocyte binding-like homologue 5 protein.Crossref | GoogleScholarGoogle Scholar |
[37] H Vu, L Pedro, T Mak, B McCormick, J Rowley, M Liu, A Di Capua, B Williams-Noonan, NB Pham, R Pouwer, B Nguyen, KT Andrews, T Skinner-Adams, J Kim, WGJ Hol, R Hui, GJ Crowther, WC Van Voorhis, RJ Quinn, Fragment-based screening of a natural product library against 62 potential malaria drug targets employing native mass spectrometry. ACS Infect Dis 2018, 4, 431.
| Fragment-based screening of a natural product library against 62 potential malaria drug targets employing native mass spectrometry.Crossref | GoogleScholarGoogle Scholar |
[38] H Vu, C Roullier, M Campitelli, KR Trenholme, DL Gardiner, KT Andrews, T Skinner-Adams, GJ Crowther, WC Van Voorhis, RJ Quinn, Plasmodium gametocyte inhibition identified from a natural-product-based fragment library. ACS Chem Biol 2013, 8, 2654.
| Plasmodium gametocyte inhibition identified from a natural-product-based fragment library.Crossref | GoogleScholarGoogle Scholar |
[39] SA Cobbold, MJ McConville, Determining the mode of action of antimalarial drugs using time-resolved LC-MS-based metabolite profiling. Methods Mol Biol 2019, 1859, 225.
| Determining the mode of action of antimalarial drugs using time-resolved LC-MS-based metabolite profiling.Crossref | GoogleScholarGoogle Scholar |
[40] SA Cobbold, HH Chua, B Nijagal, DJ Creek, SA Ralph, MJ McConville, Metabolic dysregulation induced in Plasmodium falciparum by dihydroartemisinin and other front-line antimalarial drugs. J Infect Dis 2016, 213, 276.
| Metabolic dysregulation induced in Plasmodium falciparum by dihydroartemisinin and other front-line antimalarial drugs.Crossref | GoogleScholarGoogle Scholar |
[41] G Siddiqui, C Giannangelo, A De Paoli, AK Schuh, KC Heimsch, D Anderson, TG Brown, CA MacRaild, J Wu, X Wang, Y Dong, JL Vennerstrom, K Becker, DJ Creek, Peroxide antimalarial drugs target redox homeostasis in Plasmodium falciparum infected red blood cells. ACS Infect Dis 2022, 8, 210.
| Peroxide antimalarial drugs target redox homeostasis in Plasmodium falciparum infected red blood cells.Crossref | GoogleScholarGoogle Scholar |
[42] C Giannangelo, G Siddiqui, A De Paoli, BM Anderson, LE Edgington-Mitchell, SA Charman, DJ Creek, System-wide biochemical analysis reveals ozonide antimalarials initially act by disrupting Plasmodium falciparum haemoglobin digestion. PLoS Pathog 2020, 16, e1008485.
| System-wide biochemical analysis reveals ozonide antimalarials initially act by disrupting Plasmodium falciparum haemoglobin digestion.Crossref | GoogleScholarGoogle Scholar |
[43] GW Birrell, MP Challis, A De Paoli, D Anderson, SM Devine, GD Heffernan, DP Jacobus, MD Edstein, G Siddiqui, DJ Creek, Multi-omic characterization of the mode of action of a potent new antimalarial compound, JPC-3210, against Plasmodium falciparum. Mol Cell Proteomics 2020, 19, 308.
| Multi-omic characterization of the mode of action of a potent new antimalarial compound, JPC-3210, against Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[44] G Siddiqui, A Srivastava, AS Russell, DJ Creek, Multi-omics based identification of specific biochemical changes associated with PfKelch13-mutant artemisinin-resistant Plasmodium falciparum. J Infect Dis 2017, 215, 1435.
| Multi-omics based identification of specific biochemical changes associated with PfKelch13-mutant artemisinin-resistant Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[45] DJ Creek, HH Chua, SA Cobbold, B Nijagal, JI MacRae, BK Dickerman, PR Gilson, SA Ralph, MJ McConville, Metabolomics-based screening of the malaria box reveals both novel and established mechanisms of action. Antimicrob Agents Chemother 2016, 60, 6650.
| Metabolomics-based screening of the malaria box reveals both novel and established mechanisms of action.Crossref | GoogleScholarGoogle Scholar |
[46] SA Charman, A Andreu, H Barker, S Blundell, A Campbell, M Campbell, G Chen, FCK Chiu, E Crighton, K Katneni, J Morizzi, R Patil, T Pham, E Ryan, J Saunders, DM Shackleford, KL White, L Almond, M Dickins, DA Smith, JJ Moehrle, JN Burrows, N Abla, An in vitro toolbox to accelerate anti-malarial drug discovery and development. Malar J 2020, 19, 1.
| An in vitro toolbox to accelerate anti-malarial drug discovery and development.Crossref | GoogleScholarGoogle Scholar |
[47] Y Dong, S Wittlin, K Sriraghavan, J Chollet, SA Charman, WN Charman, C Scheurer, H Urwyler, J Santo Tomas, C Snyder, DJ Creek, J Morizzi, M Koltun, H Matile, X Wang, M Padmanilayam, Y Tang, A Dorn, R Brun, JL Vennerstrom, The structure−activity relationship of the antimalarial ozonide arterolane (OZ277). J Med Chem 2010, 53, 481.
| The structure−activity relationship of the antimalarial ozonide arterolane (OZ277).Crossref | GoogleScholarGoogle Scholar |
[48] Y Dong, X Wang, S Kamaraj, VJ Bulbule, FCK Chiu, J Chollet, M Dhanasekaran, CD Hein, P Papastogiannidis, J Morizzi, DM Shackleford, H Barker, E Ryan, C Scheurer, Y Tang, Q Zhao, L Zhou, KL White, H Urwyler, WN Charman, H Matile, S Wittlin, SA Charman, JL Vennerstrom, Structure–activity relationship of the antimalarial ozonide artefenomel (OZ439). J Med Chem 2017, 60, 2654.
| Structure–activity relationship of the antimalarial ozonide artefenomel (OZ439).Crossref | GoogleScholarGoogle Scholar |
[49] JJ Moehrle, S Duparc, C Siethoff, PL van Giersbergen, JC Craft, S Arbe-Barnes, SA Charman, M Gutierrez, S Wittlin, JL Vennerstrom, First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol 2013, 75, 524.
| First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials.Crossref | GoogleScholarGoogle Scholar |
[50] MA Phillips, J Lotharius, K Marsh, J White, A Dayan, KL White, JW Njoroge, F El Mazouni, Y Lao, S Kokkonda, DR Tomchick, X Deng, T Laird, SN Bhatia, S March, CL Ng, DA Fidock, S Wittlin, M Lafuente-Monasterio, FJ Benito, LM Alonso, MS Martinez, MB Jimenez-Diaz, SF Bazaga, I Angulo-Barturen, JN Haselden, J Louttit, Y Cui, A Sridhar, AM Zeeman, C Kocken, R Sauerwein, K Dechering, VM Avery, S Duffy, M Delves, R Sinden, A Ruecker, KS Wickham, R Rochford, J Gahagen, L Iyer, E Riccio, J Mirsalis, I Bathhurst, T Rueckle, X Ding, B Campo, D Leroy, MJ Rogers, PK Rathod, JN Burrows, SA Charman, A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med 2015, 7, 296ra111.
| A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria.Crossref | GoogleScholarGoogle Scholar |
[51] MA Phillips, KL White, S Kokkonda, X Deng, J White, F El Mazouni, K Marsh, DR Tomchick, K Manjalanagara, KR Rudra, G Wirjanata, R Noviyanti, RN Price, J Marfurt, DM Shackleford, FCK Chiu, M Campbell, MB Jimenez-Diaz, SF Bazaga, I Angulo-Barturen, MS Martinez, M Lafuente-Monasterio, W Kaminsky, K Silue, AM Zeeman, C Kocken, D Leroy, B Blasco, E Rossignol, T Rueckle, D Matthews, JN Burrows, D Waterson, MJ Palmer, PK Rathod, SA Charman, A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria. ACS Infect Dis 2016, 2, 945.
| A triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with improved drug-like properties for treatment and prevention of malaria.Crossref | GoogleScholarGoogle Scholar |
[52] T Paquet, C Le Manach, DG Cabrera, Y Younis, PP Henrich, TS Abraham, MCS Lee, R Basak, S Ghidelli-Disse, MJ Lafuente-Monasterio, M Bantscheff, A Ruecker, AM Blagborough, SE Zakutansky, AM Zeeman, KL White, DM Shackleford, J Mannila, J Morizzi, C Scheurer, I Angulo-Barturen, MS Martínez, S Ferrer, LM Sanz, FJ Gamo, J Reader, M Botha, KJ Dechering, RW Sauerwein, A Tungtaeng, P Vanachayangkul, CS Lim, J Burrows, MJ Witty, KC Marsh, C Bodenreider, R Rochford, SM Solapure, MB Jiménez-Díaz, S Wittlin, SA Charman, C Donini, B Campo, LM Birkholtz, KK Hanson, G Drewes, CHM Kocken, MJ Delves, D Leroy, DA Fidock, D Waterson, LJ Street, K Chibale, Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med 2017, 9, eaad9735.
| Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase.Crossref | GoogleScholarGoogle Scholar |
[53] C Brunschwig, N Lawrence, D Taylor, E Abay, M Njoroge, GS Basarab, C Le Manach, T Paquet, DG Cabrera, AT Nchinda, C de Kock, L Wiesner, P Denti, D Waterson, B Blasco, D Leroy, MJ Witty, C Donini, J Duffy, S Wittlin, KL White, SA Charman, MB Jiménez-Díaz, I Angulo-Barturen, E Herreros, FJ Gamo, R Rochford, D Mancama, TL Coetzer, ME van der Watt, J Reader, LM Birkholtz, KC Marsh, SM Solapure, JE Burke, JA McPhail, M Vanaerschot, DA Fidock, PV Fish, P Siegl, DA Smith, G Wirjanata, R Noviyanti, RN Price, J Marfurt, KD Silue, LJ Street, K Chibale, UCT943, a next-generation Plasmodium falciparum PI4K inhibitor preclinical candidate for the treatment of malaria. Antimicrob Agents Chemother 2018, 62, e00012-18.
| UCT943, a next-generation Plasmodium falciparum PI4K inhibitor preclinical candidate for the treatment of malaria.Crossref | GoogleScholarGoogle Scholar |
[54] KA Collins, AN Abd-Rahman, L Marquart, E Ballard, N Gobeau, P Griffin, S Chalon, JJ Möhrle, JS McCarthy, Antimalarial activity of artefenomel against asexual parasites and transmissible gametocytes during experimental blood-stage Plasmodium vivax infection. J Infect Dis 2022, 225, 1062.
| Antimalarial activity of artefenomel against asexual parasites and transmissible gametocytes during experimental blood-stage Plasmodium vivax infection.Crossref | GoogleScholarGoogle Scholar |
[55] JS McCarthy, M Baker, P O’Rourke, L Marquart, P Griffin, R Hooft van Huijsduijnen, JJ Möhrle, J Antimicrob Chemother 2016, 71, 2620.
| Crossref | GoogleScholarGoogle Scholar |
[56] JS McCarthy, AN Abd-Rahman, KA Collins, L Marquart, P Griffin, A Kümmel, A Fuchs, C Winnips, V Mishra, K Csermak-Renner, JP Jain, P Gandhi, Defining the antimalarial activity of cipargamin in healthy volunteers experimentally infected with blood-stage Plasmodium falciparum. Antimicrob Agents Chemother 2021, 65, e01423-20.
| Defining the antimalarial activity of cipargamin in healthy volunteers experimentally infected with blood-stage Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[57] JS McCarthy, T Rückle, SL Elliott, E Ballard, KA Collins, L Marquart, P Griffin, S Chalon, JJ Möhrle, A single-dose combination study with the experimental antimalarials artefenomel and DSM265 to determine safety and antimalarial activity against blood-stage Plasmodium falciparum in healthy volunteers. Antimicrob Agents Chemother 2019, 64, e01371-19.
| A single-dose combination study with the experimental antimalarials artefenomel and DSM265 to determine safety and antimalarial activity against blood-stage Plasmodium falciparum in healthy volunteers.Crossref | GoogleScholarGoogle Scholar |
[58] KA Collins, T Rückle, S Elliott, L Marquart, E Ballard, S Chalon, P Griffin, JJ Möhrle, JS McCarthy, DSM265 at 400 milligrams clears asexual stage parasites but not mature gametocytes from the blood of healthy subjects experimentally infected with Plasmodium falciparum. Antimicrob Agents Chemother 2019, 63, e01837-18.
| DSM265 at 400 milligrams clears asexual stage parasites but not mature gametocytes from the blood of healthy subjects experimentally infected with Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[59] P Sinxadi, C Donini, H Johnstone, G Langdon, L Wiesner, E Allen, S Duparc, S Chalon, JS McCarthy, U Lorch, K Chibale, J Möhrle, KI Barnes, Safety, tolerability, pharmacokinetics, and antimalarial activity of the novel Plasmodium phosphatidylinositol 4-kinase inhibitor MMV390048 in healthy volunteers. Antimicrob Agents Chemother 2020, 64, e01896-19.
| Safety, tolerability, pharmacokinetics, and antimalarial activity of the novel Plasmodium phosphatidylinositol 4-kinase inhibitor MMV390048 in healthy volunteers.Crossref | GoogleScholarGoogle Scholar |
[60] JS McCarthy, C Donini, S Chalon, J Woodford, L Marquart, KA Collins, FD Rozenberg, DA Fidock, MH Cherkaoui-Rbati, N Gobeau, JJ Möhrle, A Phase 1, placebo-controlled, randomized, single ascending dose study and a volunteer infection study to characterize the safety, pharmacokinetics, and antimalarial activity of the Plasmodium phosphatidylinositol 4-kinase inhibitor MMV390048. Clin Infect Dis 2020, 71, e657.
| A Phase 1, placebo-controlled, randomized, single ascending dose study and a volunteer infection study to characterize the safety, pharmacokinetics, and antimalarial activity of the Plasmodium phosphatidylinositol 4-kinase inhibitor MMV390048.Crossref | GoogleScholarGoogle Scholar |
[61] AH Gaur, JS McCarthy, JC Panetta, RH Dallas, J Woodford, L Tang, AM Smith, TB Stewart, KC Branum, BB Freeman III, ND Patel, E John, S Chalon, S Ost, RN Heine, JL Richardson, R Christensen, PM Flynn, Y Van Gessel, B Mitasev, JJ Möhrle, F Gusovsky, L Bebrevska, RK Guy, Safety, tolerability, pharmacokinetics, and antimalarial efficacy of a novel Plasmodium falciparum ATP4 inhibitor SJ733: a first-in-human and induced blood-stage malaria phase 1a/b trial. Lancet Infect Dis 2020, 20, 964.
| Safety, tolerability, pharmacokinetics, and antimalarial efficacy of a novel Plasmodium falciparum ATP4 inhibitor SJ733: a first-in-human and induced blood-stage malaria phase 1a/b trial.Crossref | GoogleScholarGoogle Scholar |
[62] A Krause, J Dingemanse, A Mathis, L Marquart, JJ Möhrle, JS McCarthy, Pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of Actelion-451840 in an induced blood stage malaria study in healthy subjects Br J Clin Pharmacol 2016, 82, 412.
| Pharmacokinetic/pharmacodynamic modelling of the antimalarial effect of Actelion-451840 in an induced blood stage malaria study in healthy subjectsCrossref | GoogleScholarGoogle Scholar |
[63] BE Barber, M Fernandez, HB Patel, C Barcelo, SD Woolley, H Patel, S Llewellyn, AN Abd-Rahman, S Sharma, M Jain, A Ghoghari, I Di Resta, A Fuchs, I Deni, T Yeo, S Mok, DA Fidock, S Chalon, JJ Möhrle, D Parmar, JS McCarthy, K Kansagra, Safety, pharmacokinetics, and antimalarial activity of the novel triaminopyrimidine ZY-19489: a first-in-human, randomised, placebo-controlled, double-blind, single ascending dose study, pilot food-effect study, and volunteer infection study. Lancet Infect Dis 2022, 22, 879.
| Safety, pharmacokinetics, and antimalarial activity of the novel triaminopyrimidine ZY-19489: a first-in-human, randomised, placebo-controlled, double-blind, single ascending dose study, pilot food-effect study, and volunteer infection study.Crossref | GoogleScholarGoogle Scholar |
[64] RA Davis, MS Buchanan, S Duffy, VM Avery, SA Charman, WN Charman, KL White, DM Shackleford, MD Edstein, KT Andrews, D Camp, RJ Quinn, Antimalarial activity of pyrroloiminoquinones from the Australian marine sponge Zyzzya sp. J Med Chem 2012, 55, 5851.
| Antimalarial activity of pyrroloiminoquinones from the Australian marine sponge Zyzzya sp.Crossref | GoogleScholarGoogle Scholar |
[65] MSJ Arnold, JR Macdonald, RJ Quinn, TS Skinner-Adams, KT Andrews, GM Fisher, Antiplasmodial activity of the natural product compounds alstonine and himbeline. Int J Parasitol Drugs Drug Resist 2021, 16, 17.
| Antiplasmodial activity of the natural product compounds alstonine and himbeline.Crossref | GoogleScholarGoogle Scholar |
[66] R Kumar, S Duffy, VM Avery, RA Davis, Synthesis of antimalarial amide analogues based on the plant serrulatane diterpenoid 3,7,8-trihydroxyserrulat-14-en-19-oic acid. Bioorg Med Chem Lett 2017, 27, 4091.
| Synthesis of antimalarial amide analogues based on the plant serrulatane diterpenoid 3,7,8-trihydroxyserrulat-14-en-19-oic acid.Crossref | GoogleScholarGoogle Scholar |
[67] RA Davis, S Duffy, S Fletcher, VM Avery, RJ Quinn, Thiaplakortones A–D: antimalarial thiazine alkaloids from the Australian marine sponge Plakortis lita. J Org Chem 2013, 78, 9608.
| Thiaplakortones A–D: antimalarial thiazine alkaloids from the Australian marine sponge Plakortis lita.Crossref | GoogleScholarGoogle Scholar |
[68] RH Pouwer, SM Deydier, PV Le, BD Schwartz, NC Franken, RA Davis, MJ Coster, SA Charman, MD Edstein, TS Skinner-Adams, KT Andrews, ID Jenkins, RJ Quinn, Total synthesis of thiaplakortone A: derivatives as metabolically stable leads for the treatment of malaria. ACS Med Chem Lett 2013, 5, 178.
| Total synthesis of thiaplakortone A: derivatives as metabolically stable leads for the treatment of malaria.Crossref | GoogleScholarGoogle Scholar |
[69] BD Schwartz, TS Skinner-Adams, KT Andrews, MJ Coster, MD Edstein, D MacKenzie, SA Charman, M Koltun, S Blundell, A Campbell, RH Pouwer, RJ Quinn, KD Beattie, PC Healy, RA Davis, Synthesis and antimalarial evaluation of amide and urea derivatives based on the thiaplakortone A natural product scaffold. Org Biomol Chem 2015, 13, 1558.
| Synthesis and antimalarial evaluation of amide and urea derivatives based on the thiaplakortone A natural product scaffold.Crossref | GoogleScholarGoogle Scholar |
[70] S Johnson, R Rahmani, DR Drew, MJ Williams, M Wilkinson, YH Tan, JX Huang, CJ Tonkin, JG Beeson, J Baum, BJ Smith, JB Baell, Truncated latrunculins as actin inhibitors targeting Plasmodium falciparum motility and host cell invasion. J Med Chem 2016, 59, 10994.
| Truncated latrunculins as actin inhibitors targeting Plasmodium falciparum motility and host cell invasion.Crossref | GoogleScholarGoogle Scholar |
[71] T Conroy, JT Guo, NH Hunt, RJ Payne, Total synthesis and antimalarial activity of symplostatin 4. Org Lett 2010, 12, 5576.
| Total synthesis and antimalarial activity of symplostatin 4.Crossref | GoogleScholarGoogle Scholar |
[72] T Conroy, JT Guo, RG Linington, NH Hunt, RJ Payne, Total synthesis, stereochemical assignment, and antimalarial activity of gallinamide A. Chemistry 2011, 17, 13544.
| Total synthesis, stereochemical assignment, and antimalarial activity of gallinamide A.Crossref | GoogleScholarGoogle Scholar |
[73] T Conroy, JT Guo, N Elias, KM Cergol, J Gut, J Legac, L Khatoon, Y Liu, S McGowan, PJ Rosenthal, NH Hunt, RJ Payne, Synthesis of gallinamide A analogues as potent falcipain inhibitors and antimalarials. J Med Chem 2014, 57, 10557.
| Synthesis of gallinamide A analogues as potent falcipain inhibitors and antimalarials.Crossref | GoogleScholarGoogle Scholar |
[74] K Ban, S Duffy, Y Khakham, VM Avery, A Hughes, O Montagnat, K Katneni, E Ryan, JB Baell, 3-alkylthio-1,2,4-triazine dimers with potent antimalarial activity. Bioorg Med Chem Lett 2010, 20, 6024.
| 3-alkylthio-1,2,4-triazine dimers with potent antimalarial activity.Crossref | GoogleScholarGoogle Scholar |
[75] KM Katherine, L Lucantoni, M Chavchich, M Abraham, A De Paoli, MR Luth, A-M Zeeman, MJ Delves, FS-R Terán, U Straschil, J Baum, CHM Kocken, SA Ralph, EA Winzeler, VM Avery, MD Edstein, JB Baell, DJ Creek, The novel bis-1,2,4-triazine MIPS-0004373 demonstrates rapid and potent activity against all blood stages of the malaria parasite. Antimicrob Agents Chemother 65, e00311-21.
| The novel bis-1,2,4-triazine MIPS-0004373 demonstrates rapid and potent activity against all blood stages of the malaria parasite.Crossref | GoogleScholarGoogle Scholar |
[76] L Xue, D-H Shi, JR Harjani, F Huang, JG Beveridge, T Dingjan, K Ban, S Diab, S Duffy, L Lucantoni, S Fletcher, FCK Chiu, S Blundell, K Ellis, SA Ralph, G Wirjanata, S Teguh, R Noviyanti, M Chavchich, D Creek, RN Price, J Marfurt, SA Charman, ME Cuellar, JM Strasser, JL Dahlin, MA Walters, MD Edstein, VM Avery, JB Baell, 3,3′-disubstituted 5,5′-Bi(1,2,4-triazine) derivatives with potent in vitro and in vivo antimalarial activity. J Med Chem 2019, 62, 2485.
| 3,3′-disubstituted 5,5′-Bi(1,2,4-triazine) derivatives with potent in vitro and in vivo antimalarial activity.Crossref | GoogleScholarGoogle Scholar |
[77] DL Priebbenow, M Mathiew, D-H Shi, JR Harjani, JG Beveridge, M Chavchich, MD Edstein, S Duffy, VM Avery, RT Jacobs, S Brand, DM Shackleford, W Wang, L Zhong, G Lee, E Tay, H Barker, E Crighton, KL White, SA Charman, A De Paoli, DJ Creek, JB Baell, Discovery of potent and fast-acting antimalarial Bis-1,2,4-triazines. J Med Chem 2021, 64, 4150.
| Discovery of potent and fast-acting antimalarial Bis-1,2,4-triazines.Crossref | GoogleScholarGoogle Scholar |
[78] AE Williamson, PM Ylioja, MN Robertson, Y Antonova-Koch, V Avery, JB Baell, H Batchu, S Batra, JN Burrows, S Bhattacharyya, F Calderon, SA Charman, J Clark, B Crespo, M Dean, SL Debbert, M Delves, ASM Dennis, F Deroose, S Duffy, S Fletcher, G Giaever, I Hallyburton, F-J Gamo, M Gebbia, RK Guy, Z Hungerford, K Kirk, MJ Lafuente-Monasterio, A Lee, S Meister, C Nislow, JP Overington, G Papadatos, L Patiny, J Pham, SA Ralph, A Ruecker, E Ryan, C Southan, K Srivastava, C Swain, MJ Tarnowski, P Thomson, P Turner, IM Wallace, TNC Wells, K White, L White, P Willis, EA Winzeler, S Wittlin, MH Todd, Open source drug discovery: highly potent antimalarial compounds derived from the Tres Cantos arylpyrroles. ACS Cent Sci 2016, 2, 687.
| Open source drug discovery: highly potent antimalarial compounds derived from the Tres Cantos arylpyrroles.Crossref | GoogleScholarGoogle Scholar |
[79] EG Tse, L Aithani, M Anderson, J Cardoso-Silva, G Cincilla, GJ Conduit, M Galushka, D Guan, I Hallyburton, BWJ Irwin, K Kirk, AM Lehane, JCR Lindblom, R Lui, S Matthews, J McCulloch, A Motion, HL Ng, M Öeren, MN Robertson, V Spadavecchio, VA Tatsis, WP van Hoorn, AD Wade, TM Whitehead, P Willis, MH Todd, An open drug discovery competition: experimental validation of predictive models in a series of novel antimalarials. J Med Chem 2021, 64, 16450.
| An open drug discovery competition: experimental validation of predictive models in a series of novel antimalarials.Crossref | GoogleScholarGoogle Scholar |
[80] PR Gilson, W Nguyen, WA Poole, JE Teixeira, JK Thompson, K Guo, RJ Stewart, TD Ashton, KL White, LM Sanz, FJ Gamo, SA Charman, S Wittlin, J Duffy, CJ Tonkin, WH Tham, BS Crabb, BM Cooke, CD Huston, AF Cowman, BE Sleebs, Evaluation of 4-amino 2-anilinoquinazolines against Plasmodium and other apicomplexan parasites in vitro and in a P. falciparum humanized NOD-scid IL2Rγnull mouse model of malaria. Antimicrob Agents Chemother 2019, 63, e01804-18.
| Evaluation of 4-amino 2-anilinoquinazolines against Plasmodium and other apicomplexan parasites in vitro and in a P. falciparum humanized NOD-scid IL2Rγnull mouse model of malaria.Crossref | GoogleScholarGoogle Scholar |
[81] PR Gilson, C Tan, KE Jarman, KN Lowes, JM Curtis, W Nguyen, AE Di Rago, HE Bullen, B Prinz, S Duffy, JB Baell, CA Hutton, H Jousset Subroux, BS Crabb, VM Avery, AF Cowman, BE Sleebs, Optimization of 2-anilino 4-amino substituted quinazolines into potent antimalarial agents with oral in vivo activity. J Med Chem 2017, 60, 1171.
| Optimization of 2-anilino 4-amino substituted quinazolines into potent antimalarial agents with oral in vivo activity.Crossref | GoogleScholarGoogle Scholar |
[82] TD Ashton, A Ngo, P Favuzza, HE Bullen, MR Gancheva, O Romeo, M Parkyn Schneider, N Nguyen, RWJ Steel, S Duffy, KN Lowes, HJ Sabroux, VM Avery, JA Boddey, DW Wilson, AF Cowman, PR Gilson, BE Sleebs, Property activity refinement of 2-anilino 4-amino substituted quinazolines as antimalarials with fast acting asexual parasite activity. Bioorg Chem 2021, 117, 105359.
| Property activity refinement of 2-anilino 4-amino substituted quinazolines as antimalarials with fast acting asexual parasite activity.Crossref | GoogleScholarGoogle Scholar |
[83] W Nguyen, MG Dans, A Ngo, MR Gancheva, O Romeo, S Duffy, TF de Koning-Ward, KN Lowes, HJ Sabroux, VM Avery, DW Wilson, PR Gilson, BE Sleebs, Structure activity refinement of phenylsulfonyl piperazines as antimalarials that block erythrocytic invasion. Eur J Med Chem 2021, 214, 113253.
| Structure activity refinement of phenylsulfonyl piperazines as antimalarials that block erythrocytic invasion.Crossref | GoogleScholarGoogle Scholar |
[84] BL Bailey, W Nguyen, A Ngo, CD Goodman, MR Gancheva, P Favuzza, LM Sanz, FJ Gamo, KN Lowes, GI McFadden, DW Wilson, B Laleu, S Brand, PF Jackson, AF Cowman, BE Sleebs, Optimisation of 2-(N-phenyl carboxamide) triazolopyrimidine antimalarials with moderate to slow acting erythrocytic stage activity. Bioorg Chem 2021, 115, 105244.
| Optimisation of 2-(N-phenyl carboxamide) triazolopyrimidine antimalarials with moderate to slow acting erythrocytic stage activity.Crossref | GoogleScholarGoogle Scholar |
[85] MJ Buskes, KL Harvey, B Prinz, BS Crabb, PR Gilson, DJD Wilson, BM Abbott, Exploration of 3-methylisoquinoline-4-carbonitriles as protein kinase A inhibitors of Plasmodium falciparum. Bioorg Med Chem 2016, 24, 2389.
| Exploration of 3-methylisoquinoline-4-carbonitriles as protein kinase A inhibitors of Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[86] MJ Buskes, KL Harvey, BJ Richards, R Kalhor, RM Christoff, CK Gardhi, DR Littler, ED Cope, B Prinz, GE Weiss, NJ O’Brien, BS Crabb, LW Deady, PR Gilson, BM Abbott, Antimalarial activity of novel 4-cyano-3-methylisoquinoline inhibitors against Plasmodium falciparum: design, synthesis and biological evaluation. Org Biomol Chem 2016, 14, 4617.
| Antimalarial activity of novel 4-cyano-3-methylisoquinoline inhibitors against Plasmodium falciparum: design, synthesis and biological evaluation.Crossref | GoogleScholarGoogle Scholar |
[87] PR Gilson, R Kumarasingha, J Thompson, X Zhang, JS Penington, R Kalhor, HE Bullen, AM Lehane, MG Dans, TF de Koning-Ward, JK Holien, TP Soares da Costa, MD Hulett, MJ Buskes, BS Crabb, K Kirk, AT Papenfuss, AF Cowman, BM Abbott, A 4-cyano-3-methylisoquinoline inhibitor of Plasmodium falciparum growth targets the sodium efflux pump PfATP4. Sci Rep 2019, 9, 10292.
[88] B Krishnarjuna, SS Lim, SM Devine, CO Debono, R Lam, IR Chandrashekaran, G Jaipuria, H Yagi, HS Atreya, MJ Scanlon, CA MacRaild, PJ Scammells, RS Norton, Solution NMR characterization of apical membrane antigen 1 and small molecule interactions as a basis for designing new antimalarials. J Mol Recognit 2016, 29, 281.
| Solution NMR characterization of apical membrane antigen 1 and small molecule interactions as a basis for designing new antimalarials.Crossref | GoogleScholarGoogle Scholar |
[89] SM Devine, MP Challis, JK Kigotho, G Siddiqui, A De Paoli, CA MacRaild, VM Avery, DJ Creek, RS Norton, PJ Scammells, Discovery and development of 2-aminobenzimidazoles as potent antimalarials. Eur J Med Chem 2021, 221, 113518.
| Discovery and development of 2-aminobenzimidazoles as potent antimalarials.Crossref | GoogleScholarGoogle Scholar |
[90] D Hocková, A Holý, M Masojídková, DT Keough, J de Jersey, LW Guddat, Synthesis of branched 9-[2-(2-phosphonoethoxy)ethyl]purines as a new class of acyclic nucleoside phosphonates which inhibit Plasmodium falciparum hypoxanthine–guanine–xanthine phosphoribosyltransferase. Bioorg Med Chem 2009, 17, 6218.
| Synthesis of branched 9-[2-(2-phosphonoethoxy)ethyl]purines as a new class of acyclic nucleoside phosphonates which inhibit Plasmodium falciparum hypoxanthine–guanine–xanthine phosphoribosyltransferase.Crossref | GoogleScholarGoogle Scholar |
[91] P Špaček, DT Keough, M Chavchich, M Dračínský, Z Janeba, L Naesens, MD Edstein, LW Guddat, D Hocková, J Med Chem 2017, 60, 7539.
| Crossref | GoogleScholarGoogle Scholar |
[92] J Frydrych, DT Keough, M Chavchich, J Travis, M Dračínský, MD Edstein, LW Guddat, D Hocková, Z Janeba, Nucleotide analogues containing a pyrrolidine, piperidine or piperazine ring: synthesis and evaluation of inhibition of plasmodial and human 6-oxopurine phosphoribosyltransferases and in vitro antimalarial activity. Eur J Med Chem 2021, 219, 113416.
| Nucleotide analogues containing a pyrrolidine, piperidine or piperazine ring: synthesis and evaluation of inhibition of plasmodial and human 6-oxopurine phosphoribosyltransferases and in vitro antimalarial activity.Crossref | GoogleScholarGoogle Scholar |
[93] T Klejch, DT Keough, M Chavchich, J Travis, J Skácel, R Pohl, Z Janeba, MD Edstein, VM Avery, LW Guddat, D Hocková, Sulfide, sulfoxide and sulfone bridged acyclic nucleoside phosphonates as inhibitors of the Plasmodium falciparum and human 6-oxopurine phosphoribosyltransferases: Synthesis and evaluation. Eur J Med Chem 2019, 183, 111667.
| Sulfide, sulfoxide and sulfone bridged acyclic nucleoside phosphonates as inhibitors of the Plasmodium falciparum and human 6-oxopurine phosphoribosyltransferases: Synthesis and evaluation.Crossref | GoogleScholarGoogle Scholar |
[94] MM Kaiser, D Hocková, TH Wang, M Dračínský, L Poštová-Slavětínská, E Procházková, MD Edstein, M Chavchich, DT Keough, LW Guddat, Z Janeba, Synthesis and evaluation of novel acyclic nucleoside phosphonates as inhibitors of Plasmodium falciparum and human 6-oxopurine phosphoribosyltransferases. ChemMedChem 2015, 10, 1707.
| Synthesis and evaluation of novel acyclic nucleoside phosphonates as inhibitors of Plasmodium falciparum and human 6-oxopurine phosphoribosyltransferases.Crossref | GoogleScholarGoogle Scholar |
[95] DT Keough, D Rejman, R Pohl, E Zborníková, D Hocková, T Croll, MD Edstein, GW Birrell, M Chavchich, LMJ Naesens, GK Pierens, IM Brereton, LW Guddat, Design of Plasmodium vivax hypoxanthine-guanine phosphoribosyltransferase inhibitors as potential antimalarial therapeutics. ACS Chem Biol 2018, 13, 82.
| Design of Plasmodium vivax hypoxanthine-guanine phosphoribosyltransferase inhibitors as potential antimalarial therapeutics.Crossref | GoogleScholarGoogle Scholar |
[96] TS Skinner-Adams, CM Stack, KR Trenholme, CL Brown, J Grembecka, J Lowther, A Mucha, M Drag, P Kafarski, S McGowan, JC Whisstock, DL Gardiner, JP Dalton, Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials. Trends Biochem Sci 2010, 35, 53.
| Plasmodium falciparum neutral aminopeptidases: new targets for anti-malarials.Crossref | GoogleScholarGoogle Scholar |
[97] TS Skinner-Adams, J Lowther, F Teuscher, CM Stack, J Grembecka, A Mucha, P Kafarski, KR Trenholme, JP Dalton, DL Gardiner, Identification of phosphinate dipeptide analog inhibitors directed against the Plasmodium falciparum M17 leucine aminopeptidase as lead antimalarial compounds. J Med Chem 2007, 50, 6024.
| Identification of phosphinate dipeptide analog inhibitors directed against the Plasmodium falciparum M17 leucine aminopeptidase as lead antimalarial compounds.Crossref | GoogleScholarGoogle Scholar |
[98] CM Stack, J Lowther, E Cunningham, S Donnelly, DL Gardiner, KR Trenholme, TS Skinner-Adams, F Teuscher, J Grembecka, A Mucha, P Kafarski, L Lua, A Bell, JP Dalton, Characterization of the Plasmodium falciparum M17 leucyl aminopeptidase: a protease involved in amino acid regulation with potential for antimalarial drug development. J Biol Chem 2007, 282, 2069.
| Characterization of the Plasmodium falciparum M17 leucyl aminopeptidase: a protease involved in amino acid regulation with potential for antimalarial drug development.Crossref | GoogleScholarGoogle Scholar |
[99] N Drinkwater, RS Bamert, KK Sivaraman, A Paiardini, S McGowan, X-ray crystal structures of an orally available aminopeptidase inhibitor, Tosedostat, bound to anti-malarial drug targets PfA-M1 and PfA-M17. Proteins 2015, 83, 789.
| X-ray crystal structures of an orally available aminopeptidase inhibitor, Tosedostat, bound to anti-malarial drug targets PfA-M1 and PfA-M17.Crossref | GoogleScholarGoogle Scholar |
[100] S McGowan, CA Oellig, WA Birru, TT Caradoc-Davies, CM Stack, J Lowther, T Skinner-Adams, A Mucha, P Kafarski, J Grembecka, KR Trenholme, AM Buckle, DL Gardiner, JP Dalton, JC Whisstock, Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases. Proc Natl Acad Sci U S A 2010, 107, 2449.
| Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases.Crossref | GoogleScholarGoogle Scholar |
[101] S McGowan, CJ Porter, J Lowther, CM Stack, SJ Golding, TS Skinner-Adams, KR Trenholme, F Teuscher, SM Donnelly, J Grembecka, A Mucha, P Kafarski, R Degori, AM Buckle, DL Gardiner, JC Whisstock, JP Dalton, Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase. Proc Natl Acad Sci U S A 2009, 106, 2537.
| Structural basis for the inhibition of the essential Plasmodium falciparum M1 neutral aminopeptidase.Crossref | GoogleScholarGoogle Scholar |
[102] K Kannan Sivaraman, A Paiardini, M Sieńczyk, C Ruggeri, CA Oellig, JP Dalton, PJ Scammells, M Drag, S McGowan, Synthesis and structure–activity relationships of phosphonic arginine mimetics as inhibitors of the M1 and M17 aminopeptidases from Plasmodium falciparum. J Med Chem 2013, 56, 5213.
| Synthesis and structure–activity relationships of phosphonic arginine mimetics as inhibitors of the M1 and M17 aminopeptidases from Plasmodium falciparum.Crossref | GoogleScholarGoogle Scholar |
[103] NB Vinh, N Drinkwater, TR Malcolm, M Kassiou, L Lucantoni, PM Grin, GS Butler, S Duffy, CM Overall, VM Avery, PJ Scammells, S McGowan, Hydroxamic acid inhibitors provide cross-species inhibition of Plasmodium M1 and M17 aminopeptidases. J Med Chem 2019, 62, 622.
| Hydroxamic acid inhibitors provide cross-species inhibition of Plasmodium M1 and M17 aminopeptidases.Crossref | GoogleScholarGoogle Scholar |
[104] N Drinkwater, NB Vinh, SN Mistry, RS Bamert, C Ruggeri, JP Holleran, S Loganathan, A Paiardini, SA Charman, AK Powell, VM Avery, S McGowan, PJ Scammells, Potent dual inhibitors of Plasmodium falciparum M1 and M17 aminopeptidases through optimization of S1 pocket interactions. Eur J Med Chem 2016, 110, 43.
| Potent dual inhibitors of Plasmodium falciparum M1 and M17 aminopeptidases through optimization of S1 pocket interactions.Crossref | GoogleScholarGoogle Scholar |
[105] TS Skinner-Adams, GM Fisher, AG Riches, OE Hutt, KE Jarvis, T Wilson, M von Itzstein, P Chopra, Y Antonova-Koch, S Meister, EA Winzeler, M Clarke, DA Fidock, JN Burrows, JH Ryan, KT Andrews, Cyclization-blocked proguanil as a strategy to improve the antimalarial activity of atovaquone. Commun Biol 2019, 2, 166.
| Cyclization-blocked proguanil as a strategy to improve the antimalarial activity of atovaquone.Crossref | GoogleScholarGoogle Scholar |
[106] BL Howard, KL Harvey, RJ Stewart, MF Azevedo, BS Crabb, IG Jennings, PR Sanders, DT Manallack, PE Thompson, CJ Tonkin, PR Gilson, Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites. ACS Chem Biol 2015, 10, 1145.
| Identification of potent phosphodiesterase inhibitors that demonstrate cyclic nucleotide-dependent functions in apicomplexan parasites.Crossref | GoogleScholarGoogle Scholar |
[107] C Dogovski, SC Xie, G Burgio, J Bridgford, S Mok, JM McCaw, K Chotivanich, S Kenny, N Gnädig, J Straimer, Z Bozdech, DA Fidock, JA Simpson, AM Dondorp, S Foote, N Klonis, L Tilley, Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol 2015, 13, e1002132.
| Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance.Crossref | GoogleScholarGoogle Scholar |
[108] SC Xie, DL Gillett, NJ Spillman, C Tsu, MR Luth, S Ottilie, S Duffy, AE Gould, P Hales, BA Seager, CL Charron, F Bruzzese, X Yang, X Zhao, S-C Huang, CA Hutton, JN Burrows, EA Winzeler, VM Avery, LR Dick, L Tilley, Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome. J Med Chem 2018, 61, 10053.
| Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome.Crossref | GoogleScholarGoogle Scholar |
[109] C Xie Stanley, D Metcalfe Riley, H Mizutani, T Puhalovich, E Hanssen, J Morton Craig, Y Du, C Dogovski, S-C Huang, J Ciavarri, P Hales, J Griffin Robert, H Cohen Lawrence, B-C Chuang, S Wittlin, I Deni, T Yeo, E Ward Kurt, C Barry Daniel, B Liu, L Gillett David, F Crespo-Fernandez Benigno, S Ottilie, N Mittal, A Churchyard, D Ferguson, C Aguiar Anna Caroline, VC Guido Rafael, J Baum, K Hanson Kirsten, A Winzeler Elizabeth, F-J Gamo, A Fidock David, D Baud, W Parker Michael, S Brand, R Dick Lawrence, DW Griffin Michael, E Gould Alexandra, L Tilley, Design of proteasome inhibitors with oral efficacy in vivo against Plasmodium falciparum and selectivity over the human proteasome. Proc Natl Acad Sci U S A 2021, 118, e2107213118.
| Design of proteasome inhibitors with oral efficacy in vivo against Plasmodium falciparum and selectivity over the human proteasome.Crossref | GoogleScholarGoogle Scholar |
[110] SC Xie, RD Metcalfe, E Dunn, CJ Morton, SC Huang, T Puhalovich, Y Du, S Wittlin, S Nie, MR Luth, L Ma, MS Kim, CFA Pasaje, K Kumpornsin, C Giannangelo, FJ Houghton, A Churchyard, MT Famodimu, DC Barry, DL Gillett, S Dey, CC Kosasih, W Newman, JC Niles, MCS Lee, J Baum, S Ottilie, EA Winzeler, DJ Creek, N Williamson, MW Parker, S Brand, SP Langston, LR Dick, MDW Griffin, AE Gould, L Tilley, Reaction hijacking of tyrosine tRNA synthetase as a new whole-of-life-cycle antimalarial strategy. Science 2022, 376, 1074.
| Reaction hijacking of tyrosine tRNA synthetase as a new whole-of-life-cycle antimalarial strategy.Crossref | GoogleScholarGoogle Scholar |
[111] JA Boddey, AN Hodder, S Günther, PR Gilson, H Patsiouras, EA Kapp, JA Pearce, TF de Koning-Ward, RJ Simpson, BS Crabb, AF Cowman, An aspartyl protease directs malaria effector proteins to the host cell. Nature 2010, 463, 627.
| An aspartyl protease directs malaria effector proteins to the host cell.Crossref | GoogleScholarGoogle Scholar |
[112] BE Sleebs, M Gazdik, MT O’Neill, P Rajasekaran, S Lopaticki, K Lackovic, K Lowes, BJ Smith, AF Cowman, JA Boddey, Transition state mimetics of the Plasmodium export element are potent inhibitors of plasmepsin V from P. falciparum and P. vivax. J Med Chem 2014, 57, 7644.
| Transition state mimetics of the Plasmodium export element are potent inhibitors of plasmepsin V from P. falciparum and P. vivax.Crossref | GoogleScholarGoogle Scholar |
[113] BE Sleebs, S Lopaticki, DS Marapana, MT O’Neill, P Rajasekaran, M Gazdik, S Gunther, LW Whitehead, KN Lowes, L Barfod, L Hviid, PJ Shaw, AN Hodder, BJ Smith, AF Cowman, JA Boddey, Inhibition of plasmepsin V activity demonstrates its essential role in protein export, PfEMP1 display, and survival of malaria parasites. PLoS Biol 2014, 12, e1001897.
| Inhibition of plasmepsin V activity demonstrates its essential role in protein export, PfEMP1 display, and survival of malaria parasites.Crossref | GoogleScholarGoogle Scholar |
[114] AN Hodder, BE Sleebs, PE Czabotar, M Gazdik, Y Xu, MT O’Neill, S Lopaticki, T Nebl, T Triglia, BJ Smith, K Lowes, JA Boddey, AF Cowman, Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat Struct Mol Biol 2015, 22, 590.
| Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes.Crossref | GoogleScholarGoogle Scholar |
[115] W Nguyen, AN Hodder, RB de Lezongard, PE Czabotar, KE Jarman, MT O’Neill, JK Thompson, H Jousset Sabroux, AF Cowman, JA Boddey, BE Sleebs, Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P2 position of PEXEL peptidomimetics. Eur J Med Chem 2018, 154, 182.
| Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P2 position of PEXEL peptidomimetics.Crossref | GoogleScholarGoogle Scholar |
[116] M Gazdik, KE Jarman, MT O’Neill, AN Hodder, KN Lowes, H Jousset Sabroux, AF Cowman, JA Boddey, BE Sleebs, Exploration of the P3 region of PEXEL peptidomimetics leads to a potent inhibitor of the Plasmodium protease, plasmepsin V. Bioorg Med Chem 2016, 24, 1993.
| Exploration of the P3 region of PEXEL peptidomimetics leads to a potent inhibitor of the Plasmodium protease, plasmepsin V.Crossref | GoogleScholarGoogle Scholar |
[117] M Gazdik, MT O’Neill, S Lopaticki, KN Lowes, BJ Smith, AF Cowman, JA Boddey, BE Sleebs, The effect of N-methylation on transition state mimetic inhibitors of the Plasmodium protease, plasmepsin V. MedChemComm 2015, 6, 437.
| The effect of N-methylation on transition state mimetic inhibitors of the Plasmodium protease, plasmepsin V.Crossref | GoogleScholarGoogle Scholar |
[118] C Jennison, L Lucantoni, MT O’Neill, R McConville, SM Erickson, AF Cowman, BE Sleebs, VM Avery, JA Boddey, Inhibition of plasmepsin V activity blocks Plasmodium falciparum gametocytogenesis and transmission to mosquitoes. Cell Rep 2019, 29, 3796.
| Inhibition of plasmepsin V activity blocks Plasmodium falciparum gametocytogenesis and transmission to mosquitoes.Crossref | GoogleScholarGoogle Scholar |
[119] P Favuzza, M de Lera Ruiz, JK Thompson, T Triglia, A Ngo, RWJ Steel, M Vavrek, J Christensen, J Healer, C Boyce, Z Guo, M Hu, T Khan, N Murgolo, L Zhao, JS Penington, K Reaksudsan, K Jarman, MH Dietrich, L Richardson, KY Guo, S Lopaticki, WH Tham, M Rottmann, T Papenfuss, JA Robbins, JA Boddey, BE Sleebs, HJ Sabroux, JA McCauley, DB Olsen, AF Cowman, Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 2020, 27, 642.
| Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle.Crossref | GoogleScholarGoogle Scholar |
[120] AN Hodder, JJ Christensen, S Scally, T Triglia, A Ngo, RW Birkinshaw, B Bailey, P Favuzza, MH Dietrich, WH Tham, PE Czabotar, K Lowes, K Guo, N Murgolo, ML Ruiz, JA McCauley, BE Sleebs, DB Olsen, AF Cowman, Basis for drug selectivity of plasmepsin IX and X inhibition in Plasmodium falciparum and vivax. Structure 2022, 30, 947.
| Basis for drug selectivity of plasmepsin IX and X inhibition in Plasmodium falciparum and vivax.Crossref | GoogleScholarGoogle Scholar |
† Brad Sleebs is the recipient of the 2020 RACI Peter Andrews Award for Innovation in Medicinal Chemistry/Chemical Biology. The award was presented at the RACI 2022 Congress.