

Shaping the future of constrained peptides and compact proteins in drug discovery
Sven Ullrich A *
A *
A

Sven Ullrich is a Feodor Lynen Research Fellow (Humboldt Foundation) at the University of Tokyo, Japan. He holds a degree in pharmacy from Heidelberg University, Germany, and a PhD in chemistry from the Australian National University. During his doctoral studies, he was a visiting student at the University of Alberta, Canada. His research focuses on modified peptides and proteins in genetically encoded libraries for drug discovery. He received the Dr Elizabeth Schram Young Investigator Award at the 27th American Peptide Symposium (2022), was a selected participant at the Global Young Scientists Summit in Singapore (2023) and was recognised as part of the CAS Future Leaders Top 100 initiative (2024). For his graduate work, he was awarded both the John Carver Award (ACT Branch, 2024) and the Cornforth Medal (National, 2024) by the Royal Australian Chemical Institute. |
Abstract
Constrained peptides and compact proteins are potent alternatives to conventional drug modalities in academia and industry. Located in the chemical space between small molecules and classical biologics, these drug formats feature highly modular, conformationally constrained turns and loops that can be accessed through rational design, selection-based screening or computational discovery. The amino acid-derived biopolymers can engage comparatively featureless protein surfaces more effectively than low molecular weight compounds, yet at the same time offer greater potential to reach intracellular targets than larger biologics. This combination allows them to address medicinal chemistry challenges that conventional approaches struggle to solve. In this brief review, selected advances in the discovery and development of such structures are highlighted, especially those where screening of genetically encoded or synthetic libraries played a central role. Several recent examples of rational design and computationally generated peptide and protein scaffolds are also discussed, including those driven by machine learning and artificial intelligence. Across these strategies, all case studies describe the successful identification and refinement of cyclic peptides or compact proteins with antibody-like binding as promising lead structures. As the presented examples cover a wide range of structural topologies and medicinally relevant targets, they reflect the growing importance of cyclic peptides and compact proteins as new molecular modalities for drug discovery and development.
Keywords: antibody-mimetic proteins, artificial intelligence, cyclic peptides, drug design, encoded libraries, macrocyclisation, miniproteins, nanobodies, rational design, therapeutics.
Introduction
New molecular modalities are gaining traction in drug discovery.1–3 Despite the continued dominance of small molecules and antibodies in the pharmaceutical world,4,5 a variety of alternative structures have become popular in academic and industrial research,1 including proteolysis targeting chimeras (PROTACs),6,7 nucleic acid therapeutics,8,9 drug conjugates,10,11 structurally modified or constrained peptides12,13 and small, engineered proteins like antibody-mimetics.14,15 These extraordinary chemical entities promise to extend the current therapeutic reach,1 offering to diversify strategies to modulate disease-relevant biology beyond the limits of traditional medicinal chemistry,1,16,17 particularly in the context of infamously undruggable targets.18,19
Among these new drug modalities, constrained peptides and compact proteins in particular have emerged as attractive lower molecular weight alternatives to classical biologics (Fig. 1).2,3,12–15,18,19 For the purpose of this article, constrained peptides will refer to cyclic,20–22 stapled23,24 and multicyclic25–28 structures that are conformationally restricted to improve pharmaceutical properties, with a focus on their de novo discovery. Compact proteins, in turn, will refer to stably folded smaller proteins, typically below 15 kDa, that are primarily intended for high-affinity target binding. The definition of compact proteins in this context thus includes the likes of cyclotides,29 miniproteins,3,15 small grafted scaffolds30 and antibody-mimetic14,31 proteins designed for molecular recognition. Here, antibody-related structures such as nanobodies,32 which retain part of the immunoglobulin fold, are classified as antibody mimics alongside alternative binding scaffolds, as they functionally resemble full antibodies. Both constrained peptides and compact proteins are amino acid-derived structures that scaffold, stabilise and rigidify their binding epitopes to varying degrees, facilitating effective target engagement through pre-organisation.30,33–35
Continuum of constrained peptides and compact proteins, arranged broadly by increasing molecular weight. Shown are representative structures of cyclic (PDB: 1CWA),36 stapled (PDB: 8GJS)37 and multicyclic (PDB: 3QN7)34 peptides, cyclotides (PDB: 7FBP),38 miniproteins (PDB: 8UTK),39 grafted scaffolds (ColabFold, ver. 1.5.5, see https://github.com/sokrypton/ColabFold)40,41 and antibody-mimetic proteins (PDB: 7RBY).42 The following structural features are highlighted in red: the intramolecular bridge in stapled peptides, the cyclisation linchpin in multicyclic peptides, the cystine knot core in cyclotides and the inserted epitope in grafted scaffolds. Cartoon representations of the structures were rendered using ChimeraX (ver. 1.10.1, see https://www.rbvi.ucsf.edu/chimerax).43 Note that categories are not strictly discrete, that molecular size may vary within and across classes and that representations are schematic and not necessarily to scale.
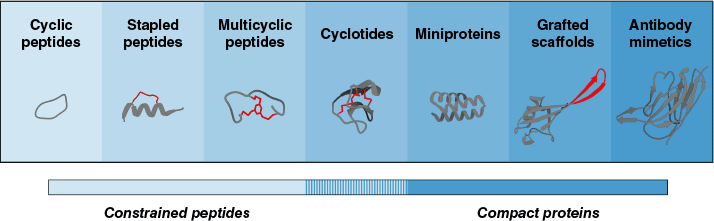
Both types of scaffolds have the potential to rival or even surpass antibodies in target-binding specificity and affinity.2,14 Remarkably, these chemical entities remain accessible through comparatively efficient chemical or biotechnological production processes.44,45 Unlike small molecules, these structures are inherently suited for engaging in protein–protein interactions, honouring in part the principles that nature has evolved for biomolecular recognition.17,46,47 Their smaller size relative to classical biologics may in some cases also permit cell penetration and even oral bioavailability, with increasing compactness and constraint thought to favour such properties, thereby conferring pharmaceutically beneficial features typically unattainable with standard antibodies.3,13,39,48–50
With growing evidence for the importance of cyclic peptides and small designer proteins, this non-exhaustive mini review aims to bring attention to selected developments in their respective fields. Emphasis is placed on methodological advances in drug discovery and development, highlighting chemical biology tools that generate intriguing early-stage lead structures. Key strategies covered often revolve around high-throughput selections and encoded libraries. Rational design efforts are also discussed, alongside the latest developments in computational methods, including artificial intelligence. The following examples from the frontiers of peptide and protein science illustrate the growing relevance and rising translational potential of constrained peptides and compact proteins, while also acknowledging the limitations and challenges that remain.
Cyclic peptides and compact proteins in drug discovery
Peptide and protein modification
Progress in peptide modification has substantially expanded the available repertoire of chemical strategies to access constrained peptides.51–56 Countless synthetic and enzymatic processes enable the construction of intramolecularly linked peptides that are cyclic,20–22 stapled23,24 and multicyclic.25–28 Biocompatible reactions, including for late-stage functionalisation, have proven to be especially valuable for screening platforms based on biological systems, as they permit introduction of moieties that strongly enhance the structural diversity and drug-likeness of peptide libraries without compromising the ensuing selection process.56–58 Although the development of such versatile and powerful chemistries is at the heart of peptide and protein chemistry, they have been reviewed in detail elsewhere and are not the central focus of this article.51–56
In early drug discovery of compact proteins, their binding epitopes can be identified through various means, including classical protein engineering, grafting or display screenings.3,30 Although screening approaches may identify de novo epitopes by randomising surface-exposed parts, modular strategies such as grafting or on-protein cyclisation allow known protein-binding epitopes to be transferred onto stably folded protein scaffolds.3,30,59 More radically, recent breakthroughs in artificial intelligence and computational design have enabled the generation of entirely new proteins, solely based on their intended target.39,60–62 Although ribosomal incorporation of non-canonical amino acids is in principle possible during recombinant protein expression,63 their use in larger biological drug leads is still considerably less widespread than in smaller peptide lead structures.64–66 In parallel, complementary chemical protein engineering approaches are emerging, where synthetic methods, often originally tailored for peptides, expand the topology of proteins and their functional properties beyond what is genetically encodable.67
Strategies using peptide libraries for drug discovery
Large-scale selection and screening platforms often form the core of cyclic peptide and compact protein drug discovery campaigns.3,13,68,69 Usually, these approaches follow typical funnel logic, conceptually drawing on Darwinian principles, whereby large variant pools are subjected to selection cycles that enrich optimal binding or function.70–72 Such library technologies can be arranged along a spectrum defined by the extent of their reliance on biological machinery.
At the biologically intensive end, phage display and messenger ribonucleic acid (mRNA) display systems exploit natural translation and replication processes to generate peptide libraries for screening, whereby sequence information of the individual compounds is stored on associated nucleotides.73,74 Phage display, for instance, uses bacteriophages that hijack bacterial machinery to produce peptides on the surface of viral coat proteins.73 Resulting phage libraries can be diversified through post-translationally applied reagents for modifications such as peptide stapling, expanding building blocks and peptide connectivity beyond what is possible with canonical amino acids.57,75,76 By contrast, mRNA display uses reconstituted translation systems in vitro and links the translated peptide covalently to puromycin and its genetic information.74 Combination with flexizyme technology77 in the random nonstandard integrated peptides discovery (RaPID) system additionally allows the direct incorporation of non-canonical building blocks into peptides during translation, most commonly reactive initiator amino acids for cyclisation.78,79 More recently, phage-displayed libraries have also been extended through genetic code expansion.75 Additionally, a range of alternative encoded library strategies has been explored across different expression frameworks.80–83
DNA-encoded libraries represent a hybrid-style approach, where chemical synthesis is used to build libraries, but a nucleic acid tag facilitates identification of the compounds.84 Construction of DNA-encoded libraries with constrained peptides can therefore be potentially more labour-intensive, as their synthesis is generally not performed by biological machinery.84,85 However, increasing independence from biological systems allows the use of harsher chemical conditions and the inclusion of structural features or functionalities that would not be compatible with the alternative systems.86,87 At the other end of the spectrum, fully synthetic, non-encoded peptide libraries have also recently received increased interest, as they similarly enable access to a wide variety of unnatural building blocks.88 Although chemically intensive library strategies can access more unusual structural spaces, their size normally remains more limited (<∼109) than that of genetically encoded libraries (>∼1010).70,89
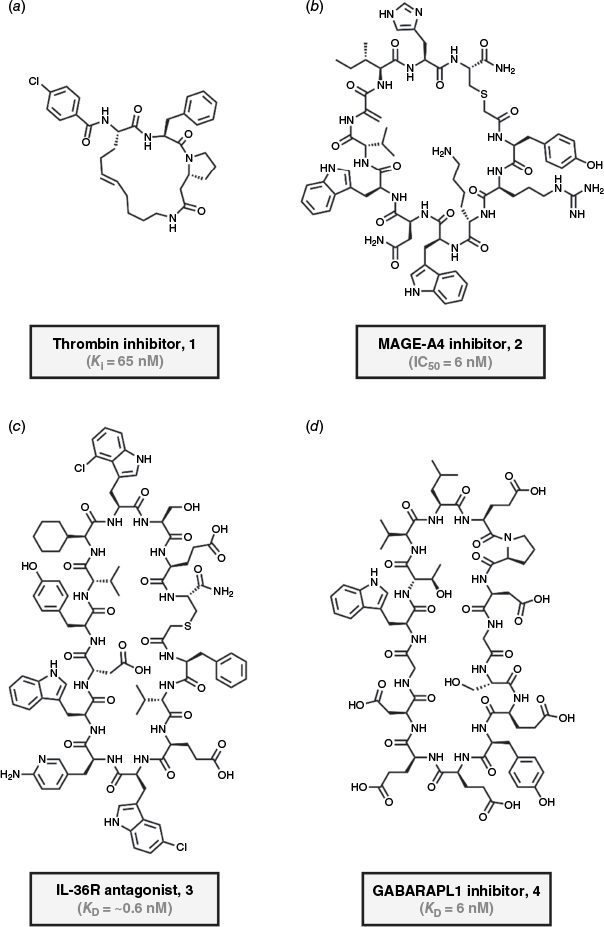
Recent work from Heinis and co-workers has focused on fully non-encoded combinatorial libraries of diverse peptide macrocycles.50,85,88,90 With the goal of obtaining orally available peptide macrocycles, the group focused on synthesising especially short peptide cycles with pronounced non-canonical character at the nanomolar scale.85 A library of ~104 thioether-cyclised peptides was generated using acoustic dispensing in microplates, whereby combinatorial diversity was created through varying sequence, cyclisation linker and acylation.85 Several macrocycles with strong inhibitory activity against thrombin were identified.85 By refining the initial and intermediary hits and screening the resulting sub-libraries for improved permeability and stability, a potent inhibitor of human thrombin (1, KD = 65 nM) with oral bioavailability in rats could be generated (Fig. 2a).85 The strategy demonstrates that orally active peptides can be obtained even from a library smaller than those typically used in genetically encoded screening endeavours by taking advantage of the modularity of non-natural cyclic peptides and the insights gained from focused screenings.85
(a) Orally bioavailable thrombin inhibitor (1; KD = 65 nM), identified from a non-encoded library strategy by Heinis and co-workers (1:2 mixture of E:Z isomers).85 (b) Covalently binding MAGE-A4 inhibitor (2, IC50 = 6 nM) identified using mRNA display by Bowers and co-workers.91 (c) IL-36R antagonist (3, KD = ~0.6 nM) obtained through medicinal chemistry optimisation of a screening hit.92 (d) De novo designed inhibitor of GABARAP (4, KD = 6 nM) discovered by Rettie et al.93
Although synthetic libraries offer high chemical variability, genetically encoded platforms still remain superior in terms of sequence space.70 A recent study by Derda and co-workers introduces a new chemical derivatisation strategy for phage-displayed libraries, as well as builds a case for late-stage reshaping during selection campaigns.94 Rather than starting the screening process with naïve libraries that are already chemically altered, as is usual in typical post-translationally modified phage library campaigns,95,96 the group only chemically diversified their libraries after initially narrowing them through conventional, unmodified phage display.94 Following such initial pre-selection, linchpin reagent 3,5-bis(bromomethyl)benzaldehyde was applied to the library to create peptide constraint directly on the phage.94 Three reactive functionalities allow the generation of monocyclic, bicyclic or pharmacophore-linked peptides through functionalisation of cysteines and the N-terminus of displayed peptides.94 Selection of the libraries against a hepatitis C virus protease variant enriched several DMT tripeptide motif-containing peptides.94 Next-generation sequencing indicated which enriched hits of the late-stage reshaping campaign could retain binding affinity after chemical modification, as exemplified by a hit peptide bicycle with nanomolar binding (KD = 30 nM) and considerable proteolytic stability.94 This alternative screening approach may be beneficial in identifying peptides that both tolerate chemical modification for enhanced drug-likeness and retain, or improve, their originally selected target binding.94
As mRNA display continues to be applied to new targets,18,97–99 recent expansions have enabled covalent inhibitor discovery through the incorporation of electrophilic warheads into cyclic peptide libraries.91,100,101 For instance, Bowers and co-workers introduced masked dehydroalanine electrophiles using phenylselenocysteine precursors into mRNA-displayed libraries.91 Covalently binding hit peptides were identified in guanidine-denaturing selections and showed binding to target-specific cysteines on proof-of-concept targets.91 The lead inhibitor of melanoma-associated antigen 4 (MAGE-A4) bound covalently with time-dependent potency (2, Fig. 2b, IC50 = 6 nM, 24 h of incubation).91 In a study using directly encoded electrophiles, Walport and co-workers incorporated unmasked fluoroamidines into RaPID libraries, selecting for covalent binders of peptidyl arginine deiminase 4 (PADI4), also through denaturing washes.100 Their approach yielded several selective inhibitors reacting at cysteines, with the most potent peptide macrocycle displaying nanomolar bioactivity (IC50 = 52 nM, 1 h of incubation) and strong selectivity over related proteins.100 With renewed interest in covalent drugs,102–105 these studies exemplify how the combination of electrophilic design elements and denaturing selection pressure enables the discovery of covalently binding cyclic peptide inhibitors from genetically encoded libraries.91,100
Beyond increasing binding mode variety through reactive warheads,91,100,101,106 recent efforts have extended genetically encoded display into the mirror-image world, allowing access to D-ligands with strongly enhanced stability.107–110 Although the concept of mirror-image screening was introduced over two decades ago for phage display,111 the chemical synthesis of D-proteins has long remained a bottleneck.112–114 However, recent improvements have lowered the entry barrier to obtaining mirror image proteins, making such campaigns slightly more accessible.112–114 Sagi, Metanis, Suga and co-workers first transferred the concept to RaPID mRNA display, where the technology was used for selection against a chemically synthesised D-protein with predominantly L-configured libraries, thereby allowing the identification of ligands of the natural protein with non-natural chirality.107 Screening a macrocyclic peptide library containing additional D- and β-amino acids against the mirror image form of matrix metalloprotease 7 (MMP-7) therefore yielded potent binders against the natural MMP-7 when synthesised in mirror form.107 The lead compound inhibited MMP-7 with nanomolar potency (IC50 = 90 nM) and remained considerably stable in serum and digestive fluids.107 In another study, Payne and co-workers targeted the C–C motif chemokine 22 (CCL22) by screening against the synthetic D-protein and a non-canonical cyclic peptide library containing sulfo-tyrosine.108 The mirrored lead macrocycle exhibited nanomolar affinity (KD = 190 nM), selective binding over related chemokines and was active in cell assays.108
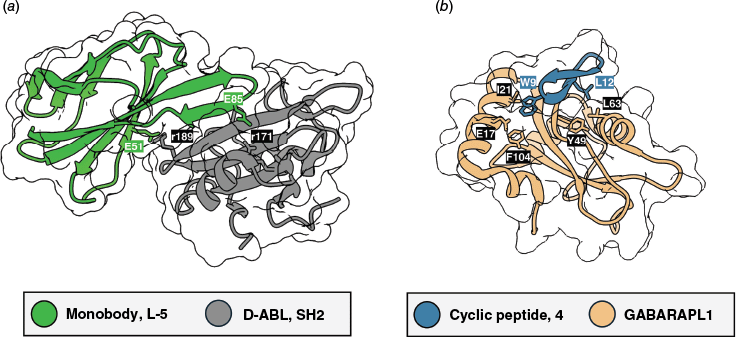
Applying the mirror-image strategy to compact protein scaffolds, Hayashi, Oishi, Murakami and co-workers used transcription-translation coupled with association of puromycin linker (TRAP) display, an mRNA display variant, to identify D-monobody ligands for cytokine monocyte chemoattractant protein-1 (MCP-1).109 Screening libraries with two randomised loops on the monobody scaffold against the D-form of the target yielded a chemically synthesised D-monobody that bound the natural L-form MCP-1 with high affinity (KD = 1.6 nM), selectively inhibited the relevant protein–protein interaction, exhibited strong proteolytic resistance and displayed undetectable immunogenicity.109 In a parallel study published back-to-back, Vázquez, Hantschel and co-workers identified D-monobodies targeting the oncogenic breakpoint cluster region–Abelson murine leukaemia viral oncogene homologue 1 SRC homology 2 domain (BCR::ABL1 SH2) through a mirror-image phage and yeast display workflow.110 Their split D-monobody constructs, assembled from two non-ligated synthetic peptide fragments,115 retained nanomolar affinity to the L-form target (D-5, KD = ~100 nM), showed considerable stability in plasma, resisted proteolysis and inhibited kinase activity in vitro (Fig. 3a).110 Both studies establish mirror-image monobodies as stable and selective compact protein scaffolds for drug discovery.109,110 More broadly, all mirror-image selections highlight the promise of non-natural chirality in peptides and proteins,107–110 which is particularly attractive for addressing targets where proteolytic degradation and immunogenicity are key factors.116
X-Ray crystallography of cyclic peptides and compact protein binders. (a) Co-crystal structure of monobody L-5 and the D-version of target ABL1 SH2 (KD = ~100 nM; PDB: 9F00).110 (b) Co-crystal structure of computationally designed GABARAP-binding cyclic peptide 4 (KD = 6 nM) with GABARAP homologue GABARAPL1 (PDB: 9HGC).93
Likewise, grafting approaches have proven valuable for generating functionalised compact proteins from pre-structured scaffolds.30 A recent example is the identification of ubiquitin-based protein-binders, stylised U-body, engineered for high-affinity target binding by Takagi, Suga and co-workers, assisted by mRNA display.40,117 Kawakami et al. used lasso-grafting to transplant MET-binding macrocyclic peptides, previously identified using the RaPID system,118 into surface-exposed loops of ubiquitin.40 However, initial naïve grafts suffered losses in target affinity, attributed to the conformations of the inserted pharmacophores.40 To restore function, the group constructed a library with randomised flanking residues around the grafted peptide and performed mRNA display selections against the original target mesenchymal to epithelial transition factor (MET).40 From this library, several affinity-optimised U-body constructs with nanomolar potency were identified, the best of which matched the binding affinity of the ancestral macrocycle (KD = 3 nM).40 Inspired by the natural occurrence of multimeric ubiquitin chains, U-bodies with peptide linker units creating up to octameric assemblies were also explored.40 Resulting multivalent U-bodies activated MET signalling independently and promoted proliferation in cell assays.40 This study establishes a generalisable strategy whereby affinity losses from grafting can be recovered through sequence optimisation guided by genetically encoded screenings.40
Even cysteine-rich proteins can be used as scaffolds in mRNA display, as Craik, Suga and co-workers demonstrated by screening an open-form variant of a highly stable cystine-knotted cyclotide.38,119 Their library design introduced randomisation into two loops of the cyclotide MCoTI-II.38,120 Screening against the coagulation protease FXIIa identified potent inhibitors, with MCoFx1 emerging as a lead candidate.38 Upon cyclisation, MCoFx1 exhibited sub-nanomolar inhibition (Ki = 0.37 nM), with X-ray crystallography revealing one loop to be especially central for target engagement.38 Biological assays verified inhibitory activity within the FXIIa-dependent intrinsic pathway and preclinical studies confirmed the compound as a promising anticoagulant.38,121
Rationally guided development of peptide and protein drug candidates
A particularly promising recent example of rationally guided drug development of cyclic peptides is enlicitide, an inhibitor targeting proprotein convertase subtilisin/kexin type 9 (PCSK9), which acts within the cholesterol metabolism pathway.122 The campaign began with mRNA display using a cyclised peptide library, where early hits showed only moderate activity against the target protein in the micromolar range.122 Through intensive iterative structure-guided design, however, these peptides were progressively refined, drastically enhancing both their affinity and pharmacokinetic properties by structure-activity relationship studies and the introduction of additional cycles.122–124 The highly potent tricyclic peptide enlicitide emerged, exhibiting picomolar inhibition of the target (Ki = ~5 pM), indicating an incredible six orders of magnitude affinity improvement from the initial hit.122–124 Additionally, the compound was orally bioavailable when formulated with a permeation enhancer and has progressed to phase III clinical trials.122,125
Another compelling demonstration of rational cyclic peptide development was reported in the context of interleukin-36 receptor (IL-36R) antagonists.92 Starting from an mRNA-displayed library, a macrocyclic peptide with similarly moderate affinity served as the starting point for optimisation.92 Through medicinal chemistry efforts, mainly driven by residue substitution with non-canonical amino acids, the group developed a highly optimised variant with sub-nanomolar binding affinity (3, Fig. 2c, KD = ~0.6 nM), improved off-rate kinetics and potent cellular inhibition of IL-36R signalling.92 The obtained pharmacophore knowledge could even inform a subsequent DNA-encoded library screen, yielding one low molecular weight compound that occupies the same binding site as the cyclic peptide.92 Both industrial studies are particularly relevant for how they demonstrate the synergy of de novo hit discovery with very intensive late-stage chemical optimisation, turning modest cyclic peptides into more viable drug candidates.92,122
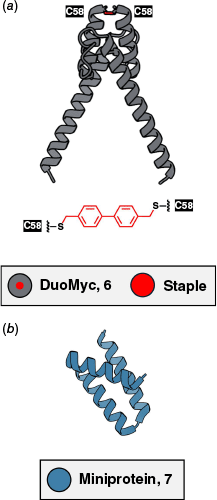
Rational scaffold design was also key to the development of DuoMYC (6), a synthetic miniprotein targeting the oncogenic myelocytomatosis transcription factor (Myc).49 Starting from the helix-loop-helix DNA-binding region of Omomyc, a slightly cell-permeable protein drug candidate,49,126 Pomplun and co-workers replaced the molecule’s leucine zipper with a short covalent bis(bromomethyl)-biphenyl linker to generate a rigid four-helix dimer (Fig. 4a).49 This strategy yielded a stable, cell-penetrant miniprotein that binds enhancer-box DNA, the typical interaction partner of heterodimerised Myc, with nanomolar affinity (KD = ~118 nM).49 DuoMYC inhibited Myc-driven transcription with nanomolar potency in a reporter assay and outperformed the larger Omomyc in serum stability and cellular uptake, demonstrating the potential of rationally designed, chemically modified miniproteins for engaging difficult-to-target proteins.49
Structural insights into compact protein therapeutics. (a) ColabFold41 model and cartoon representation of DuoMyc (6), indicating chemical posttranslational modification in red.49 (b) X-Ray crystal structure of computationally designed IL-23R minibinder (7, PDB: 8UTK).39
Nitsche and co-workers have developed methods to use heavy metal binding to forge conformationally constrained peptides.127–130 Envisioning radiopharmaceutical applications for these elements, they incorporated metal-binding motifs in nanobodies, turning these compact proteins into self-chelating structures.131,132 Nanobody scaffolds were rationally modified near their conserved disulfide bond to create local tri- and tetracysteine clusters tailored for BiIII, InIII and GaIII coordination.131 Native mass spectrometry confirmed near-quantitative binding of the metals after exposure to a brief heat and pH shock.131 The modified nanobodies remained firmly connected to the metals even under challenging conditions.131 With one BiIII-bound Lam2133,134-derived nanobody retaining its affinity to mCherry (KD = 46 nM), the study shows that incorporating subtle, rational edits to known nanobody scaffolds can confer metal-binding capability without compromising target recognition.131
Computationally assisted de novo discovery of peptide and protein lead structures
Complementary to wet lab technique discovery funnels, machine learning methods are fast becoming a viable path for de novo identification of peptide and protein lead structures.39,60,61,93,135 A topical review by Lin et al. comprehensively maps out the field from a cyclic peptide viewpoint and is recommended for further reading.135 Here, two recent breakthroughs by Baker and co-workers are briefly discussed, spanning both cyclic peptides and compact protein therapeutics.39,93
Rettie et al. developed a software workflow for the de novo design of backbone macrocyclic peptides,93 building on the success of RFdiffusion for protein design.136 Their RFpeptides pipeline allowed the computational generation of cyclic peptides solely on the basis of the structures of selected drug targets.93 The process yielded nanomolar affinity binders, whereby computationally predicted binding poses could be verified by X-ray crystallography.93 The cyclic peptides exploit several structural elements, including helices and sheet structures, but also more elaborate topologies, for target engagement.93 In one example, a designed cyclic peptide (4, Fig. 2d) binds γ-aminobutyric-acid receptor-associated protein (GABARAP) with single-digit nanomolar affinity (KD = 6 nM; Fig. 3b).93 Without the need for prior wet lab screening, this work showcases that advances in protein design allow cyclic peptide binders to now be designed entirely from structural principles.93
Berger et al. reported two de novo-designed miniproteins targeting interleukin 17A (IL-17A) and interleukin 23 receptor (IL-23R) originating from a Rosetta-based workflow, then used yeast-display libraries to enrich high-affinity variants.39 The IL-23R minibinder (7) exhibited sub-picomolar affinity (KD < 1 pM) and its compact three-helix structure also conferred exceptional heat and gastric fluid stability (Fig. 4b).39 The protein was even orally bioavailable in mice, where it matched the efficacy of a clinically used anti-IL-23 antibody in a colitis model.39 For IL-17A, linking two binding domains to form a dimer significantly increased potency to sub-picomolar cellular half-maximal inhibition values (IC50 = 0.5 pM).39 The study marks a milestone in rational protein design, showing that fully synthetic miniproteins can match or surpass established biologics in terms of stability and function, and may even achieve oral delivery.39,137
Discussion and outlook
Recent years have seen cyclic peptides and compact proteins rise to become seriously pursued drug leads.2,3,12–15,18,19,138 Both structures are highly pre-organised lower molecular weight alternatives to classical biologics, making them effective scaffolds to engage targets that evade small molecules.30,33–35 Technological innovations continue to expand the accessible design space for peptides and proteins, taking advantage of the inherent modular nature of the structures as amino acid-derived polymers. Display technologies especially facilitate the de novo discovery against challenging targets. Likewise, new strategies based on computational peptide and protein de novo design are on the rise. Complementary medicinal chemistry campaigns based on rationally guided modifications, in turn, remain ideal to refine drug hits and leads, especially regarding drug-likeness and permeability improvements or to confer theranostic functions.
Despite these advances, key challenges remain. Although cyclic peptides and compact proteins typically demonstrate impressive binding affinities in vitro, limited cell penetration often impedes access to intracellular targets. Identified as the ‘grand challenge’ of peptide and protein therapeutics, this is being actively addressed through structural innovation and delivery, including formulation technologies.139–149 Although no generalisable solution has yet emerged, the widespread success of clinically used extracellular biologics proves that cell penetration and oral availability are not prerequisites for therapeutic relevance. Multiple recent studies reporting orally bioavailable macrocyclic peptides and miniproteins, however, suggest reason for optimism.39,85,122
Manufacturing considerations also affect the translational potential of these new drug modalities with regard to clinical use. Although constrained peptides can approach small molecule accessibility,150 highly specialised and constrained designs might demand increased reagent and process complexity.125 Similarly, although compact proteins are typically easier and cheaper to access than antibodies through either recombinant or chemical synthesis,151,152 this may be dependent on the specific scaffold and likely requires case-by-case optimisation.153 Nevertheless, these challenges represent engineering rather than conceptual barriers and advances in process innovation have historically helped resolve or mitigate them.154
Looking ahead, constrained peptides and compact proteins are well positioned to carve their niche in the therapeutic landscape. The opportunity to choose or even combine computational design, experimental screenings and medicinal chemistry optimisation provides fertile ground to develop strong drug candidates. Constrained peptides and compact proteins are set to become part of the next generation of drugs, making it worthwhile to stay in the loop on their trajectory.
Declaration of funding
S. Ullrich received funding from the Alexander von Humboldt Foundation through a Feodor Lynen Research Fellowship.
Declaration of use of AI
Assistance with photo- and copyediting was provided by Photoroom and ChatGPT.
Acknowledgements
The author is grateful for a Feodor Lynen Research Fellowship granted by the Alexander von Humboldt Foundation and thanks Prof. Hiroaki Suga (University of Tokyo) and Assoc. Prof. Christoph Nitsche (Australian National University) for their mentorship during his postdoctoral research and PhD studies. S. Ullrich is the recipient of the 2024 John Carver Award and the 2024 Cornforth Medal by the Royal Australian Chemical Institute.
References
1 Valeur E, Guéret SM, Adihou H, Gopalakrishnan R, Lemurell M, Waldmann H, et al. New modalities for challenging targets in drug discovery. Angew Chem Int Ed 2017; 56: 10294-10323.
| Crossref | Google Scholar | PubMed |
2 Jing X, Jin K. A gold mine for drug discovery: strategies to develop cyclic peptides into therapies. Med Res Rev 2019; 40: 753-810.
| Crossref | Google Scholar | PubMed |
3 Crook ZR, Nairn NW, Olson JM. Miniproteins as a powerful modality in drug development. Trends Biochem Sci 2020; 45: 332-346.
| Crossref | Google Scholar | PubMed |
4 Makurvet FD. Biologics vs. small molecules: drug costs and patient access. Med Drug Discov 2021; 9: 100075.
| Crossref | Google Scholar |
5 Ngo HX, Garneau-Tsodikova S. What are the drugs of the future? MedChemComm 2018; 9: 757-758.
| Crossref | Google Scholar | PubMed |
6 Li K, Crews CM. PROTACs: past, present and future. Chem Soc Rev 2022; 51: 5214-5236.
| Crossref | Google Scholar | PubMed |
7 He S, Dong G, Cheng J, Wu Y, Sheng C. Strategies for designing proteolysis targeting chimaeras (PROTACs). Med Res Rev 2022; 42: 1280-1342.
| Crossref | Google Scholar | PubMed |
8 Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR, et al. The current landscape of nucleic acid therapeutics. Nat Nanotechnol 2021; 16: 630-643.
| Crossref | Google Scholar | PubMed |
9 Sun X, Setrerrahmane S, Li C, Hu J, Xu H. Nucleic acid drugs: recent progress and future perspectives. Signal Transduct Target Ther 2024; 9: 316.
| Crossref | Google Scholar | PubMed |
10 Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther 2022; 7: 93.
| Crossref | Google Scholar | PubMed |
11 Lindberg J, Nilvebrant J, Nygren PÅ, Lehmann F. Progress and future directions with peptide-drug conjugates for targeted cancer therapy. Molecules 2021; 26: 6042.
| Crossref | Google Scholar | PubMed |
12 Muttenthaler M, King GF, Adams DJ, Alewood PF. Trends in peptide drug discovery. Nat Rev Drug Discov 2021; 20: 309-325.
| Crossref | Google Scholar | PubMed |
13 Ji X, Nielsen AL, Heinis C. Cyclic peptides for drug development. Angew Chem Int Ed 2023; 63: e202308251.
| Crossref | Google Scholar | PubMed |
14 Zhang S, Wu L, Dang M. Antibody mimetics: the next generation antibody engineering, a retrospective and prospective analysis. Biotech J 2023; 19: e2300532.
| Crossref | Google Scholar | PubMed |
15 Ciesiołkiewicz A, Lizandra Perez J, Berlicki Ł. Miniproteins in medicinal chemistry. Bioorg Med Chem Lett 2022; 71: 128806.
| Crossref | Google Scholar | PubMed |
16 Blanco M-J, Gardinier KM. New chemical modalities and strategic thinking in early drug discovery. ACS Med Chem Lett 2020; 11: 228-231.
| Crossref | Google Scholar | PubMed |
17 Nada H, Choi Y, Kim S, Jeong KS, Meanwell NA, Lee K. New insights into protein–protein interaction modulators in drug discovery and therapeutic advance. Signal Transduct Target Ther 2024; 9: 341.
| Crossref | Google Scholar | PubMed |
18 Colas K, Bindl D, Suga H. Selection of nucleotide-encoded mass libraries of macrocyclic peptides for inaccessible drug targets. Chem Rev 2024; 124: 12213-12241.
| Crossref | Google Scholar | PubMed |
19 Swenson CS, Mandava G, Thomas DM, Moellering RE. Tackling undruggable targets with designer peptidomimetics and synthetic biologics. Chem Rev 2024; 124: 13020-13093.
| Crossref | Google Scholar | PubMed |
20 Zorzi A, Deyle K, Heinis C. Cyclic peptide therapeutics: past, present and future. Curr Opin Chem Biol 2017; 38: 24-29.
| Crossref | Google Scholar | PubMed |
21 Vinogradov AA, Yin Y, Suga H. Macrocyclic peptides as drug candidates: recent progress and remaining challenges. J Am Chem Soc 2019; 141: 4167-4181.
| Crossref | Google Scholar | PubMed |
22 Schmidt M, Toplak A, Quaedflieg PJLM, van Maarseveen JH, Nuijens T. Enzyme-catalyzed peptide cyclization. Drug Discov Today Technol 2017; 26: 11-16.
| Crossref | Google Scholar | PubMed |
23 Lau YH, de Andrade P, Wu Y, Spring DR. Peptide stapling techniques based on different macrocyclisation chemistries. Chem Soc Rev 2015; 44: 91-102.
| Crossref | Google Scholar | PubMed |
24 Moiola M, Memeo MG, Quadrelli P. Stapled peptides: a useful improvement for peptide-based drugs. Molecules 2019; 24: 3654.
| Crossref | Google Scholar | PubMed |
25 Baeriswyl V, Heinis C. Polycyclic peptide therapeutics. ChemMedChem 2013; 8: 377-384.
| Crossref | Google Scholar | PubMed |
26 Rhodes CA, Pei D. Bicyclic peptides as next‐generation therapeutics. Chem Eur J 2017; 23: 12690-12703.
| Crossref | Google Scholar | PubMed |
27 Thombare VJ, Hutton CA. Bridged bicyclic peptides: structure and function. Pept Sci 2018; 110: e24057.
| Crossref | Google Scholar |
28 Ullrich S, Nitsche C. Bicyclic peptides: paving the road for therapeutics of the future. Pept Sci 2024; 116: e24326.
| Crossref | Google Scholar |
29 de Veer SJ, Kan M-W, Craik DJ. Cyclotides: from structure to function. Chem Rev 2019; 119: 12375-12421.
| Crossref | Google Scholar | PubMed |
30 Imai M, Colas K, Suga H. Protein grafting techniques: from peptide epitopes to lasso‐grafted neobiologics. ChemPlusChem 2024; 89: e202400152.
| Crossref | Google Scholar | PubMed |
31 Akkapeddi P, Teng KW, Koide S. Monobodies as tool biologics for accelerating target validation and druggable site discovery. RSC Med Chem 2021; 12: 1839-1853.
| Crossref | Google Scholar | PubMed |
32 Muyldermans S. Applications of nanobodies. Ann Rev Anim Biosci 2021; 9: 401-421.
| Crossref | Google Scholar |
33 Cary DR, Ohuchi M, Reid PC, Masuya K. Constrained peptides in drug discovery and development. J Synth Org Chem Jpn 2017; 75: 1171.
| Crossref | Google Scholar |
34 Angelini A, Cendron L, Chen S, Touati J, Winter G, Zanotti G, Heinis C. Bicyclic peptide inhibitor reveals large contact interface with a protease target. ACS Chem Biol 2012; 7: 817-821.
| Crossref | Google Scholar | PubMed |
35 Groß A, Hashimoto C, Sticht H, Eichler J. Synthetic peptides as protein mimics. Front Bioeng Biotechnol 2016; 3: 211.
| Crossref | Google Scholar | PubMed |
36 Mikol V, Kallen J, Pflügl G, Walkinshaw MD. X-Ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 Å resolution. J Mol Biol 1993; 234: 1119-1130.
| Crossref | Google Scholar | PubMed |
37 Guerlavais V, Sawyer TK, Carvajal L, Chang YS, Graves B, Ren J-G, et al. Discovery of sulanemadlin (ALRN-6924), the first cell-permeating, stabilized α-helical peptide in clinical development. J Med Chem 2023; 66: 9401-9417.
| Crossref | Google Scholar | PubMed |
38 Liu W, de Veer SJ, Huang Y-H, Sengoku T, Okada C, Ogata K, Zdenek CN, Fry BG, Swedberg JE, Passioura T, Craik DJ, Suga H. An ultrapotent and selective cyclic peptide inhibitor of human β-factor XIIa in a cyclotide scaffold. J Am Chem Soc 2021; 143: 18481-18489.
| Crossref | Google Scholar | PubMed |
39 Berger S, Seeger F, Yu T-Y, Aydin M, Yang H, Rosenblum D, Guenin-Macé L, Glassman C, Arguinchona L, Sniezek C, Blackstone A, Carter L, Ravichandran R, Ahlrichs M, Murphy M, Swanson Pultz I, Kang A, Bera AK, Stewart L, Garcia KC, Baker D. Preclinical proof of principle for orally delivered Th17 antagonist miniproteins. Cell 2024; 187: 4305-4317.
| Crossref | Google Scholar | PubMed |
40 Kawakami N, Sato H, Terasaka N, Matsumoto K, Suga H. MET‐Activating ubiquitin multimers. Angew Chem Int Ed 2023; 62: e202307157.
| Crossref | Google Scholar | PubMed |
41 Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods 2022; 19: 679-682.
| Crossref | Google Scholar | PubMed |
42 Esparza TJ, Chen Y, Martin NP, Bielefeldt-Ohmann H, Bowen RA, Tolbert WD, et al. Nebulized delivery of a broadly neutralizing SARS-CoV-2 RBD-specific nanobody prevents clinical, virological, and pathological disease in a Syrian hamster model of COVID-19. mAbs 2022; 14: 2047144.
| Crossref | Google Scholar | PubMed |
43 Meng EC, Goddard TD, Pettersen EF, Couch GS, Pearson ZJ, Morris JH, et al. UCSF ChimeraX: tools for structure building and analysis. Prot Sci 2023; 32: e4792.
| Crossref | Google Scholar | PubMed |
44 Wang L, Wang N, Zhang W, Cheng X, Yan Z, Shao G, et al. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther 2022; 7: 48.
| Crossref | Google Scholar | PubMed |
45 Schumacher D, Helma J, Schneider AFL, Leonhardt H, Hackenberger CPR. Nanobodies: chemical functionalization strategies and intracellular applications. Angew Chem Int Ed 2018; 57: 2314-2333.
| Crossref | Google Scholar | PubMed |
46 Wang H, Dawber RS, Zhang P, Walko M, Wilson AJ, Wang X. Peptide-based inhibitors of protein–protein interactions: biophysical, structural and cellular consequences of introducing a constraint. Chem Sci 2021; 12: 5977-5993.
| Crossref | Google Scholar | PubMed |
47 Hong SH, Nguyen T, Ongkingco JF, Nazzaro A, Arora PS. From concepts to inhibitors: a blueprint for targeting protein–protein interactions. Chem Rev 2025; 125: 6819-6869.
| Crossref | Google Scholar | PubMed |
48 Philippe GJB, Craik DJ, Henriques ST. Converting peptides into drugs targeting intracellular protein–protein interactions. Drug Discov Today 2021; 26: 1521-1531.
| Crossref | Google Scholar | PubMed |
49 Ellenbroek BD, Kahler JP, Arella D, Lin C, Jespers W, Züger EAK, Drukker M, Pomplun SJ. Development of DuoMYC: a synthetic cell penetrant miniprotein that efficiently inhibits the oncogenic transcription factor MYC. Angew Chem Int Ed 2025; 64: e202416082.
| Crossref | Google Scholar | PubMed |
50 Nielsen AL, Bartling CRO, Zarda A, De Sadeleer N, Neeser RM, Schwaller P, Strømgaard K, Heinis C. Bulk measurement of membrane permeability for random cyclic peptides in living cells to guide drug development. Angew Chem Int Ed 2025; 64: e202500493.
| Crossref | Google Scholar | PubMed |
51 White CJ, Yudin AK. Contemporary strategies for peptide macrocyclization. Nat Chem 2011; 3: 509-524.
| Crossref | Google Scholar | PubMed |
52 Reguera L, Rivera DG. Multicomponent reaction toolbox for peptide macrocyclization and stapling. Chem Rev 2019; 119: 9836-9860.
| Crossref | Google Scholar | PubMed |
53 Chow HY, Zhang Y, Matheson E, Li X. Ligation technologies for the synthesis of cyclic peptides. Chem Rev 2019; 119: 9971-10001.
| Crossref | Google Scholar | PubMed |
54 Wills R, Adebomi V, Raj M. Site-selective peptide macrocyclization. ChemBioChem 2021; 22: 52-62.
| Crossref | Google Scholar | PubMed |
55 Hayes HC, Luk LYP, Tsai Y-H. Approaches for peptide and protein cyclisation. Org Biomol Chem 2021; 19: 3983-4001.
| Crossref | Google Scholar | PubMed |
56 He J, Ghosh P, Nitsche C. Biocompatible strategies for peptide macrocyclisation. Chem Sci 2024; 15: 2300-2322.
| Crossref | Google Scholar | PubMed |
57 Heinis C, Winter G. Encoded libraries of chemically modified peptides. Curr Opin Chem Biol 2015; 26: 89-98.
| Crossref | Google Scholar | PubMed |
58 Dotter H, Boll M, Eder M, Eder A-C. Library and post-translational modifications of peptide-based display systems. Biotech Adv 2021; 47: 107699.
| Crossref | Google Scholar |
59 Ullrich S, Kumaresan S, Rahman MG, Panda B, Morewood R, Nitsche C. Assembling branched and macrocyclic peptides on proteins. Chem Commun 2025; 61: 3009-3012.
| Crossref | Google Scholar | PubMed |
60 Huang P-S, Boyken SE, Baker D. The coming of age of de novo protein design. Nature 2016; 537: 320-327.
| Crossref | Google Scholar | PubMed |
61 Albanese KI, Barbe S, Tagami S, Woolfson DN, Schiex T. Computational protein design. Nat Rev Methods Primers 2025; 5: 13.
| Crossref | Google Scholar |
62 Pacesa M, Nickel L, Schellhaas C, Schmidt J, Pyatova E, Kissling L, et al. One-shot design of functional protein binders with BindCraft. Nature 2025; 646: 483-492.
| Crossref | Google Scholar | PubMed |
63 Niu W, Guo J. Cellular site-specific incorporation of noncanonical amino acids in synthetic biology. Chem Rev 2024; 124: 10577-10617.
| Crossref | Google Scholar | PubMed |
64 Rezhdo A, Islam M, Huang M, Van Deventer JA. Future prospects for noncanonical amino acids in biological therapeutics. Curr Opin Biotechnol 2019; 60: 168-178.
| Crossref | Google Scholar | PubMed |
65 Goto Y, Suga H. The RaPID platform for the discovery of pseudo-natural macrocyclic peptides. Acc Chem Res 2021; 54: 3604-3617.
| Crossref | Google Scholar | PubMed |
66 Zhou KZQ, Obexer R. Non‐canonical amino acids for engineering peptides and proteins with new functions. Isr J Chem 2024; 64: e202400006.
| Crossref | Google Scholar |
67 Grossmann TN, Durukan C. From protein structures to functional biomimetics. Synlett 2024; 36: 119.
| Crossref | Google Scholar |
68 Sohrabi C, Foster A, Tavassoli A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat Rev Chem 2020; 4: 90-101.
| Crossref | Google Scholar | PubMed |
69 You S, McIntyre G, Passioura T. The coming of age of cyclic peptide drugs: an update on discovery technologies. Expert Opin Drug Discov 2024; 19: 961-973.
| Crossref | Google Scholar | PubMed |
70 Fan Y, Feng R, Zhang X, Wang Z-L, Xiong F, Zhang S, et al. Encoding and display technologies for combinatorial libraries in drug discovery: the coming of age from biology to therapy. Acta Pharm Sin B 2024; 14: 3362-3384.
| Crossref | Google Scholar | PubMed |
71 Li X, Craven TW, Levine PM. Cyclic peptide screening methods for preclinical drug discovery. J Med Chem 2022; 65: 11913-11926.
| Crossref | Google Scholar | PubMed |
72 Dockerill M, Winssinger N. DNA‐encoded libraries: towards harnessing their full power with Darwinian evolution. Angew Chem Int Ed 2022; 62: e202215542.
| Crossref | Google Scholar | PubMed |
73 Jaroszewicz W, Morcinek-Orłowska J, Pierzynowska K, Gaffke L, Węgrzyn G. Phage display and other peptide display technologies. FEMS Microbiol Rev 2022; 46: fuab052.
| Crossref | Google Scholar | PubMed |
74 Huang Y, Wiedmann MM, Suga H. RNA display methods for the discovery of bioactive macrocycles. Chem Rev 2018; 119: 10360-10391.
| Crossref | Google Scholar | PubMed |
75 Hampton JT, Liu WR. Diversification of phage-displayed peptide libraries with noncanonical amino acid mutagenesis and chemical modification. Chem Rev 2024; 124: 6051-6077.
| Crossref | Google Scholar | PubMed |
76 Chen FJ, Pinnette N, Gao J. Strategies for the construction of multicyclic phage display libraries. ChemBioChem 2024; 25: e202400072.
| Crossref | Google Scholar | PubMed |
77 Katoh T, Suga H. Reprogramming the genetic code with flexizymes. Nat Rev Chem 2024; 8: 879-892.
| Crossref | Google Scholar | PubMed |
78 Passioura T, Suga H. A RaPID way to discover nonstandard macrocyclic peptide modulators of drug targets. Chem Commun 2017; 53: 1931-1940.
| Crossref | Google Scholar | PubMed |
79 Suga H. Max‐Bergmann award lecture: a RaPID way to discover bioactive nonstandard peptides assisted by the flexizyme and FIT systems. J Pept Sci 2018; 24: e3055.
| Crossref | Google Scholar | PubMed |
80 Tavassoli A. SICLOPPS cyclic peptide libraries in drug discovery. Curr Opin Chem Biol 2017; 38: 30-35.
| Crossref | Google Scholar | PubMed |
81 Xie J, Yap K, de Veer SJ, Edwardraja S, Durek T, Trau M, et al. High-throughput enrichment of functional disulfide-rich peptides by droplet microfluidics. Lab Chip 2025; 25: 3525-3536.
| Crossref | Google Scholar | PubMed |
82 Linciano S, Mazzocato Y, Romanyuk Z, Vascon F, Farrera-Soler L, Will E, et al. Screening macrocyclic peptide libraries by yeast display allows control of selection process and affinity ranking. Nat Commun 2025; 16: 5367.
| Crossref | Google Scholar | PubMed |
83 Brennan A, Lovell S, Vance KW, Mason JM. An intracellular peptide library screening platform identifies irreversible covalent transcription factor inhibitors. Adv Sci 2025; 12: e2416963.
| Crossref | Google Scholar | PubMed |
84 Plais L, Scheuermann J. Macrocyclic DNA-encoded chemical libraries: a historical perspective. RSC Chem Biol 2022; 3: 7-17.
| Crossref | Google Scholar | PubMed |
85 Merz ML, Habeshian S, Li B, David J, Nielsen AL, Ji X, Il Khwildy K, Duany Benitez MM, Phothirath P, Heinis C. De novo development of small cyclic peptides that are orally bioavailable. Nat Chem Biol 2023; 20: 624-633.
| Crossref | Google Scholar | PubMed |
86 Monty OBC, Simmons N, Chamakuri S, Matzuk MM, Young DW. Solution-phase Fmoc-based peptide synthesis for DNA-encoded chemical libraries: reaction conditions, protecting group strategies, and pitfalls. ACS Comb Sci 2020; 22: 833-843.
| Crossref | Google Scholar | PubMed |
87 Sunkari YK, Nguyen T-L, Siripuram VK, Flajolet M. Impact of organic chemistry conditions on DNA durability in the context of DNA-encoded library technology. iScience 2023; 26: 107573.
| Crossref | Google Scholar | PubMed |
88 Habeshian S, Merz ML, Sangouard G, Mothukuri GK, Schüttel M, Bognár Z, Díaz-Perlas C, Vesin J, Bortoli Chapalay J, Turcatti G, Cendron L, Angelini A, Heinis C. Synthesis and direct assay of large macrocycle diversities by combinatorial late-stage modification at picomole scale. Nat Commun 2022; 13: 3823.
| Crossref | Google Scholar | PubMed |
89 Koh LQ, Lim YW, Gates ZP. Affinity selection from synthetic peptide libraries enabled by de novo MS/MS sequencing. Int J Pept Res Ther 2022; 28: 62.
| Crossref | Google Scholar |
90 Nolan MD, Schüttel M, Scanlan EM, Nielsen AL. Nanomole-scale photochemical thiol-ene chemistry for high-throughput late-stage diversification of peptide macrocycles. Pept Sci 2024; 116: e24310.
| Crossref | Google Scholar |
91 Iskandar SE, Chiou LF, Leisner TM, Shell DJ, Norris-Drouin JL, Vaziri C, Pearce KH, Bowers AA. Identification of covalent cyclic peptide inhibitors in mRNA display. J Am Chem Soc 2023; 145: 15065-15070.
| Crossref | Google Scholar | PubMed |
92 Velcicky J, Cremosnik G, Scheufler C, Meier P, Wirth E, Felber R, et al. Discovery of selective low molecular weight interleukin-36 receptor antagonists by encoded library technologies. Nat Commun 2025; 16: 1669.
| Crossref | Google Scholar | PubMed |
93 Rettie SA, Juergens D, Adebomi V, Bueso YF, Zhao Q, Leveille AN, Liu A, Bera AK, Wilms JA, Üffing A, Kang A, Brackenbrough E, Lamb M, Gerben SR, Murray A, Levine PM, Schneider M, Vasireddy V, Ovchinnikov S, Weiergräber OH, Willbold D, Kritzer JA, Mougous JD, Baker D, DiMaio F, Bhardwaj G. Accurate de novo design of high-affinity protein-binding macrocycles using deep learning. Nat Chem Biol 2025; [Published online 20 June 2025].
| Crossref | Google Scholar | PubMed |
94 Yan K, Miskolzie M, Banales Mejia F, Peng C, Ekanayake AI, Atrazhev A, Cao J, Maly DJ, Derda R. Late-stage reshaping of phage-displayed libraries to macrocyclic and bicyclic landscapes using a multipurpose linchpin. J Am Chem Soc 2024; 147: 789-800.
| Crossref | Google Scholar | PubMed |
95 Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol 2009; 5: 502-507.
| Crossref | Google Scholar | PubMed |
96 Ekanayake AI, Sobze L, Kelich P, Youk J, Bennett NJ, Mukherjee R, et al. Genetically encoded fragment-based discovery from phage-displayed macrocyclic libraries with genetically encoded unnatural pharmacophores. J Am Chem Soc 2021; 143: 5497-5507.
| Crossref | Google Scholar | PubMed |
97 Alcock LJ, Gao T, Bythell-Douglas R, Gao J, Krishna Sudhakar H, Huang T, et al. Potent cyclic peptide inhibitors disrupt the FANCM–RMI interaction. J Med Chem 2025; 68: 12615-12625.
| Crossref | Google Scholar | PubMed |
98 Katzy RE, van Neer RHP, Ferraz MJ, Nicolai K, Passioura T, Suga H, et al. Development of selective nanomolar cyclic peptide ligands as GBA1 enzyme stabilisers. RSC Chem Biol 2025; 6: 563-570.
| Crossref | Google Scholar | PubMed |
99 Jiramongkol Y, Patel K, Johansen-Leete J, Maxwell JWC, Chang Y, Du JJ, et al. An mRNA-display derived cyclic peptide scaffold reveals the substrate binding interactions of an N-terminal cysteine oxidase. Nat Commun 2025; 16: 4761.
| Crossref | Google Scholar | PubMed |
100 Mathiesen IR, Calder EDD, Kunzelmann S, Walport LJ. Discovering covalent cyclic peptide inhibitors of peptidyl arginine deiminase 4 (PADI4) using mRNA-display with a genetically encoded electrophilic warhead. Commun Chem 2024; 7: 304.
| Crossref | Google Scholar | PubMed |
101 Lan T, Peng C, Yao X, Chan RST, Wei T, Rupanya A, et al. Discovery of thioether-cyclized macrocyclic covalent Inhibitors by mRNA display. J Am Chem Soc 2024; 146: 24053-24060.
| Crossref | Google Scholar | PubMed |
102 Baillie TA. Targeted covalent inhibitors for drug design. Angew Chem Int Ed 2016; 55: 13408-13421.
| Crossref | Google Scholar | PubMed |
103 Boike L, Henning NJ, Nomura DK. Advances in covalent drug discovery. Nat Rev Drug Discov 2022; 21: 881-898.
| Crossref | Google Scholar | PubMed |
104 Hillebrand L, Liang XJ, Serafim RAM, Gehringer M. Emerging and re-emerging warheads for targeted covalent inhibitors: an update. J Med Chem 2024; 67: 7668-7758.
| Crossref | Google Scholar | PubMed |
105 Kim G, Grams RJ, Hsu K-L. Advancing covalent ligand and drug discovery beyond cysteine. Chem Rev 2025; 125: 6653-6684.
| Crossref | Google Scholar | PubMed |
106 Wang S, Faucher FF, Bertolini M, Kim H, Yu B, Cao L, et al. Identification of covalent cyclic peptide inhibitors targeting protein–protein interactions using phage display. J Am Chem Soc 2025; 147(9): 7461-7475.
| Crossref | Google Scholar | PubMed |
107 Ghareeb H, Yi Li C, Shenoy A, Rotenberg N, Shifman JM, Katoh T, Sagi I, Suga H, Metanis N. Mirror‐image random nonstandard peptides integrated discovery (MI‐RaPID) technology yields highly stable and selective macrocyclic peptide inhibitors for matrix metallopeptidase 7. Angew Chem Int Ed 2024; 64: e202414256.
| Crossref | Google Scholar | PubMed |
108 Zhang BB, Harrison K, Zhong Y, Maxwell JWC, Ford DJ, Calvey LP, So SS, Peterson FC, Volkman BF, Stone MJ, Bhusal RP, Kulkarni SS, Payne RJ. Discovery of selective cyclic D-sulfopeptide ligands of the chemokine CCL22 via mirror-image mRNA display with genetic reprogramming. J Am Chem Soc 2024; 146: 34253-34259.
| Crossref | Google Scholar | PubMed |
109 Hayashi G, Naito T, Miura S, Iwamoto N, Usui Y, Bando-Shimizu M, Suzuki S, Higashi K, Nonaka M, Oishi S, Murakami H. Generating a mirror-image monobody targeting MCP-1 via TRAP display and chemical protein synthesis. Nat Commun 2024; 15: 10723.
| Crossref | Google Scholar | PubMed |
110 Schmidt N, Kumar A, Korf L, Dinh-Fricke AV, Abendroth F, Koide A, Linne U, Rakwalska-Bange M, Koide S, Essen L-O, Vázquez O, Hantschel O. Development of mirror-image monobodies targeting the oncogenic BCR::ABL1 kinase. Nat Commun 2024; 15: 10724.
| Crossref | Google Scholar |
111 Schumacher TN, Mayr LM, Minor DL, Jr, Milhollen MA, Burgess MW, Kim PS. Identification of D-peptide ligands through mirror-image phage display. Science 1996; 271: 1854-1857.
| Crossref | Google Scholar | PubMed |
112 Lander AJ, Jin Y, Luk LYP. D‐peptide and D‐protein technology: recent advances, challenges, and opportunities. ChemBioChem 2023; 24: e202200537.
| Crossref | Google Scholar | PubMed |
113 Harrison K, Mackay AS, Kambanis L, Maxwell JWC, Payne RJ. Synthesis and applications of mirror-image proteins. Nat Rev Chem 2023; 7: 383-404.
| Crossref | Google Scholar | PubMed |
114 Callahan AJ, Gandhesiri S, Travaline TL, Reja RM, Lozano Salazar L, Hanna S, et al. Mirror-image ligand discovery enabled by single-shot fast-flow synthesis of D-proteins. Nat Commun 2024; 15: 1813.
| Crossref | Google Scholar | PubMed |
115 Dutta S, Batori V, Koide A, Koide S. High‐affinity fragment complementation of a fibronectin type III domain and its application to stability enhancement. Prot Sci 2009; 14: 2838-2848.
| Crossref | Google Scholar | PubMed |
116 Robson B. Doppelgänger proteins as drug leads. Nat Biotechnol 1996; 14: 892-893.
| Crossref | Google Scholar | PubMed |
117 Mihara E, Watanabe S, Bashiruddin NK, Nakamura N, Matoba K, Sano Y, Maini R, Yin Y, Sakai K, Arimori T, Matsumoto K, Suga H, Takagi J. Lasso-grafting of macrocyclic peptide pharmacophores yields multi-functional proteins. Nat Commun 2021; 12: 1543.
| Crossref | Google Scholar | PubMed |
118 Ito K, Sakai K, Suzuki Y, Ozawa N, Hatta T, Natsume T, Matsumoto K, Suga H. Artificial human MET agonists based on macrocycle scaffolds. Nat Commun 2015; 6: 6373.
| Crossref | Google Scholar | PubMed |
119 Kan M-W, Craik DJ. Discovery of cyclotides from Australasian plants. Aust J Chem 2020; 73: 287.
| Crossref | Google Scholar |
120 Ullrich S, Philippe GJB, Li CY, Suga H. Rational design and selection of next-generation cyclotide drugs using the knotted cyclic peptide scaffold MCoTI-II. Aust J Chem 2025; 78: CH25086.
| Crossref | Google Scholar |
121 Gandini L, de Veer SJ, Chan CHH, Passmore MR, Liu K, Lundon B, et al. Anticoagulation during extracorporeal membrane oxygenation (ECMO): a selective inhibitor of activated factor XII compared to heparin in an ex vivo model. ACS Pharmacol Transl Sci 2025; 8: 1260-1269.
| Crossref | Google Scholar | PubMed |
122 Johns DG, Campeau L-C, Banka P, Bautmans A, Bueters T, Bianchi E, et al. Orally bioavailable macrocyclic peptide that inhibits binding of PCSK9 to the low density lipoprotein receptor. Circulation 2023; 148: 144-158.
| Crossref | Google Scholar | PubMed |
123 Alleyne C, Amin RP, Bhatt B, Bianchi E, Blain JC, Boyer N, et al. Series of novel and highly potent cyclic peptide PCSK9 inhibitors derived from an mRNA display screen and optimized via structure-based design. J Med Chem 2020; 63: 13796-13824.
| Crossref | Google Scholar | PubMed |
124 Tucker TJ, Embrey MW, Alleyne C, Amin RP, Bass A, Bhatt B, et al. A series of novel, highly potent, and orally bioavailable next-generation tricyclic peptide PCSK9 inhibitors. J Med Chem 2021; 64: 16770-16800.
| Crossref | Google Scholar | PubMed |
125 Li H, Thaisrivongs DA, Shang G, Chen Y, Chen Q, Tan L, et al. Total synthesis of enlicitide decanoate. J Am Chem Soc 2025; 147: 11036-11048.
| Crossref | Google Scholar | PubMed |
126 Beaulieu M-E, Jauset T, Massó-Vallés D, Martínez-Martín S, Rahl P, Maltais L, et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci Transl Med 2019; 11: eaar5012.
| Crossref | Google Scholar | PubMed |
127 Voss S, Rademann J, Nitsche C. Peptide-bismuth bicycles: in situ access to stable constrained peptides with superior bioactivity. Angew Chem Int Ed 2022; 61: e202113857.
| Crossref | Google Scholar | PubMed |
128 Voss S, Adair LD, Achazi K, Kim H, Bergemann S, Bartenschlager R, New EJ, Rademann J, Nitsche C. Cell-penetrating peptide-bismuth bicycles. Angew Chem Int Ed 2024; 63: e202318615.
| Crossref | Google Scholar | PubMed |
129 Ullrich S, Somathilake U, Shang M, Nitsche C. Phage-encoded bismuth bicycles enable instant access to targeted bioactive peptides. Commun Chem 2024; 7: 143.
| Crossref | Google Scholar | PubMed |
130 Ullrich S, Ghosh P, Shang MH, Siryer S, Kumaresan S, Panda B, Davies LJ, Somathilake U, Patel AP, Nitsche C. Synthesis and stability studies of constrained peptide–antimony bicycles. Aust J Chem 2024; 77: CH24094.
| Crossref | Google Scholar |
131 Ghosh P, Davies LJ, Nitsche C. Engineered nanobodies bind bismuth, indium and gallium for applications in theranostics. Angew Chem Int Ed 2024; 64: e202419455.
| Crossref | Google Scholar | PubMed |
132 Davies LJ, Ghosh P, Siryer S, Ullrich S, Nitsche C. Peptide–bismuth tricycles: maximizing stability by constraint. Chem Eur J 2025; 31: e202500064.
| Crossref | Google Scholar | PubMed |
133 Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, Scheid JF, et al. A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods 2014; 11: 1253-1260.
| Crossref | Google Scholar | PubMed |
134 Wang Z, Li L, Hu R, Zhong P, Zhang Y, Cheng S, et al. Structural insights into the binding of nanobodies LaM2 and LaM4 to the red fluorescent protein mCherry. Prot Sci 2021; 30: 2298.
| Crossref | Google Scholar |
135 Lin K, Zhang C, Bai R, Duan H. Cyclic peptide therapeutic agents discovery: computational and artificial intelligence-driven strategies. J Med Chem 2025; 68: 10577-10598.
| Crossref | Google Scholar | PubMed |
136 Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature 2023; 620: 1089-1100.
| Crossref | Google Scholar | PubMed |
137 Asada N, Krebs CF, Panzer U. Miniproteins may have a big impact: new therapeutics for autoimmune diseases and beyond. Signal Transduct Target Ther 2024; 9: 298.
| Crossref | Google Scholar | PubMed |
138 Gründemann C, Stenberg KG, Gruber CW. T20K: an immunomodulatory cyclotide on its way to the clinic. Int J Pept Res Ther 2018; 25(1): 9-13.
| Crossref | Google Scholar |
139 Wang CK, Craik DJ. Cyclic peptide oral bioavailability: lessons from the past. Biopolymers 2016; 106: 901-909.
| Crossref | Google Scholar | PubMed |
140 Chen K, Pei D. Engineering cell-permeable proteins through insertion of cell-penetrating motifs into surface loops. ACS Chem Biol 2020; 15: 2568-2576.
| Crossref | Google Scholar | PubMed |
141 Buckton LK, Rahimi MN, McAlpine SR. Cyclic peptides as drugs for intracellular targets: the next frontier in peptide therapeutic development. Chem Eur J 2020; 27: 1487-1513.
| Crossref | Google Scholar | PubMed |
142 Schneider AFL, Kithil M, Cardoso MC, Lehmann M, Hackenberger CPR. Cellular uptake of large biomolecules enabled by cell-surface-reactive cell-penetrating peptide additives. Nat Chem 2021; 13: 530-539.
| Crossref | Google Scholar | PubMed |
143 Zizzari AT, Pliatsika D, Gall FM, Fischer T, Riedl R. New perspectives in oral peptide delivery. Drug Discov Today 2021; 26: 1097-1105.
| Crossref | Google Scholar | PubMed |
144 Buyanova M, Pei D. Targeting intracellular protein–protein interactions with macrocyclic peptides. Trends Pharmacol Sci 2022; 43: 234-248.
| Crossref | Google Scholar | PubMed |
145 Nicze M, Borówka M, Dec A, Niemiec A, Bułdak Ł, Okopień B. The current and promising oral delivery methods for protein- and peptide-based drugs. Int J Mol Sci 2024; 25: 815.
| Crossref | Google Scholar | PubMed |
146 Chan A, Tsourkas A. Intracellular protein delivery: approaches, challenges, and clinical applications. BME Front 2024; 5: 35.
| Crossref | Google Scholar | PubMed |
147 Franke J, Arafiles JVV, Leis C, Hackenberger CPR. Intracellular delivery of native proteins by bioreversible arginine modification (BioRAM) on amino groups. Angew Chem Int Ed 2025; 64: e202506802.
| Crossref | Google Scholar | PubMed |
148 Xiao W, Jiang W, Chen Z, Huang Y, Mao J, Zheng W, et al. Advance in peptide-based drug development: delivery platforms, therapeutics and vaccines. Signal Transduct Target Ther 2025; 10: 74.
| Crossref | Google Scholar | PubMed |
149 Gare CL, White AM, Malins LR. From lead to market: chemical approaches to transform peptides into therapeutics. Trends Biochem Sci 2025; 50(6): 467-480.
| Crossref | Google Scholar | PubMed |
150 Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide‐based drugs. Chem Biol Drug Des 2012; 81: 136.
| Crossref | Google Scholar |
151 Harel O, Jbara M. Chemical synthesis of bioactive proteins. Angew Chem Int Ed 2023; 62: e202217716.
| Crossref | Google Scholar | PubMed |
152 Wu Y. Trends in nanobody technology in industrialization. Discov Nano 2025; 20: 23.
| Crossref | Google Scholar | PubMed |
153 Khakerwala Z, Gaikwad S, Kumari S, Gupta GD, Makde RD. Recombinant production of de novo designed miniprotein using various fusion protein tags. Int J Biol Macromol 2025; 319: 145041.
| Crossref | Google Scholar | PubMed |
154 Caille S, Cui S, Faul MM, Mennen SM, Tedrow JS, Walker SD. Molecular complexity as a driver for chemical process innovation in the pharmaceutical industry. J Org Chem 2019; 84(8): 4583-4603.
| Crossref | Google Scholar | PubMed |
Sven Ullrich is a Feodor Lynen Research Fellow (Humboldt Foundation) at the University of Tokyo, Japan. He holds a degree in pharmacy from Heidelberg University, Germany, and a PhD in chemistry from the Australian National University. During his doctoral studies, he was a visiting student at the University of Alberta, Canada. His research focuses on modified peptides and proteins in genetically encoded libraries for drug discovery. He received the Dr Elizabeth Schram Young Investigator Award at the 27th American Peptide Symposium (2022), was a selected participant at the Global Young Scientists Summit in Singapore (2023) and was recognised as part of the CAS Future Leaders Top 100 initiative (2024). For his graduate work, he was awarded both the John Carver Award (ACT Branch, 2024) and the Cornforth Medal (National, 2024) by the Royal Australian Chemical Institute. |