

Determination of polyoxymethylene (POM) water partition coefficients for DDT and its degradation products, with inter-laboratory comparison of the passive sampling methodology and bioaccumulation in earthworm (Eisenia fetida)
Anja Enell A # * , Stephanie Casey B # , Ayan Au Musse C , Sarah Josefsson D , Johannes Kikuchi-McIntosh A E , Greta Nilén C , Karin Wiberg B , Anna-Karin Dahlberg B F § and Maria Larsson C §
A # * , Stephanie Casey B # , Ayan Au Musse C , Sarah Josefsson D , Johannes Kikuchi-McIntosh A E , Greta Nilén C , Karin Wiberg B , Anna-Karin Dahlberg B F § and Maria Larsson C §
A
B
C
D
E
F
# Shared first authorship.
§ Shared last authorship.
Handling Editor: Peter Croot
Abstract
The widespread use of the insecticide DDT has left a legacy of pollution that still threatens ecosystems today. This study presents a method to accurately measure the bioavailability of DDT and its breakdown products in contaminated soils. This will improve risk assessments and guide sustainable land management practices, helping to protect both the environment and human health.
The insecticide dichlorodiphenyltrichloroethane (DDT) and its degradation products (collectively DDX) are persistent organic pollutants that pose significant environmental risks due to their persistence and bioaccumulation in ecosystems. Accurate quantification of DDX bioavailability in soil systems is crucial for effective land management and risk assessment.
This study utilised equilibrium passive sampling with polyoxymethylene (POM) to determine the bioavailability of DDX in soil. The sorption dynamics of 10 DDX compounds were investigated (p,p′-DDT, o,p′-DDT, p,p′-dichlorodiphenyldichloroethane (p,p′-DDD), o,p′-DDD, p,p′-dichlorodiphenyldichloroethene (p,p′-DDE), o,p′-DDE, p,p′-dichlorodiphenylmethane (p,p′-DDM), p,p′-dichlorobenzophenone (p,p′-DBP), 1-chloro-2,2-bis(4-chlorophenyl)ethylene (p,p′-DDMU) and dicofol) and their POM–water partition coefficients (KPOM) were determined. The study involved interlaboratory comparisons, using soils from nine historically contaminated sites and ecotoxicology assessments (mortality, reproduction and bioaccumulation in earthworms, Eisenia fetida) to validate the POM method.
KPOM values for 9 of the 10 DDX compounds were successfully determined, allowing for accurate quantification of freely dissolved pore water concentrations of DDX in historically contaminated soils. The interlaboratory study highlighted important considerations in extraction and gas chromatography–mass spectrometry analysis, and the ecotoxicology study demonstrated the potential of POM passive sampling as a reliable tool for assessing DDX bioavailability (bioaccumulation in Eisenia fetida).
The POM method proved to be a robust and reliable approach for quantifying freely dissolved DDX, with implications for improving the accuracy of risk assessments and guiding sustainable land management practices. The study also highlighted the need for careful consideration of analytical challenges, such as the potential degradation of DDX compounds during gas chromatography analysis, to ensure accurate quantification.
Keywords: aged soil contamination, bioavailability, earthworm toxicity and uptake, equilibrium passive sampling, persistent organic pollutants, POPs, pore water concentration, risk assessment.
Introduction
From the 1940s to the early 1970s, the insecticide dichlorodiphenyltrichloroethane (DDT) was widely used to control the spread of vector-borne diseases, particularly mosquito-borne malaria, and lice, and to prevent pests in forestry and agriculture (Mansouri et al. 2017). Technical DDT products contain primarily p,p′-DDT (65–80%) and o,p′-DDT (15–21%) and small amounts of dichlorodiphenyldichloroethene (DDE) and dichlorodiphenyldichloroethane (DDD) (European Food Safety Authority 2006). These compounds and their degradation products, e.g. 1-chloro-2,2-bis-(4-chlorophenyl)ethylene (p,p′-DDMU), p,p′-dichlorodiphenylmethane (p,p′-DDM), dicofol and p,p′-dichlorobenzophenone (p,p′-DBP), hereafter collectively referred to as DDX, are persistent organic pollutants that can still be found at elevated levels in the environment, e.g. at previously treated agricultural and forest soils (Drenning et al. 2024). In addition, dicofol is not only a transformation product of DDT, but also a commercial acaricide, which has been widely used in controlling mites after DDT was banned in 1983 in China (Huang et al. 2018).
Being hydrophobic, DDX tend to sorb to natural organic matter (NOM) in the organic-rich top layers of soil, where soil-dwelling organisms are exposed, which may cause secondary poisoning of higher trophic levels of the ecosystem (European Food Safety Authority 2006; Chattopadhyay and Chattopadhyay 2015). Toxic effects of DDTs in secondary consumers include eggshell thinning in birds of prey and endocrine disruption in fish (Tubbs 2016), whereas long-term (chronical) effects for humans include endocrine disruption, cancer and immunodeficiency (Mansouri et al. 2017).
In Sweden, technical DDT has been extensively used in forestry to control pine weevils (Hylobius abietis), for example, treating plants in forest nurseries. Over time, as the insects developed resistance to DDT, gradually higher doses were required and applied until DDT was eventually fully banned in 1975, which has resulted in more than 750 old forest nurseries being classified as polluted (Ekelund and Hamilton 2001). Many of these sites are, besides the contamination, arable land of high quality, consisting of mainly sandy soils. Conventional remediation, i.e. excavation and landfilling, is not an environmentally sustainable option, considering the large volumes of contaminated soil and the need for extensive masses of uncontaminated soil for restoration. Sustainable remediation alternatives and accurate risk assessments are thus highly needed. Generic risk assessments, where soil contaminant concentrations are compared with national soil quality standards (also known as soil guideline values, soil screening values or soil target levels) (Swartjes 2011) may, however, result in overestimation of risks and unnecessary soil excavations. By considering the bioavailability of the contaminants, more accurate assessments can be made (Alexander 2000; Volchko et al. 2020). Several tools for assessing the bioavailability and mobility of hydrophobic organic compounds (HOC) have been developed, and their use in site-specific risk assessment projects is evolving. Equilibrium passive sampling allows for measurement of the freely dissolved concentration in pore water (CW,free), which is considered to reflect the bioavailable concentration (Hawthorne et al. 2011; Gomez-Eyles et al. 2012; Mayer et al. 2014), in contrast to the total porewater concentration (CW,total), which also includes compounds sorbed to dissolved organic matter (DOM) and particulate and colloidal matter (Enell et al. 2016).
By the use of pre-determined compound-specific equilibrium polymer–water partition coefficients, Kpolymer/water (L kg–1), and the quantification of the concentration of HOC in the polymer (Cpolymer) the water concentration (CW,free) can be calculated using Eqn 1:
In order to turn equilibrium passive sampling into a standardised and routine analysis of CW,free, it is crucial to have well-calibrated Kpolymer/water values and to use a polymer that is commercially available, inexpensive and robust during handling, e.g. does not tear, is easy to clean and is chemically stable in both aqueous media and organic solvents. Polyoxymethylene (POM) meets these criteria (Jonker and Koelmans 2001; Hawthorne et al. 2009) and is already used in commercial laboratories in Sweden for equilibrium passive sampling of polyaromatic hydrocarbons (PAHs), using 76-µm POM strips and the method described in Arp et al. (2014). POM–water partition coefficients (KPOM) have been determined for 76-µm strips for several HOC, e.g. polychlorinated biphenyls (PCBs) (Hawthorne et al. 2009; Perron et al. 2013a), polycyclic aromatic compounds (PACs) (Hawthorne et al. 2011; Kupryianchyk et al. 2011; Perron et al. 2013a; Josefsson et al. 2015) and polybrominated diphenyl ethers (PBDEs) (Perron et al. 2013b), but also for more polar substances, e.g. organophosphate esters (OPEs) (Qin et al. 2023). However, for DDX, KPOM values have, to our knowledge, only been reported for p,p′-DDE and p,p′-DDT (Endo et al. 2011). To meet the need for tools that can be used to improve risk assessments of DDX contaminated sites, we therefore opted to use POM in this study and to investigate this polymer’s applicability as a prediction tool for bioavailability for 10 environmentally relevant DDT related compounds: p,p′-DDT, o,p′-DDT, p,p′-DDD, o,p′-DDD, p,p′-DDE, o,p′-DDE, p,p′-DDM, p,p′-DBP, p,p′-DDMU and dicofol. For IUPAC names see Supplementary Table S1.
The objectives of the study were to (i) derive KPOM values for the selected DDX (using 76-µm POM strips) to enable determination of CW,free, (ii) perform an interlaboratory comparison study for determining CW,free in historically contaminated soils using the derived KPOM values, and (iii) compare the results with bioaccumulation and ecotoxicological studies (bioaccumulation of DDX, mortality, growth and reproduction) of the earthworm Eisenia fetida to verify POM as a biomimetic method. The results are discussed in terms of practical strategies for improving the risk assessment of historically DDT-contaminated soils by considering bioavailability.
Materials and methods
Part 1 of this study involved determining the KPOM values in Lab A. In Part 2, the reproducibility and applicability of the POM method were tested in an interlaboratory study involving two labs (Lab A and Lab B) analysing DDX in contaminated soils. Part 3 assessed the POM method as a biomimetic method by comparing it to the bioaccumulation of DDX by Eisenia fetida (Lab A). Detailed descriptions for the three parts of the study are given in the ‘Materials and Methods’ in the Supplementary material.
Part 1 – determination of KPOM (Lab A)
KPOM was determined for 10 DDX: o,p′-DDT, p,p′-DDT, o,p′-DDD, p,p′-DDD, o,p′-DDE, p,p′-DDE, p,p′-DDMU, p,p′-DDM, p,p′-DBP and dicofol (Supplementary Table S1). Fortification standards of native DDX compounds were prepared in methanol to obtain four mixtures (Cf1–Cf4; Supplementary Table S2). The aqueous solutions for the POM–water experiments were prepared using Milli-Q Ultrapure water, NaN3 and salts of NaH2PO4 and Na2HPO4. Two internal standard (IS) toluene solutions were prepared: (i) 13C-labelled o,p′-DDT, p,p′-DDT, o,p′-DDD, p,p′-DDD, o,p′-DDE and p,p′-DDE and (ii) deuterium-labelled dicofol (Supplementary Table S3). 13C-labelled PCB101 was used as a recovery standard (RS) (Supplementary Table S3).
The influence of analyte concentrations on KPOM was tested using 28-day end-over-end tumbling (Gerhardt Laboshake, Germany) with four different DDX concentration levels, chosen to cover the expected range of concentrations to be found in soils at historical contaminated sites but not exceeding aqueous solubilities: C1, C2, C3 and C4 in methanol (Table 1). The time required for the analytes to reach equilibrium between the aqueous phase and the POM (at pH 7.0), and the potential effect of pH (at 28 days tumbling), were investigated using the C2-level concentration, at tumbling times of 3, 7, 14, 28 and 56 days, and in buffer solutions titrated to pH 4.1 and pH 8.4 with 3 mol L–1 HCl and 1 mol L–1 NaOH (Table 1). All tests were performed in triplicate.
| Parameters | Concentration levels | ||||
|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | ||
| Concentration of DDX (µg L–1) | |||||
| p,p′-DBP | 0.003 | 0.9 | 10 | 100 | |
| p,p′-DDM | 0.03 | 1.6 | 9 | 90 | |
| Dicofol | 0.01 | 0.4 | 9 | 90 | |
| o,p′-DDD | 0.2 | 0.5 | 9 | 90 | |
| p,p′-DDD | 0.2 | 0.5 | 9 | 90 | |
| p,p′-DDMU | 0.04 | 3 | 9 | 90 | |
| o,p′-DDT | 0.3 | 0.6 | 9 | 90 | |
| o,p′-DDE | 0.2 | 0.5 | 9 | 90 | |
| p,p′-DDT | 0.3 | 0.6 | 9 | 90 | |
| p,p′-DDE | 0.2 | 0.4 | 9 | 90 | |
| Testing scheme | |||||
| Tumbling time (days) at pH = 7 | 28 | 3, 7, 14, 28 and 56 | 28 | 28 | |
| pH at 28 days of tumbling | 7.0 | 4.1, 7.0 and 8.4 | 7.0 | 7.0 | |
In brief, 76-μm-thick POM (CS Hyde, Lake Villa, IL, USA) was cut into 4- × 5-cm strips, weighed and placed in a 100-mL bottle with 100 g of 1 g L–1 NaN3 and 10 mmol L–1 PO4-buffer solution. For tests using concentration levels C1 and C2, 50 µL of the DDX fortification mixture (Cf1–Cf2; Supplementary Table S2) was added. For concentration levels C3 and C4, a staggered fortification approach was used to avoid exceedance of water solubilities. Once fortified, bottles were tumbled end-over-end. At the end of each test, POM strips were removed, wiped dry and placed in glass amber vials. POM strips and sample solutions were stored at +4°C until analysis.
Water samples were extracted twice with n-hexane using liquid–liquid extraction, with IS added prior to extraction. Extracts were combined, evaporated to 2 mL, treated with anhydrous Na2SO4 and concentrated to 100 µL. RS solution (Supplementary Table S3) was added, the solvent exchanged to toluene, and the sample stored in gas chromatography (GC) vials at −20°C for gas chromatography–mass spectrometry (GC-MS) analysis. POM strips were extracted twice with 40 mL of acetone–n-hexane (1:1, v/v) using ultra-sonication, with IS added beforehand. Combined extracts were evaporated to ~0.5 mL and extracts of low concentrations (C1, C2) were cleaned up with deactivated silica. All extracts were concentrated to 100 µL, solvent was exchanged to toluene, RS was added, and the sample stored at −20°C for GC-MS analysis. Further details are in ‘POM Concentrations, CPOM (Part 1)’ in the Supplementary material.
All measurements were performed in the selected ion monitoring mode. Identification and quantification of the target compounds were done using quantification standard solutions that included all compounds, in addition to IS and RS. More details are given in ‘POM Concentrations, CPOM (Part 1)’ in the Supplementary material.
Target compounds were quantified by use of a 10-point calibration curve. If a calibration point was below 80% or above 120% of expected linearity, the point was excluded to a minimum of eight calibration points. Quantification standards were analysed after every tenth sample to monitor instrumental performance and ensure calibration validity, with solvent blanks run at the start and end of each sequence to check for carry-over effects. Liners were replaced after 50 injections or upon sign of decomposition. Concentrations were calculated using isotope dilution. Owing to lack of an IS for p,p′-DDMU, p,p′-DDM and p,p′-DBP, calculations were based on 13C-o,p′-DDE peak areas (Supplementary Table S3). The limit of detection (LOD) was defined as the mean blank concentration plus 3× standard deviation (s.d.) and, in the absence of blank contamination, the LOD was set to the lowest concentration of the calibration curve. Samples with concentrations exceeding the range of the calibration curve were diluted, re-spiked and reanalysed. Sample blanks (n = 3) and process blanks (aqueous and POM) were processed in parallel to the experiments.
No DDX compounds were detected in the blanks, except for p,p′-DDMU (found in both sample blanks and process blanks), traces of p,p′-DDM (in one sample blank) and p,p′-DBP (in one process blank) (Supplementary Table S4). If the amount in the blank was >10% of the amount in the corresponding sample, the data for this sample was excluded from further evaluation (Supplementary Table S5).
Since the concentrations of DDX in both the POM and the water phase were measured, KPOM can be obtained even if there was a mass loss of the analyte during the experiment (e.g. volatilisation, transformation, glassware sorption). Nevertheless, to ensure good data quality, mass balance calculations were performed (amount in water + amount in POM) ÷ added amount), and if the recovery was <50 or >130%, the data were excluded (Supplementary Table S6). To check for potential degradation of DDT (to DDD) and dicofol (to DBP), which can occur in the GC injector (Foreman and Gates 1997; Fujii et al. 2011), mass balance calculations (recovery, i.e. added v. measured amounts) were compared between p,p′-DDT and ∑p,p′-DDT/D, o,p′-DDT and ∑o,p′-DDT/D, and dicofol and ∑p,p′-dicofol + DBP. These calculations indicated very low, or acceptably low, degradation in all experiments, except for dicofol at pH 8.4 as expected, due to the instability of this substance under alkaline conditions (Yin et al. 2017). The recovery was generally higher for ∑DDT/D than DDT, but the difference between DDT and ∑DDT/D recoveries were on average only 7% (s.d. ± 3) and 10% (s.d. ± 3) for p,p′- and o,p′-DDT/D respectively, which is well below the 20% threshold set by the United States Environmental Protection Agency (1995) guidelines for methods for the determination of organic compounds in drinking water. The difference between the recovery of dicofol and ∑p,p′-dicofol + DBP were on average 16% (s.d. ± 16), indicating a higher degradation of dicofol to DBP than DDT to DDD.
Part 2 – interlaboratory comparison of the POM method (Lab A and B)
In this exercise, we opted to use in-house methods without harmonisation and with special attention to potential degradation of DDX (in the injector) and large differences in concentration levels between different DDX compounds. The two laboratories performed parallel passive sampling with POM to determine CW,free (in triplicate) in soil samples from nine DDX-contaminated forest plant nurseries in Sweden (Supplementary Table S7), together with analyses of Csoil.
Soil samples were collected at a depth of 0–20 cm. The samples were sieved (<2 mm) and homogenised in the field before dividing into two subsamples sent to Lab A and B. After arrival at the lab, the soil sample was further homogenised and divided into subsamples for POM tests and analyses for soil characterisation. Details on the sampling, sample preparation and determination of soil properties (total organic carbon, TOC, content; pH; water holding capacity, WHC; and grain size distribution) and levels of potential soil contaminants other than DDX (pesticides and metals) are described in section ‘S1.3.1 Soil sampling and analyses of soils’ in the Supplementary material. Soil properties are listed in Table 2. None of the soils contained elevated levels of inorganic contaminants, but low to medium levels of pesticides other than DDT were found at some sites (Supplementary Table S8).
| Site | Deje Syd | Jakobsbyn | Ljungaskog | Kolleberga | Deje Nord | Sya | Stakheden | Klockatorp | Åby | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sample depth (cm) | 0–20 | 0–20 | 0–20 | 0–20 | 0–20 | 0–20 | 0–20 | 0–20 | 0–20 | |
| Sand 2–0.063 mm (%) | 29 | 88 | 84 | 91 | 20 | 89 | 62 | 81 | 85 | |
| Silt 63–2 µm (%) | 67 | 7.0 | 12 | 6.2 | 75 | 7.7 | 34 | 16 | 12 | |
| Clay <2 µm (%) | 4.7 | 4.7 | 3.7 | 3.2 | 4.9 | 3.7 | 3.8 | 2.7 | 2.9 | |
| pH (H2O) | 5.98 | 5.99 | 5.76 | 6.32 | 5.61 | 5.45 | 5.25 | 4.73 | 5.63 | |
| Total-N (%) | 0.12 | 0.11 | 0.12 | 0.081 | 0.14 | 0.074 | 0.092 | 0.12 | 0.088 | |
| Total-C (%) | 1.64 | 2.04 | 1.97 | 1.92 | 1.94 | 1.07 | 1.73 | 2.66 | 1.58 | |
| TOC (%) | 1.63 | 2.04 | 1.97 | 1.91 | 1.93 | 1.07 | 1.73 | 2.65 | 1.57 | |
| WHC (%) | 73.1 | 51.0 | 53.3 | 40.8 | 53.4 | 37.4 | 78.9 | 59.7 | 48.3 |
TOC, total organic carbon; WHC, water holding capacity.
In brief, 1 g of wet soil was mortared with anhydrous Na2SO4, IS was added and the soil extracted using accelerated solvent extraction (ASE) with in-cell clean up based on the method in Kim et al. (2019). A procedure blank containing anhydrous Na2SO4 and active silica was included in every batch of samples. The sample extract was evaporated to near dryness using nitrogen and solvent exchanged to toluene (final volume 0.4 mL). RS (Supplementary Table S3) was added to GC vials after transferring the extracts. More details are given in section ‘S1.3.1 Soil sampling and analyses of soils’ in the Supplementary material.
Freeze-dried soil (0.5–1 g) was Soxhlet extracted with dichloromethane (DCM) for 24 h, similar to Huang et al. (2018). Prior to extraction, IS was added to the sample (Supplementary Table S3) and a procedural solvent blank was included in every batch of samples. One of the soil samples (Deje Nord) was analysed in triplicate. After Soxhlet extraction, the extract was evaporated to near dryness using nitrogen and the solvent changed to n-hexane (1 mL). Activated granular copper was added to remove sulfur. The sample was then further cleaned up on an alumina–silica column and analytes were eluted with DCM–n-hexane similar to Huang et al. (2018). The sample was evaporated to near dryness using nitrogen, and the solvent changed to isooctane (final volume 2 mL). RS was added (Supplementary Table S3), and the sample was vortexed before an aliquot was transferred to a GC vial for analysis. More details are given in section ‘S1.3.1 Soil sampling and analyses of soils’ in the Supplementary material.
The tests were performed the same way in Lab A and B. In brief, 76-μm POM strips (4 × 5 cm) were placed in amber glass bottles together with 25 g of homogenised soil (calculated as dry weight, DW) and then mixed with 93 mL of water containing CaCl2 and NaN3. Vials were tumbled end-over-end for 28 days in the dark. Thereafter, the POM strips were removed, rinsed with ultrapure water, and wiped dry before being placed in clean scintillation vials and frozen (−20°C) until extraction.
To study variance caused by differences in the extraction and analysis steps of the two labs and thereby exclude any other variance (e.g. caused by heterogeneous contaminant dispersion in the soil samples and differences in conducting the shaking experiment), six of the POM strips at each lab were cut into halves (‘I’ and ‘II’) after the shake-test (in total 24 halves). The ‘I’ halves (n = 12) were then extracted and analysed by Lab B whereas Lab A extracted and analysed the ‘II’ halves (n = 12) (Supplementary Table S9).
The CW,free (ng L–1) of the DDX were calculated using the derived KPOM (L kg–1 POM) (part 1 of this study) and Eqn 2:
where CPOM is the concentration of DDX in the POM strip (ng kg–1), calculated from the amount of DDX in the strip divided by the weight of the POM strip (Supplementary Table S9).
To avoid underestimation of CW,free during equilibrium passive sampling, a criterion of <5% depletion of the HOC from the soil or sediment has been recommended (Mayer et al. 2014). With the exception of dicofol, the highest depletion (defined in the Supplementary material, see ‘S1.3.3 Control of depletion of DDX from the soil during the POM-test’) reported for Lab A and B was 31 and 36%, both found for o,p′-DDE, for which the Csoil was close to the limit of quantification (LOQ). Soils with TOC content >2% came close to meeting the 5% criterion for all DDX (Supplementary Table S10), except for dicofol analysed by Lab A, for which the depletion was 35%. Lab A also obtained very high depletion of dicofol (85 to >100%) for four forest plant nursery soils, whereas Lab B received ≤17% for all soils. Dicofol in these four samples was thus excluded from further evaluation in both Parts 2 and 3.
POM strips were extracted as described above (Lab A) with minor adjustments for Lab B; using 24 h of shaking at low speed instead of sonication, and isooctane instead of toluene as final solvent. Lab A performed clean up of all POM extracts using deactivated silica. More details are given in ‘S1.3 Interlaboratory comparison of the POM-method (Part 2)’ in the Supplementary material.
Lab A analysed soils and POM extracts using GC-MS as described in Part 1. Lab B analysed soil and POM extracts using GC-MS/MS and multiple reaction monitoring (MRM) mode. Identification and quantification were performed using authentic reference standards (Supplementary Table S3). For data evaluation, the software Agilent MassHunter Quantitative Analysis (for QQQ) was used.
In Lab A, the QA+QC of the GC-MS analysis is described in Part 1. POM sample blanks (i.e. POM in aqueous solution, with no soil added; n = 3) and a process blank per nine POM samples were included in the sample preparation stage and run together with the POM samples. Three process blanks were included in the preparation and analysis of the soil samples. The LOQ was defined as the mean blank concentration plus 10× s.d. and, in the absence of blank contamination, the LOQ was set to a signal-to-noise (S/N) ratio of 10. The average + s.d. of the recoveries for IS added to the POM samples was 98 ± 24% (n = 252). For IS added to soil samples, the average + s.d. of the recovery was 103 ± 18% (n = 63).
In Lab B, POM sample blanks (i.e. POM in aqueous solution, with no soil added; n = 3) and a process blank per six POM samples were included in the sample preparation stage and run together with the POM samples. Target compounds were quantified using an 11-point calibration curve. If a calibration point was below 80% or above 120% of expected linearity, the point was excluded to a minimum of seven calibration points. Concentrations of target compounds were calculated as described for Lab A. If the amount in the blank was 10% of the amount in the corresponding sample, the sample was excluded from further evaluation. Recoveries for IS added to the POM samples were occasionally found to be high, with six exceptional values. Excluding these six, the average ± s.d. was 106 ± 55 (n = 246). The average ± s.d. of the recovery for the soil samples was 104 ± 58 (n = 98). When samples were below the LOQ, replacement values were used for statistical analysis. The LOQ replacement values were half of the lowest point used on the calibration curve. If the compound was below the limit of detection (S/N ratio <3), the replacement value was three times the value reported, divided by the S/N ratio.
The statistical evaluation was performed using Excel (ver. 2407 Build 16.0.17830.20210, Microsoft) and the Excel add-in statistical program Data Analysis Toolpack.
Part 3 – verification of POM as a biomimetic method (Lab A)
Ecotoxicity tests with Eisenia fetida as model organism following the protocol of ISO 11268-1:2012 (mortality after 28 days of exposure) and ISO 11268-2:2023 (reproduction after 56 days of exposure) were performed at room temperature (20 ± 2°C) on sub-samples for the nine soils from the field sites and one ISO control soil (in single samples), and the earth worm bioaccumulation of DDX was analysed. The surviving worms from the mortality test had their gut content purged, and their bodies analysed for lipid content and bioaccumulation of DDX. GC-MS analysis was carried out as described in Part 1. Details are given in ‘S1.4 Verification of POM as a biomimetic method (Part 3)’ in the Supplementary material.
Results and discussion
Part 1 – determination of KPOM
Analysed DDX concentrations in water (CW), in POM (CPOM) and calculated KPOM values for the 10 studied DDTs are given in Supplementary Tables S11, S12 and S13. At the end of the experiment, the water concentrations were always <0.3% of the saturated water solubilities (Supplementary Table S1), even in high level treatments.
No correlation between pH and log KPOM was observed (Supplementary Fig. S1). There was an increase in mean KPOM between pH 7.0 and 8.4 for p,p′-DDM and p,p′-DDT, but the reverse was found for p,p′-DDD, and no other significant differences were observed at P < 0.05 (Supplementary Table S14). The pH effect was not evaluated for p,p′-DDMU and dicofol due to lack of data (blank/sample ratios >10% and CW < LOD respectively). Given the small sample size (n = 3) and varying results, no trend was deemed strong enough to separate the data set. Thus, data from the tests at the three pH levels (pH 4.1, 7.0 and 8.4) were combined into one data set for the evaluation of equilibrium time and the potential effect of analyte concentrations on KPOM.
At 28 days, after shaking POM with DDX-spiked water (using the C2 concentration), a steady state indicating chemical equilibrium (i.e. no apparent change in KPOM values over time) was achieved for all the studied compounds (Fig. 1). Previous kinetic studies on POM of 76-µm thickness have shown that HOC with log KOW < 4.5 can reach equilibrium with POM after 14 days of shaking, whereas compounds with higher hydrophobicity (log KOW between 4.5 and 6.8) may need approximately double the time (Hawthorne et al. 2011; Josefsson et al. 2015). The same trend was observed in our experiment, where compounds with high KOW values seemed to need longer to reach equilibrium, although most of the compounds appeared to have reached equilibrium already at 14 days. For example, p,p′-DDM, with the second lowest KOW value of the studied DDX (log KOW 5.3), may have reached equilibrium already after 7 days, whereas p,p′-DDE, which has the highest KOW value (log KOW 6.9) needed at least 28 days (statistics shown in Supplementary Table S15). There was also no difference between mean log KPOM values for p,p′-DDD, and o,p′- and p,p′-DDT derived at 14 and 28 days, indicating that 14 days of shaking will be enough for these compounds too. Dicofol (log KOW 5.8) and DDMU (log KOW 6.2) could not be statistically evaluated due to lack of data at several or all time points (Supplementary Fig. S2), but as the hydrophobicity of these compounds are lower than many of the other DDX compounds studied, they are assumed to reach equilibrium within the same time frame (i.e. within 28 days). Consequently, it was concluded that an exposure time of 28 days should be sufficient to reach equilibrium for all the investigated DDX. For evaluation of the potential effect of analyte concentrations on KPOM, the replicates with equilibration times of 28 and 56 days were used.
Mean log KPOM for the DDX as a function of tumbling time (days) derived for the C2 concentration level. Error bars represent the standard deviation (n = 3). Green circles, blue triangles, and red crosses indicate the pH of the test; 7.0, 8.4 and 4.1 respectively. Dicofol was excluded due to lack of data. DDMU is shown in Supplementary material; Fig. S2.
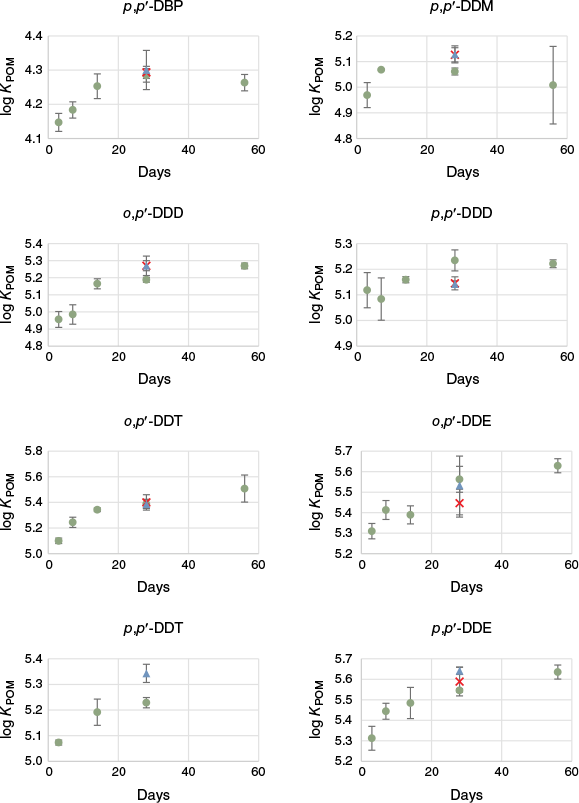
There were no consistent trends in KPOM values for three of the four tested concentrations (using the 28- and 56-day replicates; Supplementary Fig. S3 and Supplementary Table S16). The lowest concentration level (C1) gave, however, a noticeably lower mean log KPOM for the DDDs and DDEs but not for the DDTs (the other DDX could not be evaluated at this level due to data not meeting quality requirements or having CW < LOD). These lower KPOM values at the C1 level for some compounds were likely due to overestimation of areas for peaks with low signal-to-noise ratios. The DDTs had 50% higher starting concentrations than DDDs and DDEs in C1 (Table 1) and should theoretically reach a higher CW at equilibrium than the compounds with higher hydrophobicity (i.e. o,p′- and p,p′-DDE). Since the opposite was observed (CWo,p′-DDE > CWo,p′-DDT and CWp,p′-DDE ≈ CWp,p′-DDT (Supplementary Table S11), this indicates overestimation of peak area, leading to overestimation of CW at this very low concentration level. Therefore, it was decided to exclude C1 data for DDE and DDD in the final determination of log KPOM.
The mean KPOM values obtained at C3 did not differ significantly from those obtained at C4 for all compounds that could be evaluated at these two levels (i.e. dicofol, p,p′-DDM, and p,p′- and o,p′-DDE). Between C2 and C3, o,p′- and p,p′-DDE had an increased log KPOM by 0.38 and 0.39 log units respectively. Between C2 and C4, there was a slightly lower mean log KPOM for p,p′-DDM (−0.24 log unit), but a slightly higher value for p,p′-DDE (+0.25 log unit). Regressions of log CPOM v. log CW for the compounds with valid data (i.e. data that met our stated quality criteria) from three C levels (Supplementary Fig. S4) gave linear isotherms, with r2 values of 0.99, 0.97 and 0.96, for p,p′‑DDM, p,p′‑DDE and o,p′‑DDE respectively. Hence, we concluded that KPOM is not dependant on the analyte concentrations within our investigated interval. Since no general trend with concentration levels was observed, data from all concentration levels were used to calculate the final KPOM values.
The final log KPOM values for the 10 DDX compounds studied are listed in Table 3. Our measurements of log KPOM were highly repeatable (relative standard deviation, r.s.d., was 0.9–3.1%) except for p,p′-DDMU (r.s.d. = 16%), for which the final mean log KPOM was based on only three valid measurements. The final KPOM values for p,p′-DDT and p,p′-DDE are in the same range as values previously reported by Endo et al. (2011), with a deviation of −0.3 and +0.3 log units (i.e. a factor 0.5 and 2) respectively, but with better or equal r.s.d. (Table 3).
| Average log K POM this study ± s.d. | Average log K POM reported by Endo et al. (2011) | log K POM derived using the linear regression derived by Endo et al. (2011); log K POM = 1.01 × log K OW − 0.60 (±difference to our study) | ||
|---|---|---|---|---|
| p,p′-DBP | 4.30 ± 0.038 (n = 13; r.s.d. = 0.88%) | 3.78 (+0.51) | ||
| p,p′-DDM | 5.04 ± 0.11 (n = 16; r.s.d. = 2.1%) | 4.80 (+0.24) | ||
| Dicofol | 5.47 ± 0.061 (n = 5; r.s.d. = 1.1%) | 5.29 (+0.19) | ||
| o,p′-DDD | 5.26 ± 0.048 (n = 13; r.s.d. = 0.92%) | 5.44 (−0.18) | ||
| p,p′-DDD | 5.18 ± 0.050 (n = 11; r.s.d. = 1.0%) | 5.57 (−0.39) | ||
| p,p′-DDMU | 4.43 ± 0.71 (n = 3; r.s.d. = 16%) | 5.63 (−1.2) | ||
| o,p′-DDT | 5.40 ± 0.065 (n = 14; r.s.d. = 1.2%) | 6.15 (−0.75) | ||
| o,p′-DDE | 5.63 ± 0.17 (n = 16; r.s.d. = 3.1%) | 6.29 (−0.66) | ||
| p,p′-DDT | 5.32 ± 0.078 (n = 8; r.s.d. = 1.5%) | 5.66 ± 0.24 (n = 6; r.s.d. = 4.2%) | 6.31 (−0.99) | |
| p,p′-DDE | 5.70 ± 0.18 (n = 16; r.s.d. = 3.1%) | 5.44 ± 0.17 (n = 6; r.s.d. = 3.1%) | 6.36 (−0.65) |
For the average log KPOM reported by Endo et al. (2011), the sorption isotherms for the average log KPOM used a batch method where POM (76 μm thick) was mixed with water fortified with organochlorine pesticides (end-over-end at 10 rpm) for 37 days at a concentration range covering three orders of magnitude. Data derived for log KPOM derived using the linear regression derived by Endo et al. (2011) used the single-parameter linear free energy relationship (SP-LFER) model, i.e. log KPOM = 1.01 × log KOW − 0.60, and KOW values obtained with the SPARC model (Supplementary Table S1).
The relationship between the final log KPOM and the log KOW of the DDX (DDMU excluded) conformed to a linear regression (log KPOM = 0.44 × log KOW + 2.6) with an explanatory power of r2 = 0.81 (Supplementary Fig. S5). This regression has a lower slope (and different intercept) than the single-parameter linear free energy relationship (SP-LFER) model derived by Endo et al. (2011), which was based on log KPOM values for 43 compounds (not including any organochlorine pesticides (OCPs). Endo et al. also derived KPOM for 13 OCPs, including p,p′-DDT and p,p′-DDE, and noted that these deviated from the SP-LFER model; their experimentally determined KPOM values for p,p′-DDT and p,p′-DDE were lower (Table 3). However, their determined KPOM values matched well with our final values, and our regression between log KPOM and log KOW displayed a good linearity. The KOW values used here are from the SPARC model (ARCHem, see http://www.archemcalc.com/HTML/index.html). Few reliable experimental log KOW data exist for DDX (Pontolillo and Eganhouse 2001). The choice of using SPARC is based on the results in Eganhouse et al. (2018), who evaluated five of the most widely used and currently available computational methods and concluded that SPARC provided results that best matched the few available controlled experimental DDX data. However, we note that values derived from SPARC in 2024 gave higher values for o,p′-DDT (+0.8 log unit) and p,p-DDT (+0.9 log unit) compared to those reported by Eganhouse et al. (2018).
As experimental data are generally considered more reliable than predicted data, we recommend using the experimental final log KPOM values rather than the ones derived from log KPOM − log KOW regressions. The KPOM for DDMU should, though, be used with caution as the value is based on only n = 3 and the r.s.d. is high.
Other equilibrium passive sampler materials have been used for the determination of CW,free of DDX, such as low-density polyethylene (PE) (Hale et al. 2009; Borrelli et al. 2018) and poly(dimethylsiloxane) (PDMS) (Maruya et al. 2009; Xing et al. 2009; Eganhouse 2016). The precision we obtained (s.d. < 0.18 and s.e. < 0.05 log units, excluding DDMU), was of equal quality to data compiled in the study by Eganhouse (2016), where s.d. was generally <0.2 for log KPDMS/water derived from PDMS between 7 and 100 µm thick. It also compared well to data from Hale et al. (2009) where s.e. <0.16 for log KPE/water, using 51-µm-thick PE. The higher s.d. of our DDMU data, along with the much lower KPOM value than Kpolymer/water values reported by both Eganhouse (2016) and Hale et al. (2009) for this compound, indicates a potential underestimation. A comparison of our log KPOM data against log KPDMS and log KPE data from these studies is shown in Supplementary Fig. S6. In addition, all three materials are also regarded equivalent in terms of the affinity for hydrophobic non-polar chemicals (Endo et al. 2011). However, POM offers benefits when aiming to extract polar compounds from environmental phases since POM has H-bond acceptor sites in its molecular structure in contrast to PDMS and PE, and thus strong H-bond donor compounds generally have higher KPOM/water than KPDMS/water and KPE/water (Endo et al. 2011). Hence, POM is expected to be a more sensitive sorbent than PDMS and PE for, e.g. dicofol, which possesses an OH group, and thus has H-bond donor properties.
Part 2 – application of the POM method on soil samples from DDX-contaminated sites – interlaboratory comparison
As no monitoring of potential degradation of DDT to DDD in the GC injector was done in this part of the study, and as comparative Lab A and Lab B data suggested that degradation occurred (Supplementary Tables S20 and S22), sum levels of DDT and DDD (∑DDT/D) were used for both labs. This approach is also motivated by findings in Foreman and Gates (1997), which showed that even with monitoring through a Performance Evaluation Standard (PES) containing DDT but not DDD or DDE (US EPA methods, see references 14–17 in Foreman and Gates 1997), the degree of degradation and formation is uncertain and particularly DDT and DDD cannot be quantified individually, i.e. only as a sum, as has been applied in e.g. Wiberg et al. (2002). In the case of dicofol and DBP, comparative Lab A and Lab B data did not show consistent trends and the compounds were therefore not summed.
Soil concentrations of ∑p,p′-DDT/D, ∑o,p′-DDT/D, p,p′-DDE, o,p′-DDE, dicofol and p,p′-DBP are shown in Fig. 2 (Csoil for all individual compounds can be found in Supplementary Tables S17–S18). The Csoil values of p,p′-DDMU and p,p′-DDM were below the LOQ for all soils in Lab B’s analyses, and just above the LOQ in Lab A’s analyses. These two compounds were thus excluded from further evaluation. The mean Csoil (μg g–1 DW) of ∑DDX of Lab A and B ranged from ~5 to ~30 μg g–1 DW for eight of the nine studied sites, whereas one site (Åby) showed significantly higher concentrations (110 μg g–1 DW). The most dominant DDX in all soil samples was ∑p,p′-DDT/D, which accounted for 60–76% of the total concentration, followed by ∑o,p′-DDT/D (10–23%), p,p′-DDE (2.1–18%), dicofol (0.77–8.0%), p,p′-DBP (0.30–3.7%) and o,p′-DDE (0.0006–1.9%) (Supplementary Table S19).
Concentrations (μg g–1 DW) of DDX in soil samples from nine forest plant nurseries analysed by Lab A and Lab B. Åby concentrations as shown are divided by 10. Deje Nord in Lab B was analysed in triplicate (s.d. = 0.34 μg g–1 DW).
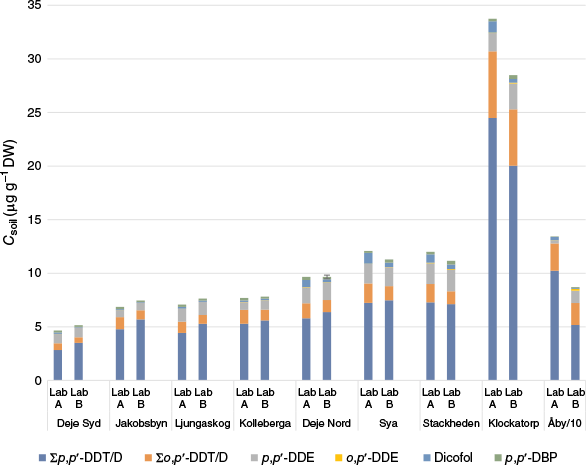
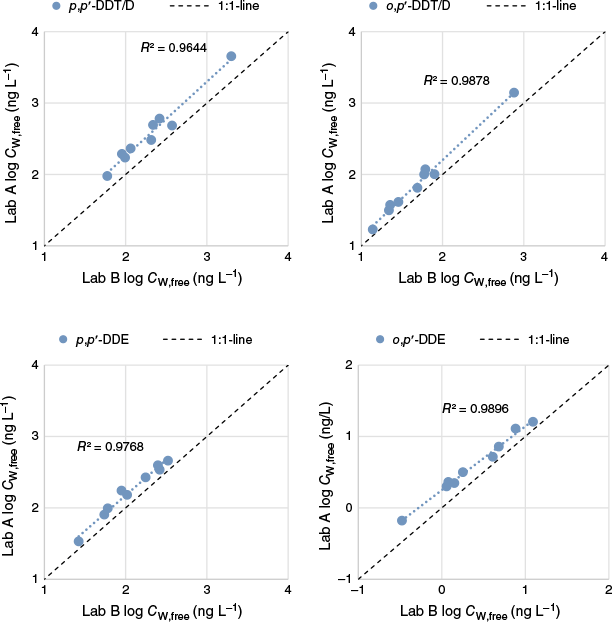
Both labs quantified similar Csoil for ∑p,p′-DDT/D, ∑o,p′-DDT/D, p,p′-DDE and p,p′-DBP, with concentration ratios of the labs (Csoil,LabA/Csoil,LabB) between 0.5 and 2 (Supplementary Table S20). Higher ratios, up to 4.8, were found for dicofol, but with a mean of 2.3. For this compound, Lab A found mostly higher levels (9 out of 10 sites), whereas for p,p′-DBP, Lab B found higher levels in 4 of 10 sites. This implies that a higher level of degradation in the GC injector of dicofol to p,p′-DBP occurred in Lab B. However, Lab B seemed to have had less conversion of DDT into DDD compared to Lab A. Differences in extraction procedure, clean up method, reference compound solutions and instrumental analysis can also be reasons for the different Csoil, which are expanded upon in Materials and methods, Part 2. For o,p′-DDE, one outlier was identified (negligible levels detected at Lab B) and excluded from the comparison. Overall, results showed relatively good agreement with mean ratios (Supplementary Table S20) ranging from 0.8 to 2.3, and the precision of the analysis (Lab B triplicate) was high, with r.s.d. ranging from 2.2 to 11%, with a mean of 6.4%.
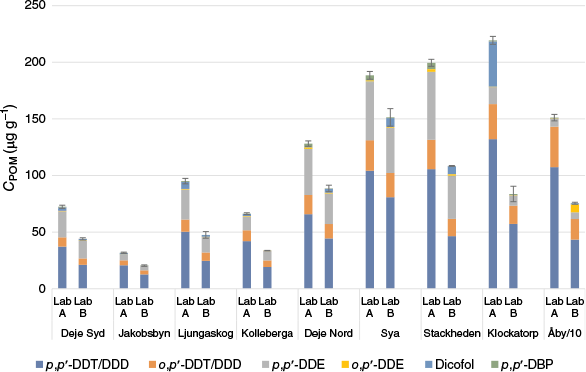
The levels of DDX in the POM strips (CPOM) are shown in Fig. 3, all CPOM data, means and standard deviations are in Supplementary Table S21, and ratios between CPOM analysed by Lab A and Lab B are in Supplementary Table S22. Lab A consistently quantified equal or higher CPOM than Lab B for all compounds. For the POM analyses, both labs experienced major challenges with very high MS signals. This required dilution of the extracts, further addition of IS and reanalysis, often more than once. This procedure is not optimal for good accuracy and is likely to have caused some of the observed differences between the labs. The CPOM mean ratios between labs were below 2, except for dicofol and p,p′-DBP with CPOM mean ratios of 35 and 10 respectively. Although the degradation of dicofol to DBP in the injector was deemed to be at an acceptably low level for soil samples, the ratios for POM suggest that this could have been an issue in these analyses. This highlights the importance of monitoring the degradation or adjusting the injection method to avoid such degradation, e.g. as suggested by Yin et al. (2017). Another potential explanation for the differences could be that 13C-labelled internal standards were not available for these two compounds; a deuterated internal standard was used for dicofol, and DBP was quantified using 13C-o,p′-DDE. In spite of analytical challenges, the precision of the analyses was good for both labs, with a mean r.s.d. of 5.1 and 11% respectively for Lab A and B (Supplementary Table S21).
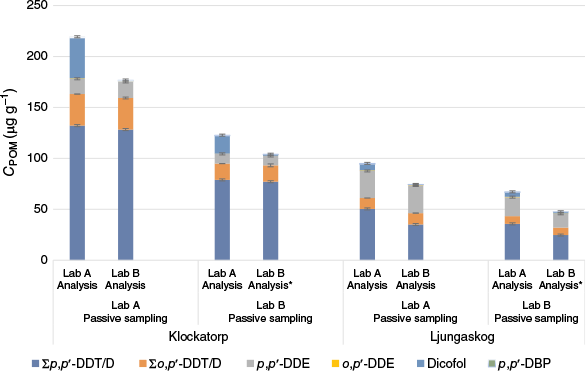
The strips shaken in soil from Klockatorp and Ljungaskog (by both labs) were cut in half and sent to the other lab for DDX analysis (n = 3 for each soil except Klockatorp shaken by Lab B, n = 2). This way, there were no differences in homogenisation and subsampling of the soil, and the comparison was focused on the analytical procedure from POM extraction onwards. Concentrations, standard deviations and lab data ratios are shown in Fig. 4 and Supplementary Table S22. The results showed that analysing the same POM strip resulted in similar precision, with a mean r.s.d. of 4.9 and 11% for Lab A and B respectively (Supplementary Table S23) (5.1 and 11% in the full CPOM comparison). Lab ratios improved, however, some high ratios were still found. The CPOM results for both isomers of ΣDDT/D and DDE were not significantly different between labs (t-test: P > 0.05), with all ratios between labs ranging from 0.8 to 1.4 (as compared with 1.2–2.6). CPOM values for dicofol and p,p′-DBP, however, were significantly different for every strip for nearly every site, with Lab A consistently quantifying higher values than Lab B, i.e. the same trend as for Csoil. This could be attributed to the analytical procedure from POM extraction onwards. As the agreement between labs was good for most substances (ratios close to one), and only unacceptably high for the low level compounds, dicofol and DPB, this points to the instrumental analysis as the major cause for the differences, e.g. the need for several dilution steps and reanalysis, lack of some 13C-labelled IS, and the potential degradation of dicofol to DPB in the GC injector. The somewhat higher precision of Lab A might partly be attributed to well established homogenisation and subsampling procedures.
Based on the concentrations in the POM strips (CPOM), the CW,free values for the nine field sites were determined (Supplementary Table S24 and Fig. 5) using KPOM established in Part 1 of this study (Table 3). The compound pairs p,p′-DDT/p,p′-DDD and o,p′-DDT/o,p′-DDD have different KPOM values, with DDTs having higher values. In our conversions of CPOM into CW,free, we opted for using individual levels of DDT and DDD and their respective KPOM. Consequently, the following CW,free results for DDT and DDD are approximations, with potential underestimation of DDT and overestimation of DDD. As we cannot be sure of the exact deviation from a true ΣDDT/D, the quantification results are to be seen as relatively valid with respect to each other, but not accurate. For every compound, Åby showed, as expected, significantly higher CW,free than the other soils, due to the much higher Csoil, which reflects the land use history of the sampling area as a DDT dipping station for saplings. This, however, also resulted in higher variation in the analysis due to multiple dilution of the sample being necessary for the value to be within the range of the calibration curve. Åby also generally showed bad lab ratios for Csoil and CPOM. Although there was some variation in reported concentrations, the CW,free data were significantly correlated between labs (P < 0.05, r2 > 0.96 for log–log relationships) (Fig. 5), excluding p,p′-DBP (r2 = 0.93) and dicofol (r2 = 0.43) (Supplementary Fig. S5). Overall, the r.s.d. remained low for both labs (mean r.s.d. Lab A = 3.9%; mean r.s.d. Lab B = 10%).
Part 3 – verification of POM as a biomimetic method (Lab A)
Normalising worm DDX concentrations (Cworm) to the individual fat content of the worms resulted in lipid concentrations (Cworm_lipid) ranging for ∑DDX-10 from 558 to 3840 µmol kg–1 lipid with Jakobsbyn at the low end and Sya at the top (Table 4). As mortality was 100% in the soil with the highest Csoil (Åby), no worms could be analysed for this soil. All Cworm and Cworm_lipid data are presented in Supplementary Tables S25 and S26.
| Σ10-DDX | C soil (mg kg–1 soil DW) | C worm_lipid (µmol kg–1 lipid) | Mortality after 28 days (%) | Adult biomass after 28 days (%) | Reproduction 56 days | |
|---|---|---|---|---|---|---|
| Number of juveniles hatched | ||||||
| ISO control | 0 | 0 | 0 | 112 | 81 | |
| Deje Syd | 4.7 | 760 | 0 | 130 | 146 | |
| Jakobsbyn | 6.9 | 560 | 0 | 133 | 49 | |
| Ljungaskog | 7.1 | 1 030 | 0 | 118 | 93 | |
| Kolleberga | 7.7 | 79 | 0 | 123 | 32 | |
| Deje Nord | 9.7 | 907 | 75 | 118 | 15 | |
| Sya | 12 | 3 800 | 33 | 118 | 47 | |
| Stakheden | 12 | 2 200 | 0 | 135 | 83 | |
| Klockatorp | 34 | 2 900 | 0 | 120 | 86 | |
| Åby | 130 | NA | 100 | NA | 3 | |
| Pearsons correlation for C soil v. C worm_lipid & ecotoxicity results | 0.60 | 0.72 | −0.28 | −0.48 | ||
| Pearsons correlation for C worm_lipid v. ecotoxicity results | 0.09 | 0.08 | −0.03 |
log–log correlations between Cworm_lipid v. CPOM, Csoil and CTOC (i.e. the soil concentration normalised to the fraction of TOC content; Csoil/fTOC), were conducted for the ∑DDX-10 (Fig. 6), the individual DDX, ∑p,p′-DDT/D and ∑o,p′-DDT/D (Supplementary Table S27). The worst log–log correlations were observed for Cworm_lipid v. Csoil (r2 of 0.60–0.93), and the best for Cworm_lipid v. CPOM (r2 of 0.82–0.97) and Cworm_lipid v. CTOC (r2 from 0.89 to 0.97), excluding results for p,p′-DBP, the compound with the lowest KOW value, which had r2 of 0.84, 0.24 and 0.00012 for CPOM, CTOC and Csoil respectively.
Correlations of log Cworm_lipid with various chemical measurements, including log CPOM (top left), log Csoil (top right) and log CTOC (bottom left).
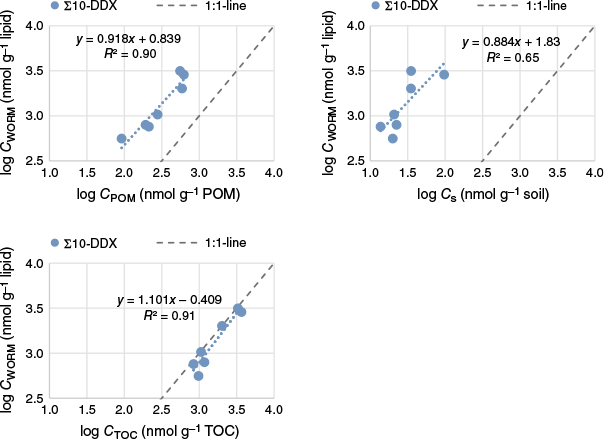
In addition to the strong linear log–log correlations between Cworm_lipid and CPOM, the slopes of these relationships were also close to one (between 0.80 and 1.05) for all DDX, except for dicofol and o,p′-DDD, which had slightly lower slopes of 0.66 and 0.77 respectively (Fig. 6 and Supplementary Table S27). The fact that the linear regression slopes were near one and exhibited better r2 values than log–log correlations of Cworm_lipid v. Csoil demonstrates that the POM method has the capacity to accurately predict the bioavailability of DDX in soil and is superior for predicting bioaccumulation compared to analysing total DDX content in the soil.
The strong correlations and feasibility of using POM as a prediction tool for bioaccumulation align with other studies on POM and HOC bioaccumulation in earthworms. Arp et al. (2014) reported a log–log correlation (r2 = 0.94) for PAH bioaccumulation by Enchytraeus crypticus using 76 μm POM membranes, as in this study. Similarly, Wang et al. (2018) found strong correlations for DDX bioaccumulation by Eisenia fetida using PE (r2 = 0.93) and solid-phase microextraction (SPME) (r2 = 0.91). By contrast, poorer r2 values were observed for bioaccessibility methods such as Tenax (r2 = 0.79), isotope dilution (r2 = 0.77) and supercritical fluid extraction (r2 = 0.47). The only study, to our knowledge, that has investigated the correlation between POM and earthworms is the study by Denyes et al. (2016), who also used 76 μm POM to predict E. fetida bioaccumulation of p,p′-DDT and p,p′-DDE. They did not report any r2 values but concluded that their mean worm bioaccumulation factors (BAFs) were within 50% of the POM-derived BAFs.
It can be argued, based on the good correlation between Cworm_lipid and CTOC (Fig. 6), that this analysis can be used instead of POM analyses. One possibility, however, is that the good correlation can be attributed to the fact that all soils included in the study originated from plant nurseries situated in rural–agrarian regions with comparable NOM and similar contaminant age. Inclusion of soils from more urban environments would likely have resulted in greater variation and a poorer correlation, as anthropogenic carbon, such as soot, has been demonstrated to have a significantly greater sorption capacity for HOC than NOM (Cornelissen et al. 2005). The presence of types of black carbon, such as soot and charcoal, may therefore reduce the predictive power of CTOC and lead to overestimation of bioavailability. It is therefore recommended that POM analysis and the here derived Cworm_lipid v. CPOM correlations are used in preference to Csoil and TOC determinations to predict the bioaccumulation of DDX in worms.
As illustrated in Fig. 6, CPOM can serve as a prediction tool for Cworm_lipid. Consequently, passive sampling with POM may also be employed to predict ecotoxicity, provided that a correlation exists between the organism’s bioaccumulation of DDX and the toxic response. However, the endpoints investigated here (growth, reproduction and mortality) were not sufficiently sensitive for the concentration range investigated. No significant correlation was found for mortality or reproduction against Cworm_lipid or CPOM. It should be noted, though, that the sample with the highest expected Cworm_lipid (Åby; with Csoil of 134 mg DDX kg–1 DW), was not analysed, due to 100% mortality of worms, and that the Csoil of Σp,p′-DDX showed a moderate and significant (P < 0.05) linear correlation with mortality percentage at 28 days (Pearsons correlation: 0.72, r2 = 0.52).
DDT is an insecticide, and low levels of DDT in soil are expected to have toxic effects on terrestrial organisms, for example, an acute 50% lethal concentration (LC50) for the cricket Gryllus pennsylvanicus has been reported to be 10 mg DDT kg–1 DW in soil with 10% NOM. There are, however, few data for terrestrial organisms available in ecological databases. Available data for Eisenia fetida in the scientific literature suggest that this species can tolerate high DDX concentrations, which is in line with our results (Table 4); the half maximal effective concentration (EC50) for reproduction has been reported to be 588 mg DDT kg–1 DW, in a sandy soil with 1% TOC (Hund-Rinke and Simon 2005) and Shi et al. (2016) reported an acute LC50 of 274 mg DDT kg–1 DW (14 days exposure) and a chronic LC50 of 146 mg kg–1 for historical DDT-contaminated soils. In addition, Shi et al. (2016) showed that DDT in concentrations lower than 100 mg kg–1, in both historical contaminated soils and DDT-spiked natural soils, had minimal effects on the mortality of mature earthworms (Eisenia fetida), whereas few survivors were found in spiked artificial soils at DDT soil concentrations >200 mg kg–1. Further investigations with more sensitive species or endpoints are thus required to determine whether CPOM can be used to predict toxicity.
POM can also be a tool to investigate the feasibility of remediation options and to check the achievement of objectives after remedial actions have been taken, especially in the case of stabilisation measures, which involve the sorption of contaminants onto a sorbent, e.g. biochar (Denyes et al. 2016). As the amount of contaminant is not removed from the soil, but stabilised, it is not possible to measure the effectiveness of remediation by Csoil analysis, and methods are needed to demonstrate how well the sorbent is working and whether the solubility, bioavailability and dispersion of the contaminants have been reduced to acceptable levels (Wang et al. 2018). Complementary measurements of CW,free and TOC enable calculations of soil–porewater distribution coefficients (Kd) and TOC–normalised soil–water distribution coefficients (KTOC), see Supplementary Eqn S3, which are useful for this purpose, i.e. to assess how the sorption of DDX can improve after stabilisation measures, but also for risk assessment purposes, e.g. in modelling transport between different environmental compartments.
The KTOC values of this study can be found in Supplementary Table S28, and ratios of log KTOC derived by Lab A and Lab B are presented in Supplementary Table S28. The ratios were close to one in experiments where depletion was <10%, with the exception of the thermolabile compounds (mainly dicofol and its degradation product p,p′-DBP). The thermoinstability and potential matrix effect (Foreman and Gates 1997) may result in overestimations and underestimations of log KTOC, which is reflected in the absence of good linear correlations between log KTOC and log KOW in our study (examples given in Supplementary Fig. S7).
In conclusion, this study shows that POM is a reliable tool for measuring CW,free, and can be used for further calculations of bioaccumulation, using the here derived correlations for earthworms. The importance of controlling for degradation during GC analysis to accurately determine individual DDX analytes is highlighted, which is important when deriving site and compound-specific distribution coefficients needed for dispersion modelling.
Conclusions
In this study, KPOM values were determined for 10 environmentally relevant DDX compounds. The novel KPOM data presented here will allow for assessing freely dissolved pore water concentrations in soils and sediments, as well as other environmental media using POM as a passive sampler, which will facilitate investigations and risk assessments on the bioavailability and ecotoxicity of DDX. As the POM method is similar for other organic contaminants (e.g. PAH), the same POM sampling of contaminated soil or sediment can be used for measuring a multitude of compounds simultaneously.
Our experiments show that POM, as a quantitative method for freely dissolved DDX, seems to be a robust and reliable method with generally high precision (low r.s.d.) and linear isotherms. By contrast, the interlaboratory study showed that the analysis of DDX in the POM strip can be challenging, as several of the DDX compounds are thermolabile and can be converted or degraded during GC analysis, which can lead to both under- and overestimations of the levels. We also found that another major challenge is when DDX compounds occur at high or highly different levels, which may require pre-screening and adjustment of the analytical method so as to avoid dilution and reanalysis of sample extracts.
Soil-to-POM ratios of 100–200 have previously been proven to be enough for meeting the suggested criterion of <5% depletion when analysing HOC in historical urban contaminated soils and sediments. In this study we used a soil-to-POM ratio of 110:1 (on DW soil basis), which led to ≤36% depletion (excluding dicofol). To meet the <5% criterion in future tests of agricultural soils, where the sorbent domain consists of NOM and little or no anthropogenic carbon is present, we recommend that the soil-to-POM ratio is increased to 800:1. Owing to the strong sorption of POM for polar compounds, even higher ratios may be needed to bring down the depletion of dicofol to <5%.
Data availability
The data used to generate the results in this paper are available in the Supplementary material and at the Swedish National Data Service’s (SND) research data catalogue (Researchdata.se; doi:10.5878/efnj-xv18).
Declaration of funding
This study was financially supported by the Swedish Geotechnical Institute (reference number 1.1-1801-0039 to A. Enell), FORMAS (grant number 2019-01166 to M. Larsson), and the Geological Survey of Sweden (SGU) and Sveaskog Timber AB through the research project Myco-DDT (reference number 3415-1660/2021 to A. K. Dahlberg).
References
Alexander M (2000) Aging, bioavailability, and overestimation of risk from environmental pollutants. Environmental Science and Technology 34, 4259-4265.
| Crossref | Google Scholar |
Arp HPH, Lundstedt S, Josefsson S, Cornelissen G, Enell A, Allard AS, Kleja DB (2014) Native oxy-PAHs, N-PACs, and PAHs in historically contaminated soils from Sweden, Belgium, and France: their soil-porewater partitioning behavior, bioaccumulation in Enchytraeus crypticus, and bioavailability. Environmental Science and Technology 48, 11187-11195.
| Crossref | Google Scholar | PubMed |
Borrelli R, Tcaciuc AP, Verginelli I, Baciocchi R, Guzzella L, Cesti P, Zaninetta L, Gschwend PM (2018) Performance of passive sampling with low-density polyethylene membranes for the estimation of freely dissolved DDx concentrations in lake environments. Chemosphere 200, 227-236.
| Crossref | Google Scholar | PubMed |
Chattopadhyay S, Chattopadhyay D (2015) Remediation of DDT and its metabolites in contaminated sediment. Current Pollution Reports 1, 248-264.
| Crossref | Google Scholar |
Cornelissen G, Gustafsson Ö, Bucheli TD, Jonker MTO, Koelmans AA, Van Noort PCM (2005) Extensive sorption of organic compounds to black carbon, coal, and kerogen in sediments and soils: Mechanisms and consequences for distribution, bioaccumulation, and biodegradation. Environmental Science and Technology 39, 6881-6895.
| Crossref | Google Scholar |
Denyes MJ, Rutter A, Zeeb BA (2016) Bioavailability assessments following biochar and activated carbon amendment in DDT-contaminated soil. Chemosphere 144, 1428-1434.
| Crossref | Google Scholar | PubMed |
Drenning P, Volchko Y, Enell A, Berggren Kleja D, Larsson M, Norrman J (2024) A method for evaluating the effects of gentle remediation options (GRO) on soil health: demonstration at a DDX-contaminated tree nursery in Sweden. Science of The Total Environment 948, 174869.
| Crossref | Google Scholar | PubMed |
Eganhouse RP (2016) Determination of polydimethylsiloxane–water partition coefficients for ten 1-chloro-4-[2,2,2-trichloro-1-(4-chlorophenyl)ethyl]benzene-related compounds and twelve polychlorinated biphenyls using gas chromatography/mass spectrometry. Journal of Chromatography A 1438, 226-235.
| Crossref | Google Scholar | PubMed |
Eganhouse RP, DiFilippo E, Pontolillo J, Orem W, Hackley P, Edwards BD (2018) DDT and related compounds in pore water of shallow sediments on the Palos Verdes Shelf, California, USA. Marine Chemistry 203, 78-90.
| Crossref | Google Scholar |
Endo S, Hale SE, Goss K-U, Arp HPH (2011) Equilibrium partition coefficients of diverse polar and nonpolar organic compounds to polyoxymethylene (POM) passive sampling devices. Environmental Science and Technology 45, 10124-10132.
| Crossref | Google Scholar | PubMed |
Enell A, Lundstedt S, Arp HPH, Josefsson S, Cornelissen G, Wik O, Berggren Kleja D (2016) Combining Leaching and Passive Sampling to Measure the Mobility and Distribution between Porewater, DOC, and Colloids of Native Oxy-PAHs, N-PACs, and PAHs in Historically Contaminated Soil. Environmental Science and Technology 50, 11797-11805.
| Crossref | Google Scholar | PubMed |
European Food Safety Authority (2006) Opinion of the Scientific Panel on contaminants in the food chain on a request from the commission related to DDT as an undesirable substance in animal feed. EFSA Journal 433, 1-69.
| Crossref | Google Scholar |
Foreman WT, Gates PM (1997) Matrix-enhanced degradation of p,p′-DDT during gas chromatographic analysis: a consideration. Environmental Science and Technology 31, 905-910.
| Crossref | Google Scholar |
Fujii Y, Haraguchi K, Harada KH, Hitomi T, Inoue K, Itoh Y, Watanabe T, Takenaka K, Uehara S, Yang HR, Kim MY, Moon CS, Kim HS, Wang P, Liu A, Hung NN, Koizumi A (2011) Detection of dicofol and related pesticides in human breast milk from China, Korea and Japan. Chemosphere 82, 25-31.
| Crossref | Google Scholar | PubMed |
Gomez-Eyles JL, Jonker MTO, Hodson ME, Collins CD (2012) Passive samplers provide a better prediction of PAH bioaccumulation in earthworms and plant roots than exhaustive, mild solvent, and cyclodextrin extractions. Environmental Science and Technology 46, 962-969.
| Crossref | Google Scholar | PubMed |
Hale SE, Tomaszewski JE, Luthy RG, Werner D (2009) Sorption of dichlorodiphenyltrichloroethane (DDT) and its metabolites by activated carbon in clean water and sediment slurries. Water Research 43, 4336-4346.
| Crossref | Google Scholar | PubMed |
Hawthorne SB, Miller DJ, Grabanski CB (2009) Measuring low picogram per liter concentrations of freely dissolved polychlorinated biphenyls in sediment pore water using passive sampling with polyoxymethylene. Analytical Chemistry 81, 9472-9480.
| Crossref | Google Scholar |
Hawthorne SB, Jonker MTO, Van Der Heijden SA, Grabanski CB, Azzolina NA, Miller DJ (2011) Measuring picogram per liter concentrations of freely dissolved parent and alkyl PAHs (PAH-34), using passive sampling with polyoxymethylene. Analytical Chemistry 83, 6754-6761.
| Crossref | Google Scholar | PubMed |
Huang H, Zhang Y, Chen W, Chen W, Yuen DA, Ding Y, Chen Y, Mao Y, Qi S (2018) Sources and transformation pathways for dichlorodiphenyltrichloroethane (DDT) and metabolites in soils from northwest Fujian, China. Environmental Pollution 235, 560-570.
| Crossref | Google Scholar | PubMed |
Hund-Rinke K, Simon M (2005) Terrestrial ecotoxicity of eight chemicals in a systematic approach. Journal of Soils and Sediments 5, 59-65.
| Crossref | Google Scholar |
Josefsson S, Arp HPH, Kleja DB, Enell A, Lundstedt S (2015) Determination of polyoxymethylene (POM) – water partition coefficients for oxy-PAHs and PAHs. Chemosphere 119, 1268-1274.
| Crossref | Google Scholar | PubMed |
Jonker MTO, Koelmans AA (2001) Polyoxymethylene solid phase extraction as a partitioning method for hydrophobic organic chemicals in sediment and soot. Environmental Science & Technology 35(18), 3742-3748.
| Crossref | Google Scholar |
Kim I, Lee S, Kim SD (2019) Determination of toxic organic pollutants in fine particulate matter using selective pressurized liquid extraction and gas chromatography–tandem mass spectrometry. Journal of Chromatography A 1590, 39-46.
| Crossref | Google Scholar | PubMed |
Kupryianchyk D, Reichman EP, Rakowska MI, Peeters ETHM, Grotenhuis JTC, Koelmans AA (2011) Ecotoxicological effects of activated carbon amendments on macroinvertebrates in nonpolluted and polluted sediments. Environmental Science and Technology 45, 8567-8574.
| Crossref | Google Scholar | PubMed |
Mansouri A, Cregut M, Abbes C, Durand MJ, Landoulsi A, Thouand G (2017) The environmental issues of DDT pollution and bioremediation: a multidisciplinary review. Applied Biochemistry and Biotechnology 181, 309-339.
| Crossref | Google Scholar | PubMed |
Maruya KA, Zeng EY, Tsukada D, Bay SM (2009) A passive sampler based on solid‐phase microextraction for quantifying hydrophobic organic contaminants in sediment pore water. Environmental Toxicology and Chemistry 28, 733-740.
| Crossref | Google Scholar | PubMed |
Mayer P, Parkerton TF, Adams RG, Cargill JG, Gan J, Gouin T, Gschwend PM, Hawthorne SB, Helm P, Witt G, You J, Escher BI (2014) Passive sampling methods for contaminated sediments: scientific rationale supporting use of freely dissolved concentrations. Integrated Environmental Assessment and Management 10, 197-209.
| Crossref | Google Scholar | PubMed |
Perron MM, Burgess RM, Suuberg EM, Cantwell MG, Pennell KG (2013a) Performance of passive samplers for monitoring estuarine water column concentrations: 1. Contaminants of concern. Environmental Toxicology and Chemistry 32, 2182-2189.
| Crossref | Google Scholar |
Perron MM, Burgess RM, Suuberg EM, Cantwell MG, Pennell KG (2013b) Performance of passive samplers for monitoring estuarine water column concentrations: 2. Emerging contaminants. Environmental Toxicology and Chemistry 32, 2190-2196.
| Crossref | Google Scholar |
Pontolillo J, Eganhouse RP (2001) The search for reliable aqueous solubility (Sw) and octanol–water partition coefficient (KOW) data for hydrophobic organic compounds: DDT and DDE as a case study. Water-Resources Investigations Report 01-4201, US Geological Survey, US Department of the Interior, Reston, VA, USA.
Qin Z, Bian R, Liu LY, Stubbings WA, Zhao X, Li F, Wu F, Wang S (2023) Determination of polyoxymethylene–water partition coefficients for diverse organophosphate esters (OPEs) and prediction of the free-dissolved OPEs in OPE-contaminated soil. Science of The Total Environment 875, 162528.
| Crossref | Google Scholar | PubMed |
Shi Y, Zhang Q, Huang D, Zheng X, Shi Y (2016) Survival, growth, detoxifying and antioxidative responses of earthworms (Eisenia fetida) exposed to soils with industrial DDT contamination. Pesticide Biochemistry and Physiology 128, 22-29.
| Crossref | Google Scholar | PubMed |
Swartjes FA (2011) ‘Dealing with Contaminated Sites. From Theory towards practical application.’ (Springer Science+Business Media: Dordrecht, Netherlands) 10.1007/978-90-481-9757-6
Tubbs CW (2016) California condors and DDT: examining the effects of endocrine disrupting chemicals in a critically endangered species. Endocrine Disruptors 4, e1173766.
| Crossref | Google Scholar |
Volchko Y, Berggren Kleja D, Back P-E, Tiberg C, Enell A, Larsson M, Jones CM, Taylor A, Viketoft M, Åberg A, Dahlberg A-K, Weiss J, Wiberg K, Rosén L (2020) Assessing costs and benefits of improved soil quality management in remediation projects: a study of an urban site contaminated with PAH and metals. Science of The Total Environment 707, 135582.
| Crossref | Google Scholar | PubMed |
Wang J, Taylor A, Xu C, Schlenk D, Gan J (2018) Evaluation of different methods for assessing bioavailability of DDT residues during soil remediation. Environmental Pollution 238, 462-470.
| Crossref | Google Scholar | PubMed |
Wiberg K, Bergman A, Olsson M, Roos A, Blomkvist G, Haglund P (2002) Concentrations and enantiomer fractions of organochlorine compounds in baltic species hit by reproductive impairment. Environmental Toxicology and Chemistry 21, 2542-2551.
| Crossref | Google Scholar | PubMed |
Xing YN, Guo Y, Xie M, Shen RL, Zeng EY (2009) Detection of DDT and its metabolites in two estuaries of south China using a SPME-based device: First report of p,p′-DDMU in water column. Environmental Pollution 157, 1382-1387.
| Crossref | Google Scholar | PubMed |
Yin G, Athanassiadis I, Bergman Å, Zhou Y, Qiu Y, Asplund L (2017) A refined method for analysis of 4,4′-dicofol and 4,4′-dichlorobenzophenone. Environmental Science and Pollution Research 24, 13307-13314.
| Crossref | Google Scholar | PubMed |