

Cocrystals and Salts of 3,5-Bis(pyridinylmethylene)piperidin-4-one with Aromatic Poly-Carboxylates and Resorcinols: Influence of Stacking Interactions on Solid-State Luminescence Properties
Debarati Das A and Kumar Biradha A BA Department of Chemistry, Indian Institute of Technology, Kharagpur 721302, India.
B Corresponding author. Email: kbiradha@chem.iitkgp.ac.in
Australian Journal of Chemistry 72(10) 742-750 https://doi.org/10.1071/CH19062
Submitted: 7 February 2019 Accepted: 16 April 2019 Published: 17 May 2019
Abstract
Two bis-pyridyl-substituted α,β-unsaturated ketones were shown to form complexes with carboxylic acids and resorcinol derivatives. The neutral acid–acid homosynthon was observed in only one complex out of the five acid-bis-pyridyl containing complexes studied here, while the –COO−⋯HOOC– synthon was found to be dominant as it was observed in four complexes. The carboxylates self-assembled to form discrete dimeric, anionic, 1D chains and also exhibited mixed ionic hydrogen bonds. On the other hand, resorcinol derivatives displayed O–H⋯N hydrogen bonding to form tetrameric aggregates of bis-pyridyl ketone molecules and respective co-formers, while 3,5-dihydroxy benzoic acid (DHBA) molecules formed 1D chains by clipping two molecules of ketones with three DHBA molecules. Such clipping by the resorcinol derivatives promoted continuous π–π stacking interactions. Consequently, these materials emitted at higher wavelengths compared with the parent bis-pyridyl-substituted α,β-unsaturated ketones.
Introduction
The design and syntheses of supramolecular architectures[1–5] are an alluring part of research owing to their promising applications in molecular recognition,[6–8] solid-state reactions,[9–12] host–guest chemistry,[13–16] medicinal chemistry,[17–19] molecular sensing,[20,21] polymorphism,[22–24] and as optical materials,[25,26] and for the judicious design of new crystalline materials.[27] Several supramolecular architectures have been designed and analysed by the aid of non-covalent and multiple intermolecular interactions.[28–30] The extensive use of salts is manifested by a vast number of marketed salts of active pharmaceutical ingredients (APIs).[31–33] In the case of organic salts, charge-assisted hydrogen bonding is generally a dominant interaction given its strength and directionality.[34–37] The acid ionisation constant, Ka, is commonly employed as the tool for predicting the nature of resultant solids of acid–base reactions in the pharmaceutical industry.[38,39] Practically, the pKa values depend on reaction conditions making it difficult to predict the nature of the product of acid–base reactions.
The synthetic analogues of curcumin are known to exhibit interesting fluorescent properties and biological activities. Adams et al. have synthesised and analysed a series of curcumin analogues that inhibited tumour cell growth more effectively compared with curcumin itself and cis-platin. Among the compounds reported, 3,5-bis-(2-fluorobenzylidene)-piperidin-4-one was the more efficient anti-cancer and anti-angiogenesis agent, and was shown to have implications for clinical developments.[40] Similarly, 3,5-bis(benzylidene)-4-piperidones derivatives were also studied by Lagisetty et al. for anti-cancer and anti-inflammatory properties.[41] Furthermore, some 3,5-bis(arylidene)-4-piperidone derivatives were found as two-photon sensitisers in photodynamic therapy.[42] The cocrystals of bispyridyl-substituted α,β-unsaturated ketones were also explored for their luminescence properties and anti-tumour activities.[43,44]
Carboxylic acids also play a crucial role in crystal engineering studies, notably in the formation of acid–pyridine hetero synthons in cocrystals or salts and are well studied in the literature.[30,37,45,46] The importance of combinations of carboxylate–pyridinium synthons and cation–π interactions in creating functional materials with semiconductivity, photochromism, and photoluminescence on a switchable basis has been elucidated by us in the recent past.[47] The tetrameric aggregate formed between 2,2′,6,6′-tetracarboxybiphenyl (H4TCA) and 2,6-bis(4-pyridylmethylene)cyclohexanone (BPCH) was shown to play an important role in generating such switchable properties. On the other hand, the hydroxy groups of resorcinols were shown to hydrogen bond to bis-pyridyl olefin derivatives to form discrete tetrameric aggregates and template solid-state [2 + 2] photodimerisation.[48–50]
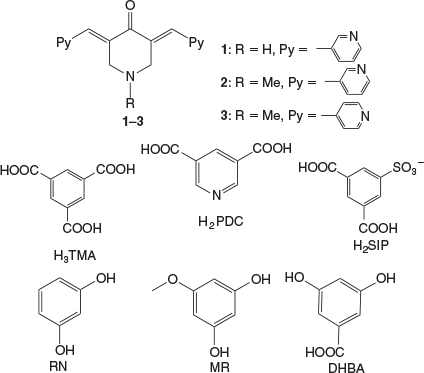
Furthermore, the host–guest complexes of trimesic acid (H3TMA) with BPCH were shown by us to form two-component hosts for inclusion of aromatics.[36] In this manuscript molecules similar to BPCH, 1–3, but containing one –NH−/−NMe functionality in the cyclohexanone in place of –CH2–, have been considered to study complexation reactions with various carboxylic acids and resorcinols. The co-partners for 1–3 considered are carboxylic acids such as H3TMA, pyridine-3,5-dicarboxylic acid (H2PDC), 3,5-dicarboxybenzenesulfonate (H2SIP), and 1,3-dihydroxy aromatics such as resorcinol (RN), 5-methoxy resorcinol (MR), and 3,5-dihydroxybenzoic acid (DHBA) (Chart 1). The complexation reactions of acids with 1 resulted in salts given the high basicity of the amine functional group, whereas that of 3 with di-hydroxy derivatives resulted in the formation of cocrystals with anticipated π–π stacking aggregation of moieties of 3. Such stacking interactions between the moieties of 3 were found to result in high wavelength emissions in the solid state compared with that of 3 alone.
|
Results and Discussion
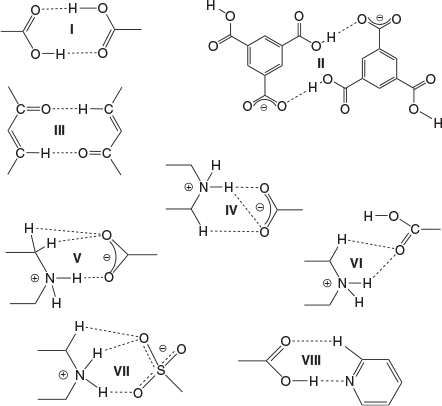
The bis-pyridyl-substituted α,β-unsaturated ketones 1, 2, and 3 were prepared by condensation reaction between the corresponding pyridine carboxaldehyde and piperidones.[51,52] The synthesised compounds were recrystallised from CH3OH before cocrystallisation reactions. The ketone 1 is found to have more propensity towards the formation of salts with acids whereas 3 having an N-methyl substituent formed one salt and three cocrystals. However, the complexation reactions of 2 resulted in precipitates. The H3TMA was found to form crystals of salts with both 1 and 3, while H2PDC and H2SIP formed ionic complexes with 1. The cocrystals with resorcinol derivatives were obtained only with 3. The crystal structures were analysed in terms of their protonation/deprotonation, supramolecular synthons, and hydrogen-bonding networks. The crystallographic parameters and C–O bond distances of carboxylates for all of the complexes are tabulated in Tables 1 and 2. The hydrogen-bonding synthons that are observed in this study are illustrated in Chart 2.

|

|
|
Complexes of 1 and 3 with H3TMA, H2PDC, and H2SIP
The complexation reactions of the bispyridyl-substituted α,β-unsaturated ketone 1 with H3TMA, H2PDC, and H2SIP in CH3OH resulted in single crystals of three complexes [(H1)2(H2TMA)2]·2CH3OH (4), [(H1)(HPDC)]·(H2PDC)·H2O (5), and [(H1)(H2SIP)]·CH3OH (6). Ketone 3 with H3TMA under similar reaction conditions resulted in [(H3)(H2TMA)]·CH3OH (7). The complexes 4, 5, and 7 crystallised in a triclinic space group P-1 whereas 6 crystallised in a monoclinic P21/c space group. The asymmetric unit of 4 contains two units each of H2TMA, H1, and a methanol molecule, whereas that of 7 has one unit each of H2TMA, H3, and a methanol molecule. The asymmetric unit of 5 consists of one unit of H1 and one each of HPDC, H2PDC, and a water molecule. That of complex 6 contains one unit each of H1, H2SIP, and methanol molecules. The C–O bond lengths of H3TMA and H2PDC molecules in 4 and 5, respectively (Table 2) and S–O bond distances in 6 (S–O: 1.441(2), 1.449(2), 1.460(2) Å) indicate that the acid molecules are mono-deprotonated.
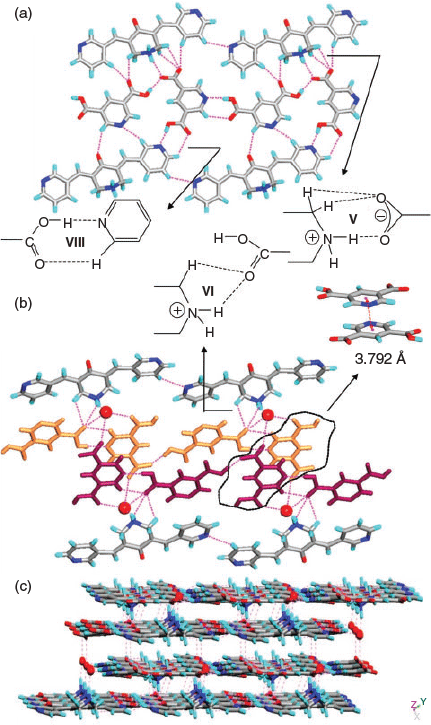
In 4, two H2TMA moieties form a dimer via –COO−⋯HOOC hydrogen bonding (synthon II, R22(14), encircled, in Fig. 1b). The two symmetrically independent moieties of H1 differ in their molecular geometry with respect to their pyridyl N-atoms: one exhibits syn-orientation of N-atoms and the other exhibits anti-orientation (upper one in Fig. 1a). Out of the four pyridine moieties two interact with –COOH groups via O–H⋯N hydrogen bonding, one with –NH2+ via N–H⋯N hydrogen bonding, whereas the fourth one forms a O–H⋯N hydrogen bond (Fig. 1a) with a methanol molecule. Furthermore, the NH2+ groups of both moieties form strong hydrogen bonds with –COO−, a cyclic synthon IV (Fig. 1b). Apart from this synthon, NH2+ groups further hydrogen bond with –COO− and –COOH groups to form N–H⋯O hydrogen bonds. Furthermore, the –C=O moieties of H1 form dimers via C–H⋯O synthon III (C–H⋯O: 2.30 Å, C⋯O: 2.722(8) Å, 107°; C–H⋯O: 2.46 Å, C⋯O: 2.358(8) Å, 162°) (Fig. 1b). The structure of 4 can be described as a 2D layer in which each dimer of H2TMA is surrounded by four dimers of H1 (Fig. 1c). These layers are interconnected via methanol molecules and piperdinium ions to form a 3D network (Fig. 1d).

|
In the case of 5, the carboxylates HPDC and H2PDC are assembled by –COOH⋯−OOC hydrogen bonding and synthon VIII to form 1D chains (Fig. 2a). These 1D chains of carboxylates further hydrogen bond to NH2+ groups of H1 on both sides of the chain via synthons V and VI (Fig. 2a, b) to form a corrugated 2D layer. The pyridine N-atoms of H1 exhibit syn-geometry and one of them participates in acid–pyridine heterosynthon VIII while the other forms weak C–H⋯N hydrogen bonding (Table S1, Supplementary Material). The 2D layers further connect with each other via H2O molecules and a plethora of N–H⋯O and C–H⋯O hydrogen bonding (Fig. 2b) and π–π stacking interactions (centroid-to-centroid distance of two H2PDC units: 3.792 and 3.680 Å) to form a 3D network (Fig. 2c).
|
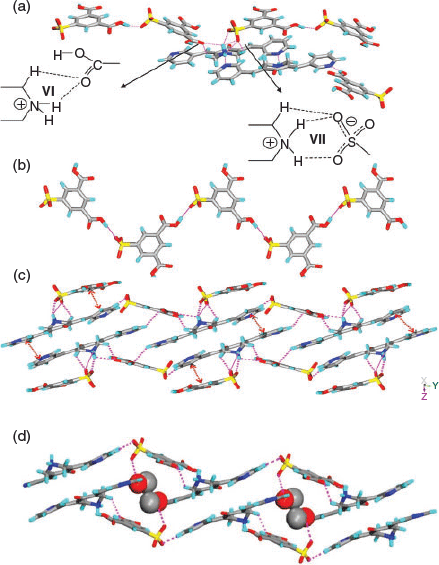
In complex 6, similar to 5, the H2SIP molecules form 1D chains by –COOH⋯−OOC hydrogen bonding. The carboxylic acid moieties of H2SIP also exhibit synthon VI, whereas the sulfonate moiety forms synthon VII via charge-assisted hydrogen bonding with –NH2+ of H1 (Fig. 3a, c). On the other hand, the pyridine N-atoms of H1 exhibit anti-geometry and form N–H⋯N and O–H⋯N hydrogen bonds with the –NH2+ of H1 molecules and –COOH of H2SIP respectively. Furthermore, the solvent molecules occupy the cavities and are hydrogen bonded to the sulfonate ion (O–H⋯O: 2.04 Å, O⋯O: 2.852(4) Å, 171°) (Fig. 3d).
|
In complex 7, the H2TMA anions form 1D chains via –COO−⋯H⋯−OOC– mixed ionic hydrogen bonds (O⋯O: 2.471(2) Å) (encircled in Fig. 4c). The pyridyl moieties exhibit acid–pyridine heterosynthon VIII and weak C–H⋯N hydrogen bonds with a piperidone moiety while the –NH2+ moiety participates in N+–H⋯O− hydrogen bonding with –COO− (N–H⋯O: 1.77(2) Å, N⋯O: 2.676(2) Å, 162°(2)) (Fig. 4a). In addition, the ketone H3 forms a dimer via cyclic C–H⋯O synthon-III (Fig. 4b) similar to H1 in 4. The MeOH molecule and H3 moieties, via a O–H⋯O hydrogen bond (Table S1, Supplementary Material) and synthon VIII respectively, hang on both sides of the 1D chain of acid molecules (Fig. 4c). These 1D chains are interconnected by N+–H⋯O− hydrogen bonding (Fig. 4d).

|
Complexes of 3 with RN, MR, and DHBA
It was shown by us earlier that resorcinol derivatives act as a clip for bis-pyridyl α,β-unsaturated ketones and template the solid-state double [2 + 2] reactions within tetrameric aggregates.[49,50] Molecules 1 and 3 are found to be photo-inactive in all the above complexes as well as in their pure solid forms. Therefore, the resorcinol derivatives were used to complex with 1–3 to examine their complex aggregates and possible templating of a solid state [2 + 2] reaction. However, single crystals of complexes 8–10 were obtained only in the case of 3 with RN, MR, and DHBA, respectively. The complexes (3)·(RN) (8) and (3)2·(DHBA)2 (10) crystallise in the P-1 space group whereas (3)·(MR) (9) is in the P21/c space group. The asymmetric unit of 8 contains one unit each of 3 and RN and that of 9 contains one unit each of 3 and MR molecules. The complex 10 contains two units each of 3 and DHBA molecules. The crystal structure analysis of 8 and 9 reveals that both O–H groups in the resorcinol moieties are pointing in the same direction and thus clip two units of 3 together via O–H⋯N hydrogen bonding (Table S1, Supplementary Material; Fig. 5a, b) to give tetrameric aggregates. However in 10, such clipping of 3 by DHBA results in a 1D network (Fig. 5c). In all these aggregates, the double bonds of 3 are criss-cross aligned making them photo-inactive. A similar observation was made in the BPCH derivative earlier. In two of these aggregates (8 and 10) the piperidone units are found to have a head-to-tail arrangement which is constituted by the electrostatic interaction between the N-atom and C-atom of C=O (N⋯C: 3.002 Å in 8; 2.970 and 3.010 Å in 10). Apart from strong O–H⋯N hydrogen bonding by resorcinol, in all the complexes 8–10, the aromatic –CH that is in between two –OH groups exhibits additional C–H⋯N hydrogen bonding with pyridyl groups (Fig. 5a–d). In 8, the plethora of C–H⋯O hydrogen bonds, C–H⋯π (of 3.193 and 3.228 Å, between ketone and RN molecules), and π–π stacking interactions connect the tetrameric aggregates of 3 (3.824 and 4.073 Å) to construct a 1D network. In 9, the MR molecules form 1D chains (Fig. 6b) via C–H⋯O hydrogen bonding (Table S1, Supplementary Material) between methoxy groups. These chains are further interconnected by a plethora of C–H⋯O hydrogen bonding, C–H⋯π interactions (2.709 and 3.207 Å between ketone and MR units), and π–π stacking interactions (3.881 Å, between pyridyl moieties of two neighbouring ketone molecules) to result in a 2D layer (Fig. 6c). In the case of 10, the 1D chain of π-stacked molecules of 3 propagated via homodimers are further connected by C–H⋯O hydrogen bonds between the –COOH and –NMe group of 3 and CH⋯π interactions (2.671 Å and 3.436 Å) to result in a 3D hydrogen-bonded network (Fig. 7b, c).

|

|

|
Although the centroids of double bonds are found to be within the photo-reactive distances (8: 4.037, 4.145 Å; 9: 3.592 Å; 10: 3.794, 3.908 and 4.157, 4.154 Å), they are photo-inactive due to their criss-cross alignment as the –C=C–C=C– torsions range from 56° to 67°.
Luminescent Properties of Complexes 4–10
The bis-pyridyl-substituted ketones are luminescent in nature owing to their conjugation. Solid-state luminescence properties of bis-pyridyl ketones and their complexes have been recorded at room temperature. The ketone 1 and its complexes 4–6 were excited at 320 nm whereas 3 and its complexes 7–10 were at 350 nm. The emission spectra clearly indicate that the resorcinol conformers alter the emission peak position in the case of 3. The bispyridyl substituted ketone 1 emitted at 500 nm and its salts (4–6) mainly showed α,β-unsaturated ketone based luminescence (Fig. 8a) (λem: 504 nm) while 4 exhibited emission at 487 nm which is blue-shifted by 13 nm due to weak edge-to-face π–π stacking interactions of 4.034 and 4.085 Å between pyridyl moieties compared with 1. The complex 7 emitted at 507 nm and at the same position as 3 (Fig. 8b). The complexes of 3 (8–10) displayed red-shifted emission peaks: 523, 534, and 557 nm for complexes 8–10 respectively (Fig. 8b). In the case of ionic complexes, the aromatic moieties participated in segregated edge-to-face π–π and C–H⋯π stacking interactions and lacked strong cation–π interactions. In the case of the three cocrystals, the extensive π–π and C–H⋯π stacking interactions facilitated intermolecular electron transport compared with the parent compound as evident from their packings hence a red-shift is obtained. In the case of 8, clipping of resorcinols brought the molecules of ketone in close proximity to form tetrameric aggregates. As a result, the pyridyl moieties interacted with each other by π–π stacking interactions (3.824 and 4.073 Å). The continuous stacking interactions happened with the help of C–H⋯O=C hydrogen bonds (C⋯O: 3.577(3) Å) and successive clipping by resorcinols. Likewise in complex 9, the pyridyl moieties experienced π–π stacking interactions of 3.881 Å and C–H⋯π interactions (2.709 and 3.207 Å) between the MR and ketone molecules. Whereas in 10, continuous columns of bis-pyridyl-substituted ketones can be observed by π–π stacking interactions (3.717 to 4.117 Å) between terminal pyridyl moieties. The 1D stacks further formed 2D layers via homosynthon I and thus exhibited extensive π–π stacking interactions. Furthermore, the DHBA molecules formed a 3D supramolecular architecture via C–H⋯π interactions (2.671 Å and 3.436 Å) with molecules of the ketone. Thus stacking interactions between the molecules of ketone and conformers influenced the solid-state emission profile.

|
The Role of ΔpKa in Predicting the Nature of Solids
As discussed previously, the acidity or basicity of the components plays an important role in predicting the outcome of the solids of acid–base reactions. Often the reaction conditions and extent of ionisation makes the process challenging. The calculated ΔpKa values (Table S2, Supplementary Material) obtained using MarvinSketch software[53] for complexes 4–7 and 10 are found to all be positive. The complexes 4–6 have higher positive values (+3.22, +2.98, and +9.24, respectively) and resulted in salts. For 7, the value (+0.79) suggests partial transfer of protons in principle. It has also produced a salt and exhibited a mixed ionic synthon. In the case of 10, where DHBA is used as cocrystallising agent, the small positive value of ΔpKa (+0.32) also endorsed the cocrystal formation.
Conclusions
In this study, the reactions of two bispyridyl-substituted α,β-unsaturated ketones 1 and 3 with carboxylates and resorcinol derivatives resulted in complexes 4–6 and 7–10 respectively. Notably, ketone 1 formed salts only with carboxylic acids. Out of the seven complexes, the homosynthon I was only observed in 10, while the acid–pyridine neutral heterosynthon VIII was observed in two ionic complexes 5 and 7. The H2TMA molecules formed acid dimers via charge-assisted hydrogen bonding in 4, while in 7 the same acid in the presence of ketone 3 exhibited mixed ionic hydrogen bonding to form 1D chains. Furthermore, the sulfonate group in 6 participated in charge-assisted hydrogen bonding with a –NH2+ moiety to form synthon VII. The presence of additional hydrogen bonding sites (secondary and tertiary amine groups), compared with previously studied similar types of bispyridyl-substituted α,β-unsaturated ketones resulted in the formation of different synthons IV–VII and hydrogen-bonding networks. Both the RN and MR exhibited O–H⋯N hydrogen bonding to form tetrameric aggregates of bis-pyridyl ketone 3 and respective conformers while DHBA molecules formed 1D chains by clipping two molecules of 3 with three DHBA molecules. The stacking interactions of the resorcinol conformers influenced the electron transfer and altered the emission profile of the ketone 3. The complexes 8–10 were found to be photo-inactive due to their criss-cross alignment in the crystal structures. The ΔpKa values were in agreement with the observed salt/cocrystal formations. This study provides an easy approach to construct luminescent hydrogen-bonded architectures.
Experimental
FT-IR spectra were recorded with a PerkinElmer Instrument spectrum Rx Serial No. 73713. The X-ray powder diffraction (XRPD) patterns were recorded with a BRUKER-AXS-D8-ADVANCE diffractometer (Cu target). Melting points were taken using a Fisher Scientific melting point apparatus (cat. no. 12-144-1). The solid-state luminescence spectra were collected with a Spex Fluorolog-3 (model FL3-22) spectrofluorimeter. The bispyridyl α,β-unsaturated ketones were synthesised following previously reported procedures.[51,52]
Crystallisation of 4–6
To a 1 mL methanolic solution of 1 (0.015 g, 0.052 mmol), a methanolic solution of H3TMA (0.010 g, 0.052 mmol) was added to obtain 4, a H2PDC solution for 5 (0.0086 g, 0.052 mmol in MeOH), and a NaH2SIP solution (0.013 g, 0.052 mmol in MeOH) for 6. After 4–5 days light yellow crystals formed in 40–50 % yield with respect to 1.
Crystallisation of 7–10
To a 2 mL methanolic solution of 3 (0.010 g, 0.052 mmol) was added a 1 mL acetonitrile solution of H3TMA (0.015 g, 0.052 mmol) for 7, RN (0.0057 g, 0.052 mmol) for 8, a 5MR solution for 9 (0.0072 g, 0.052 mmol), and DHBA (0.0080 g, 0.052 mmol) for 10. The mixtures were left for slow evaporation. Light yellow coloured crystals of 7, dark yellow coloured crystals of 8 and 9, and orange coloured crystals of 10 were formed after 5 days. Yield: ~50 % with respect to 3.
Crystals of 4. Mp 220–222°C. νmax (KBr)/cm−1 3449.71(m), 1701.57(s), 1617.37(s), 1265.35(w), 1243.69(w), 1093.03(s), 1050.84(s), 818.41(s), 748.34(s), 698.78(s), 472.31(w).
Crystals of 5. Mp 240–242°C. νmax (KBr)/cm−1 3411.89(s), 2371.16(w), 1706.67(s), 1612.55(s), 1409.51(s), 1341.20(s), 1314.61(s), 1247.86(s), 1203.99(s), 1136.60(s), 815.98(s), 703.46(s), 482.57(s).
Crystals of 6. Mp 234–236°C. νmax (KBr)/cm−1 3422.74(m), 3024.75(m), 2810.49(m), 1903.57(m), 1685.18(m), 1617.85(s), 1429.66(s), 1316.00(w), 1279.19(w), 1246.78(s), 1197.22(s), 1172.53(s), 1036.42(s), 811.62(s), 626.78(s).
Crystals of 7. Mp 210–212°C. νmax (KBr)/cm−1 3441.08(s), 2495.11(m), 1707.72(w), 1617.60(s), 1383.97(s), 1266.52(s), 1170.24(s), 820.63(s), 751.93(s), 684.71(s), 531.75(s).
Crystals of 8. Mp 180–182°C. νmax (KBr)/cm−1 3031.05(s), 2731.00(w), 2611.52(m), 1675.35(s), 1596.22(m), 1414.01(s), 1278.68(s), 1221.91(s), 1182.35(s), 1143.39(s), 1056.37(s), 820.89(s), 771.87(s), 525.91(s).
Crystals of 9. Mp 182–184°C. νmax (KBr)/cm−1 1950.11(m), 1674.98(s), 1599.05(m), 1546.45(s), 1498.91(s), 1329.83(s), 1274.83(s), 1008.88(m), 818.24(s), 684.64(s), 537.97(s).
Crystals of 10. Mp 200–202°C. νmax (KBr)/cm−1 1696.48(s), 1589.11(s), 1419.49(m), 1171.68(s), 1060.88(s), 1011.75(s), 820.78(s), 774.25(s), 730.09(s), 529.10(s).
Crystal Structure Determination
Bruker-APEX-II CCD X-ray diffractometer was used to collect single-crystal data using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at room temperature (293 K) and for 10 only at low temperature (100 K) by the hemisphere method. Direct methods was used to solve the structure and least-squares methods on F2 was adapted for the refinement procedure using SHELX-14.[54] The hydrogen atoms were fixed at calculated positions and refined using an adapting riding model, whereas non-hydrogen atoms were refined anisotropically.
Supplementary Material
IR spectra, X-ray powder patterns, geometrical parameters of hydrogen bonding of the complexes, ΔpKa values of the complexes, and crystallographic information for complexes 4–10 (CIF) are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank DST(SERB), New Delhi, India for financial support and DST-FIST for single-crystal X-ray diffractometer. D. D. thanks IITKgp for a research fellowship.
References
[1] G. R. Desiraju, Crystal Engineering: The Design of Organic Solids 1989 (Elsevier Science Publishers B. V.: Amsterdam).[2] M. J. Zaworotko, Chem. Commun. 2001, 1.
| Crossref | GoogleScholarGoogle Scholar |
[3] F. Diederich, M. Gómez-López, Chem. Soc. Rev. 1999, 28, 263.
| Crossref | GoogleScholarGoogle Scholar |
[4] L. Maggini, D. Bonifazi, Chem. Soc. Rev. 2012, 41, 211.
| Crossref | GoogleScholarGoogle Scholar | 21748186PubMed |
[5] D. B. Amabilino, J. F. Stoddart, Pure Appl. Chem. 1993, 65, 2351.
| Crossref | GoogleScholarGoogle Scholar |
[6] G. Zhang, M. Mastalerz, Chem. Soc. Rev. 2014, 43, 1934.
| Crossref | GoogleScholarGoogle Scholar | 24336604PubMed |
[7] M. Wenzel, J. R. Hiscock, P. A. Gale, Chem. Soc. Rev. 2012, 41, 480.
| Crossref | GoogleScholarGoogle Scholar | 22080279PubMed |
[8] G. W. Gokel, L. J. Barbour, R. Ferdani, J. Hu, Acc. Chem. Res. 2002, 35, 878.
| Crossref | GoogleScholarGoogle Scholar | 12379140PubMed |
[9] K. Biradha, R. Santra, Chem. Soc. Rev. 2013, 42, 950.
| Crossref | GoogleScholarGoogle Scholar | 23223680PubMed |
[10] J. W. Lauher, F. W. Fowler, N. S. Goroff, Acc. Chem. Res. 2008, 41, 1215.
| Crossref | GoogleScholarGoogle Scholar | 18712885PubMed |
[11] B. R. Bhogala, B. Captain, A. Parthasarathy, V. Ramamurthy, J. Am. Chem. Soc. 2010, 132, 13434.
| Crossref | GoogleScholarGoogle Scholar | 20815400PubMed |
[12] T. Friščić, L. R. MacGillivray, Chem. Commun. 2005, 5748.
| Crossref | GoogleScholarGoogle Scholar |
[13] D. J. Cram, J. M. Cram, Science 1974, 183, 803.
| 17780761PubMed |
[14] M. Zhang, X. Yan, F. Huang, Z. Niu, H. W. Gibson, Acc. Chem. Res. 2014, 47, 1995.
| Crossref | GoogleScholarGoogle Scholar | 24804805PubMed |
[15] Y. Wan, H. Yang, D. Zhao, Acc. Chem. Res. 2006, 39, 423.
| Crossref | GoogleScholarGoogle Scholar | 16846206PubMed |
[16] J. J. Rebek, Chem. Commun. 2000, 637.
| Crossref | GoogleScholarGoogle Scholar |
[17] M. J. Webber, R. Langer, Chem. Soc. Rev. 2017, 46, 6600.
| Crossref | GoogleScholarGoogle Scholar | 28828455PubMed |
[18] D. Tan, L. Lootsa, T. Friščić, Chem. Commun. 2016, 52, 7760.
| Crossref | GoogleScholarGoogle Scholar |
[19] D.-S. Guo, Y. Liu, Acc. Chem. Res. 2014, 47, 1925.
| Crossref | GoogleScholarGoogle Scholar | 24666259PubMed |
[20] C. B. Aakeröey, D. J. Salmon, CrystEngComm 2005, 7, 439.
| Crossref | GoogleScholarGoogle Scholar |
[21] S. K. McNeil, S. P. Kelley, C. Beg, H. Cook, R. D. Rogers, D. E. Nikles, ACS Appl. Mater. Interfaces 2013, 5, 7647.
| Crossref | GoogleScholarGoogle Scholar | 23859489PubMed |
[22] W. W. Porter, S. C. Elie, A. J. Matzger, Cryst. Growth Des. 2008, 8, 14.
| Crossref | GoogleScholarGoogle Scholar | 19367342PubMed |
[23] D. Braga, F. Grepioni, Chem. Commun. 2005, 3635.
| Crossref | GoogleScholarGoogle Scholar |
[24] S. Bhattacharya, B. K. Saha, Cryst. Growth Des. 2011, 11, 2194.
| Crossref | GoogleScholarGoogle Scholar |
[25] S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner, S. Y. Park, J. Am. Chem. Soc. 2013, 135, 4757.
| Crossref | GoogleScholarGoogle Scholar | 23458412PubMed |
[26] H. Wu, Y. Zhou, L. Yin, C. Hang, X. Li, H. Ågren, T. Yi, Q. Zhang, L. Zhu, J. Am. Chem. Soc. 2017, 139, 785.
| Crossref | GoogleScholarGoogle Scholar | 28027639PubMed |
[27] M. Paul, S. Chakraborty, G. R. Desiraju, J. Am. Chem. Soc. 2018, 140, 2309.
| Crossref | GoogleScholarGoogle Scholar | 29353480PubMed |
[28] G. R. Desiraju, Angew. Chem. Int. Ed. 2007, 46, 8342.
| Crossref | GoogleScholarGoogle Scholar |
[29] M. W. Hosseini, Chem. Commun. 2005, 47, 5825.
| Crossref | GoogleScholarGoogle Scholar |
[30] B. K. Saha, S. Bhattacharya, CrystEngComm 2010, 12, 2369.
| Crossref | GoogleScholarGoogle Scholar |
[31] S. M. Berge, L. D. Bighley, D. C. Monkhouse, J. Pharm. Sci. 1977, 66, 1.
| Crossref | GoogleScholarGoogle Scholar | 833720PubMed |
[32] G. S. Paulekuhn, J. B. Dressman, C. Saal, J. Med. Chem. 2007, 50, 6665.
| Crossref | GoogleScholarGoogle Scholar | 18052022PubMed |
[33] D. L. Prohotsky, F. Zhaoa, J. Pharm. Sci. 2012, 101, 1.
| Crossref | GoogleScholarGoogle Scholar | 21905029PubMed |
[34] S. Chen, Y. Yuan, L. Pan, S. Xu, H. Xia, L. Yuan, Cryst. Growth Des. 2009, 9, 874.
| Crossref | GoogleScholarGoogle Scholar |
[35] K. Suresh, A. Nangia, Cryst. Growth Des. 2014, 14, 2945.
| Crossref | GoogleScholarGoogle Scholar |
[36] R. Santra, K. Biradha, Cryst. Growth Des. 2009, 9, 4969.
| Crossref | GoogleScholarGoogle Scholar |
[37] T. R. Shattock, P. Vishweshwar, Z. Wang, M. J. Zaworotko, Cryst. Growth Des. 2005, 5, 2046.
| Crossref | GoogleScholarGoogle Scholar |
[38] S. S. A. Abidi, Y. Azim, A. K. Gupta, C. P. Pradeep, J. Mol. Struct. 2018, 1166, 202.
| Crossref | GoogleScholarGoogle Scholar |
[39] A. Lemmerer, S. Govindraju, M. Johnston, X. Motloung, K. L. Savig, CrystEngComm 2015, 17, 3591.
| Crossref | GoogleScholarGoogle Scholar |
[40] B. K. Adams, E. M. Ferstl, M. C. Davis, M. Herold, S. Kurtkaya, R. F. Camalier, M. G. Hollingshead, G. Kaur, E. A. Sausville, F. R. Rickles, J. P. Snyder, D. C. Loitta, M. Shoji, Bioorg. Med. Chem. 2004, 12, 3871.
| Crossref | GoogleScholarGoogle Scholar | 15210154PubMed |
[41] P. Lagisetty, P. Vilekar, K. Sahoo, S. Anant, V. Awasthi, Bioorg. Med. Chem. 2010, 18, 6109.
| Crossref | GoogleScholarGoogle Scholar | 20638855PubMed |
[42] K. W. Short, T. L. Kinnibrugh, D. M. Sammeth, T. V. Timofeeva, Proc. SPIE 2009, 7164,
| Crossref | GoogleScholarGoogle Scholar |
[43] L.-D. Liu, S.-L. Liu, Z.-X. Liu, G.-G. Hou, J. Mol. Struct. 2016, 1112, 1.
| Crossref | GoogleScholarGoogle Scholar |
[44] H.-J. Li, L. Wang, J.-J. Zhao, J.-F. Sun, J.-L. Sun, C.-H. Wang, G.-G. Hou, J. Mol. Struct. 2015, 1079, 414.
| Crossref | GoogleScholarGoogle Scholar |
[45] S. Mohamed, D. A. Tocher, M. Vickers, P. G. Karamertzanis, S. L. Price, Cryst. Growth Des. 2009, 9, 2881.
| Crossref | GoogleScholarGoogle Scholar |
[46] P. Vishweshwar, A. Nangia, V. M. Lynch, J. Org. Chem. 2002, 67, 556.
| Crossref | GoogleScholarGoogle Scholar | 11798330PubMed |
[47] S. Roy, S. P. Mondal, S. K. Ray, K. Biradha, Angew. Chem. Int. Ed. 2012, 51, 12012.
| Crossref | GoogleScholarGoogle Scholar |
[48] L. R. MacGillivray, J. L. Reid, J. A. Ripmeester, J. Am. Chem. Soc. 2000, 122, 7817.
| Crossref | GoogleScholarGoogle Scholar |
[49] R. Santra, K. Biradha, CrystEngComm 2008, 10, 1524.
| Crossref | GoogleScholarGoogle Scholar |
[50] R. Santra, K. Biradha, CrystEngComm 2011, 13, 3246.
| Crossref | GoogleScholarGoogle Scholar |
[51] E. S. Leonova, M. V. Makarov, E. Y. Rybalkina, S. L. Nayani, P. Tongwa, A. Fonari, T. V. Timofeeva, I. L. Odinets, Eur. J. Med. Chem. 2010, 45, 5926.
| Crossref | GoogleScholarGoogle Scholar | 21035234PubMed |
[52] D. Das, K. Biradha, Inorg. Chem. Front. 2017, 4, 1365.
| Crossref | GoogleScholarGoogle Scholar |
[53] ChemAxon, MarvinSketch (version 17.13.0) 2008 (ChemAxon: Budapest). Available at http://www.chemaxon.com
[54] G. M. Sheldrick, SHELXL-2014 2014 (University of Göttingen: Göttingen).