

Synthesis and Characterization of Zn-Carboxylate Metal–Organic Frameworks Containing Triazatruxene Ligands
Adil Alkaş A and Shane G. Telfer A BA MacDiarmid Institute for Advanced Materials and Nanotechnology, Institute of Fundamental Sciences, Massey University, Palmerston North 4442, New Zealand.
B Corresponding author. Email: s.telfer@massey.ac.nz
Australian Journal of Chemistry 72(10) 786-796 https://doi.org/10.1071/CH19213
Submitted: 14 May 2019 Accepted: 7 June 2019 Published: 3 July 2019
Abstract
Reactions between triazatruxene-based tricarboxylate ligands, H3tat-R, and zinc nitrate under solvothermal conditions afforded new metal–organic frameworks (MOFs) with the general formula [Zn3(tat-R)2(H2O)2], MUF-tat-R (R = a hydrocarbon substituent on the triazatruxene nitrogen atoms). Single-crystal X-ray diffraction analysis revealed that these frameworks are 3D networks with a (10,3)-a topology. Linear trinuclear zinc clusters are connected to tat ligands to form chiral channels that accommodate the substituents on the tat ligands. MUF-tat and MUF-tat-benzyl crystallize in a cubic crystal system whereas MUF-tat-butyl and MUF-tat-hexyl are tetragonal. MUF-tat-benzyl retains its porosity on activation, which was confirmed by gas adsorption studies.
Introduction
The structural and chemical features of organic ligands play a central role in the properties and potential uses of metal–organic frameworks (MOFs). Important ligand variables include their number and type of donor groups, symmetries, and lengths. Introducing functional groups into ligands is a common strategy for tuning the chemical and physical properties of the pores of the resulting frameworks, especially if the isoreticular principle[1] is adhered to. This requires designing ligand backbones that can be easily derivatized. Symmetrically substituted ligands are also generally desired as they can lead to uniform crystal growth that is free from symmetry-related disorder. Ligand functionalization has been extensively studied in MOFs containing ditopic linkers for a variety of applications, such as gas sorption,[2] separation,[3] catalysis,[4] and photoluminescence.[5,6]
Tritopic carboxylate ligands are commonly used organic linkers in the chemistry of MOFs.[1,7] The classic member of this series is benzene-1,3,5-tricarboxylic acid, which generates HKUST-1 on reaction with copper(ii) ions.[8] Researchers have used extended tritopic linkers, such as 1,3,5-benzenetribenzoic acid (btb), to construct MOFs with high porosity.[9] It was later demonstrated that using the btb ligand along with its functionalized derivatives could result in multivariate MOF-177 analogues with enhanced gas adsorption behaviour.[10] Moreover, chiral frameworks containing achiral tritopic linkers have been reported.[11–13] Chiral MOFs have potential applications in chiral separation,[14,15] asymmetric catalysis,[16–18] and non-linear optics.[19] However, common tritopic linkers[8,20–22] used in the field of MOFs are usually unsubstituted, perhaps owing to the difficult and costly preparation of their derivatives that can be substituted in a symmetric way. Previously, we developed a convenient method for the synthesis of tritopic tris(carboxylic acid) ligands based on a triazatruxene core. They are referred to as H3tat-R (tat = triazatruxene; R = a hydrocarbon substituent on the triazatruxene nitrogen atoms), and we previously used them to prepare a series of quaternary multicomponent MOFs in conjunction with a pair of linear bis(carboxylic acid) linkers.[23] Here, we focus on the use of these ligands to build zinc(ii) MOFs on their own. Different functional groups appended to the nitrogen sites of the triazatruxene core were found to dictate the chemistry of these frameworks.
Results and Discussion
Synthesis of Ligands
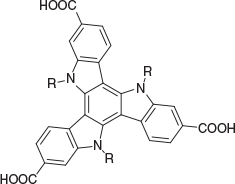
In this study, six triazatruxene ligands were used for constructing new MOFs (Fig. 1). The synthetic routes to five of these ligands have been described previously.[23] These routes initially involve the trimerization of 1H-indole-6-carboxylic acid ethyl ester followed by the introduction of a substituent at the nitrogen sites (if required). Finally, hydrolysis produces the corresponding carboxylic acids. For the synthesis of H3tat-benzyl, another synthetic procedure was developed. As illustrated in Scheme 1, H3tat-benzyl was synthesized starting from the previously reported compound Br3tat.[24] Br3tat-benzyl was prepared by alkylation of Br3tat with benzyl bromide, and purified by column chromatography. Similar synthetic procedures for benzylation of triazatruxene and its derivatives have previously been reported.[25,26] H3tat-benzyl was obtained by carboxylation of Br3tat-benzyl. Although this procedure worked well for the synthesis H3tat-benzyl, it is not ideal for functional groups that are sensitive to conditions used in the carboxylation step. Because the procedure described previously does not involve a carboxylation step, it can be used for the preparation of more diverse triazatruxene derivatives. Detailed synthetic procedures and the characterization of Br3tat-benzyl and H3tat-benzyl are available in the Experimental section.
|

|
MUF-tat-benzyl, [Zn3(tat-benzyl)2(H2O)2]
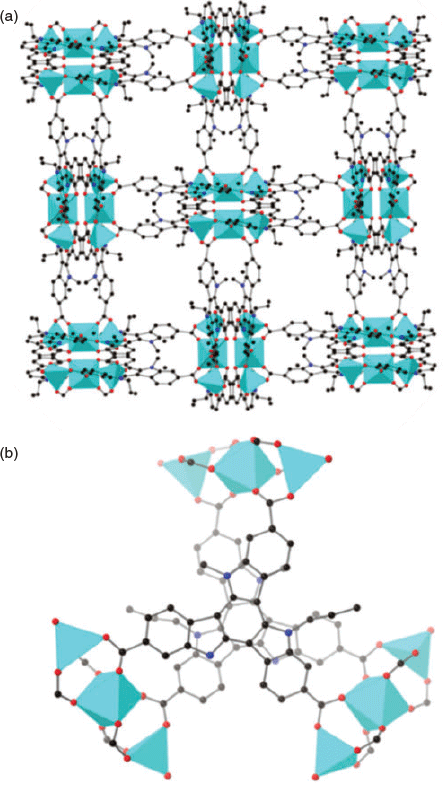
The solvothermal reaction of H3tat-benzyl with Zn(NO3)2 in DMF at 85°C afforded yellow crystals of MUF-tat-benzyl, [Zn3(tat-benzyl)2(H2O)2] (Fig. 2a). Benzoic acid (BA) was used as a modulator to grow large crystals. The structure of MUF-tat-benzyl was determined by single-crystal X-ray diffraction (SCXRD), which was carried on a solvated crystal (in N,N′-dibutylformamide (DBF)) at room temperature. The data revealed that MUF-tat-benzyl crystallizes in the cubic space group P4132, which is chiral. The Flack parameter of 0.04(5), indicates that the crystal selected for diffraction is a genuine enantiomorph even though achiral precursors were used. Presumably crystals belonging to the P4332 space group are formed to an equal extent. As shown in Fig. 2, pairs of ligands are connected through a linear trinuclear zinc cluster to form a three-dimensional chiral (10,3)-a network. Each asymmetric unit contains three ZnII ions, a third of the tat-benzyl ligand, and two water molecules. The two peripheral metal ions of the trinuclear cluster are tetrahedrally coordinated to three carboxylate oxygen atoms and one water molecule each, while the central metal ion is octahedrally coordinated to six carboxylate oxygen atoms (Fig. 2c). MOFs containing this type of cluster have been reported in the literature.[27–32] MUF-tat-benzyl has a low crystallographic density (0.70 g cm−3) and high calculated[33] porosity (void fraction of 69 %). As shown in Fig. 3, the framework possesses two different types of channels. A chiral helical channel extends along the crystallographic b axis direction and is 12 Å across (including van der Waals radii) (Fig. 3a). The length of each side of the triangular channel, along the [111] direction, is ~21 Å (distance between two zinc atoms at the corner of the triangle) (Fig. 3b). The tat-benzyl ligands are nearly planar, and pairs of ligands come into close contact. The distance between the centroids of their central phenyl rings is ~3.95 Å, which is in the range of π–π interactions.[34] Also, the two ligands in a pair do not have a perfect alignment; instead, they are offset with a rotational angle of ~30°. The three benzyl groups of each ligand embrace the adjacent ligand and are located in the pores of the framework (Fig. 3).

|

|
The chiral structure of MUF-tat-benzyl is a result of two possible assembly modes of pairs of tat-benzyl ligands. The two ligands can associate in either a clockwise or counterclockwise fashion, which results in the formation of two enantiomers (Fig. 4). Although individual crystals of MUF-tat-benzyl incorporate only one of these enantiomers, the bulk sample is a racemic mixture because during framework formation, each of the two enantiomers has an equal chance of crystallization. However, in future work, the preparation of homochiral MUF-tat-benzyl may be possible by using a chiral additive (modulating agent) during synthesis.[35] Researchers have also reported homochiral frameworks that can be formed through spontaneous symmetry-breaking crystallization without using any chiral additives.[36]

|
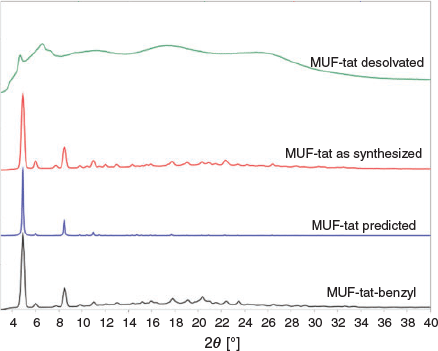
The bulk identity and thermal stability of MUF-tat-benzyl were investigated by powder X-ray diffraction (PXRD) measurements and thermogravimetric analysis (TGA). All major peaks in the experimental PXRD pattern of MUF-tat-benzyl match the pattern simulated from the SCXRD model well (Fig. 5a). This result indicates that the single crystal is representative of the pure bulk sample. The desolvated crystals become opaque when exposed to air and lose their crystallinity (Fig. 5a). They presumably adopt this amorphous form during the gas adsorption measurements (see below). But the framework connectivity is maintained: crystallinity is restored when the MOF is heated in DMF overnight at 85°C (Fig. 5). Interestingly, crystallinity does not emerge from the sample standing in DMF at room temperature. The TGA curve shows that MUF-tat-benzyl has a weight loss of 3 % from 20 to 100°C, corresponding the loss of free solvent molecules from the pores. The weight loss between 100 and 200°C has a value of 2 %, which matches up with two coordinated water molecules (2.08 %, Fig. 5b). After this point, the curve does not plateau, and the weight loss becomes more pronounced, indicating framework instability. Limited stability was also observed in some other frameworks containing trinuclear zinc(ii) clusters and tritopic linkers.[30]

|
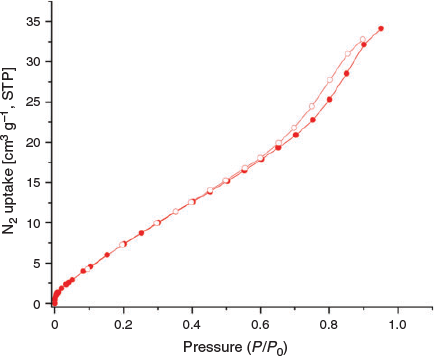
The porous nature of MUF-tat-benzyl was investigated by carrying out adsorption studies with gaseous adsorbates at different temperatures. Prior to the adsorption measurements, MUF-tat-benzyl was washed with dichloromethane to remove the DMF. Then, the sample was activated at a temperature of 80°C under vacuum for 20 h to generate a solvent-free framework. First, adsorption experiments for N2 at low temperatures were performed. The experimental data showed a low uptake for N2 at 77 K (Fig. 6). This is an unexpected result, considering that the structural features of MUF-tat-benzyl indicate high porosity. As the framework is still porous (see below), this can be ascribed to diffusion limitations that prevent the N2 molecules from entering the 1D pore at low temperature.[37–39]
|
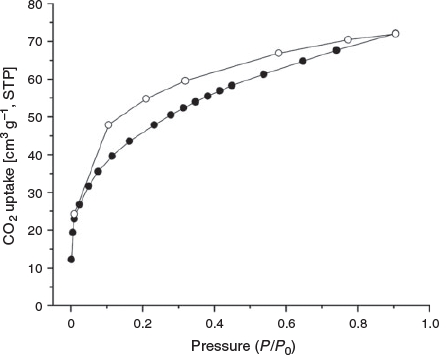
Next, the CO2 isotherm of MUF-tat-benzyl was measured at 195 K. The isotherm shows a fairly steep rise at low pressure followed by a gradual increase, which reaches a maximum amount of 70 cm3 g−1. This result indicates that MUF-tat-benzyl retains its porosity on removal of solvent molecules and the structure does not collapse. As can be seen in Fig. 7, there is a significant degree of hysteresis between the adsorption and desorption isotherms. This will be discussed in the following section.
|
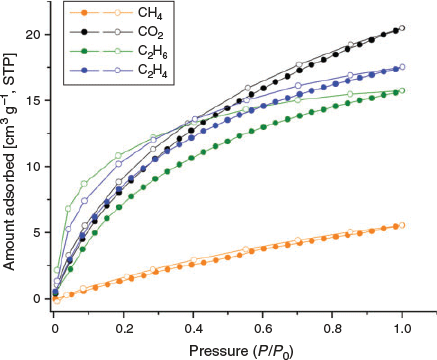
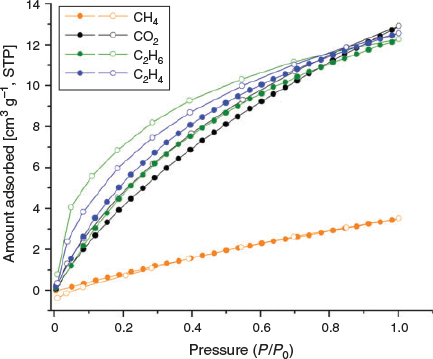
Further, CO2, CH4, C2H6, and C2H4 isotherms were measured at both 273 and 293 K (Figs 8 and 9). As was observed in the adsorption isotherm of CO2 at 195 K, hysteresis was also observed in adsorption isotherms of CO2, C2H4, and C2H6 at 273 and 293 K. The hysteresis may be related to insufficient equilibration time.[40] Therefore, we investigated the optimal equilibration time for gas sorption measurements by conducting tests on MUF-tat-benzyl at 293 K using CO2 as the adsorbate. We performed CO2 adsorption measurement with 4, 6, 8, and 12 min of equilibration time. Although some improvement was observed as the equilibration time increased, a completely reversible isotherm could not be obtained. Based on this observation, it is clear that longer times are needed to completely remove the hysteresis. To collect a dataset in a reasonable amount of time, we decided to perform sorption measurements with 6-min equilibration time.
|
|
The adsorption isotherms of CO2 show a gradual increase and reach a maximal amount of 22 and 13 cm3 g−1 at 273 and 293 K respectively. MUF-tat-benzyl takes up similar amounts of C2H6 and C2H4 at 293 K, whereas the adsorption amount of C2H4 is slightly higher than that of C2H6 at 273 K. The high C2H6 and C2H4 adsorption capacity of MUF-tat-benzyl as compared with CH4 is compatible with the larger size and polarizability of C2H6 and C2H4 relative to CH4.[41–43]
MUF-tat, [Zn3(tat)2(H2O)2]
MUF-tat was obtained as orange crystals by a solvothermal reaction of H3tat with Zn(NO3)2 in DMF at 85°C (Fig. 10). SCXRD data reveal that MUF-tat crystallizes in the cubic chiral space group P4332 (Table 1). The diffraction pattern simulated from the single-crystal model is in agreement with the experimental pattern, demonstrating the phase purity of the bulk sample. The PXRD pattern of MUF-tat also matches the pattern of MUF-tat-benzyl (Fig. 11). This result indicates that these two MOFs are isostructural. MUF-tat has a much lower crystallographic density (0.47 g cm−3) and higher porosity (void fraction of 81 %) relative to MUF-tat-benzyl. This is because the pores of MUF-tat-benzyl are partially occupied by benzyl groups. Another difference between these two frameworks is the distance that separates the two ligands in a pair. In the case of MUF-tat-benzyl, this distance is 3.95 Å. This might be the shortest distance allowed owing to the orientation of methylene carbons on the benzyl group, which are not planar to the triazatruxene core. However, in the structure of MUF-tat, substituent-free ligands are just 3.43 Å apart.

|
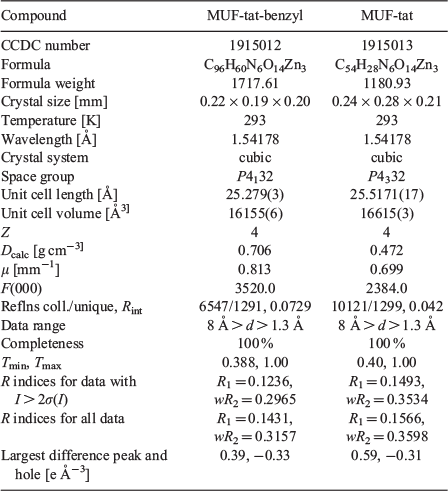
|
|
To obtain solvent-free MUF-tat, the crystals were washed with dichloromethane or acetone before being heated at 80°C under vacuum for 20 h. Activated samples were then used for gas sorption measurements. Unlike MUF-tat-benzyl, MUF-tat took up a negligible amount of gas (CO2, CH4, C2H4, and C2H6) at both 273 and 293 K, which suggests that the framework does not retain its porosity after removal of solvent. The PXRD pattern of an activated sample of MUF-tat shows some weak peaks, which are at different positions relative to the pattern of freshly synthesized sample (Fig. 11). It seems that MUF-tat collapses into a poorly crystalline phase.
MUF-tat-butyl, MUF-tat-hexyl, MUF-tat-allyl, and MUF-tat-cyclohexyl
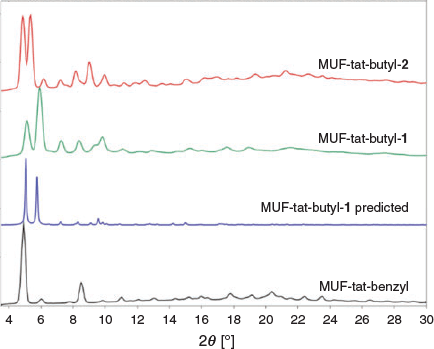
After syntheses of MUF-tat and MUF-tat-benzyl, we intended to prepare an isoreticular family of MOFs using the ligands H3tat-allyl, H3tat-butyl, H3tat-hexyl, and H3tat-cyclohexyl. We expected that these ligands could form MOFs that are isostructural with MUF-tat-benzyl because these linkers share its symmetry and size parameters. Under similar conditions to those under which MUF-tat was synthesized, H3tat-butyl produced two types of crystal that differed in their morphologies and sizes (Fig. 12). The smaller crystals had a rectangular prismatic shape, and they were termed MUF-tat-butyl-1. The bigger crystals had a pseudo-cubic shape, and were termed MUF-tat-butyl-2. Both MUF-tat-butyl-1 and MUF-tat-butyl-2 did not extinguish under crossed polarizers on an optical microscope, indicating that they do not belong to a cubic crystal system, unlike MUF-tat-benzyl. PXRD measurements of these crystals showed two distinct patterns, indicating that they are not identical frameworks (Fig. 13). These patterns also do not match the PXRD pattern of MUF-tat-benzyl (Fig. 13). We observed that increasing the amount of zinc nitrate and modulator (benzoic acid) increased the formation of phase 1, yet neither of the two phases could be synthesized in pure form. Both types of crystals become opaque shortly after exposure to air even when immersed in solvent (DMF).
|
|
Single-crystal X-ray diffraction analysis of MUF-tat-butyl-1 and MUF-tat-butyl-2 was not easy owing to their fragile nature. The first data collection for MUF-tat-butyl-1 was performed using a solvated crystal (in DMF). The crystal started to demonstrate poor diffraction shortly after measurement started. Similar behaviour was also observed for a DBF-solvated crystal. Next, the crystal was mounted in a sleeve that had a small amount of solvent (DBF) in it. Using a sleeve prevents air exposure, while the solvent in the sleeve creates a solvent atmosphere that prevents the evaporation of the solvent from the pores of the crystal. However, even with a sleeve, the sample started to lose its crystallinity after ~3 h; therefore a complete dataset could not be collected. Crystallographic details of the unit cell are given in Table 2. Using the available amount of data, however, we were able to partially identify the structure of MUF-tat-butyl-1. It shows similarities to the structure of MUF-tat-benzyl – the ligand pairs are connected through trinuclear zinc clusters (Fig. 14). The PXRD pattern calculated from this dataset matches the experimental pattern for this phase (Fig. 13). Similar procedures were also used for the SCXRD analysis of MUF-tat-butyl-2. However, the data collected were insufficient to fully refine its structure.

|
|
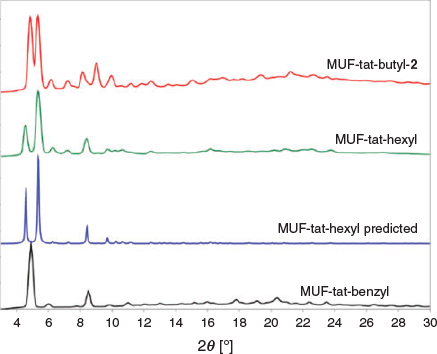
The synthesis of MUF-tat-hexyl produced only one type of crystal (Fig. 12). MUF-tat-hexyl crystals have a rectangular prismatic shape and do not extinguish under crossed polarizers on an optical microscope. The PXRD pattern of these crystals is different than that of MUF-tat-benzyl (Fig. 15). However, the pattern shows similarities to the pattern of MUF-tat-butyl-2 (Fig. 15). The stability of MUF-tat-hexyl was insufficient to collect a complete dataset for SCXRD analysis. Its structure was partially solved using a part of the data collected. The PXRD pattern calculated from these data matches the experimental pattern (Fig. 15). Crystallographic details of the unit cell of MUF-tat-hexyl are given in Table 2.
|
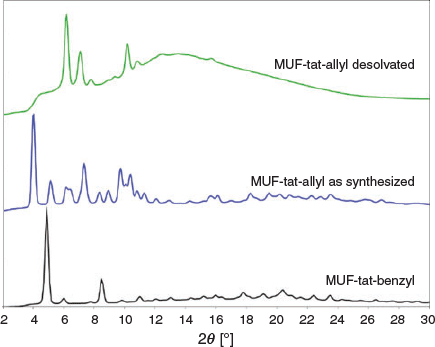
MUF-tat-allyl was obtained as yellow plate-like crystals (Fig. 11). The crystals extinguish under crossed polarizers on an optical microscope, indicating that they belong to a non-cubic crystal system. Its PXRD pattern is different than all the abovementioned frameworks. To activate MUF-tat-allyl, the crystals were washed with acetone and then heated at 100°C under vacuum for 20 h. The activated sample was then subjected to gas adsorption measurements, which resulted in insignificant amounts of gas uptake. This might be due to either the collapse of the framework or insufficient activation. The PXRD pattern of the desolvated sample is different than that of an as-synthesized sample, indicating that a phase transition occurred or a new material was formed (Fig. 16). SCXRD analysis was carried out on both as-synthesized and desolvated samples. The desolvated sample diffracted poorly, whereas the solvated sample showed good diffraction only at the beginning of measurement. Unfortunately, good-quality SCXRD data could not be collected for this sample as the crystal diffracted for only a short period of time. Crystallographic details of the unit cell of MUF-tat-allyl are given in Table 2.
|
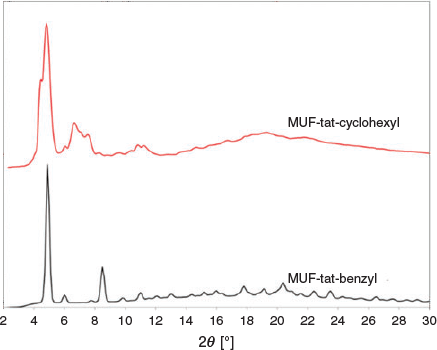
MUF-tat-cyclohexyl was obtained as colourless cube-like crystals. Clearly, these crystals belong to a cubic crystal system as they are fully extinguished at all rotational positions under crossed polarizers on an optical microscope. Although its PXRD pattern shows some similarities with the structure of MUF-benzyl, it has some additional peaks (Fig. 17). Therefore, it might be either a mixture of two phases or a completely different phase. In addition, all single-crystal samples of this MOF showed nearly no diffraction during SCXRD measurements. No further analysis was done on this framework.
|
It is interesting that these ligands with similar geometries can produce MOFs with distinct phases. The different behaviour of these ligands may be related to the size and orientation of side groups on each ligand. These groups can cause a certain degree of bending in the triazatruxene core. They can also have an effect on face-to-face interaction of ligand pairs, which can produce dimers with different stacking modes (Fig. 18). Each stacking mode may then lead to formation of a different phase. In addition, the trinuclear zinc cluster is not the only possible cluster. Formation of different clusters will also give rise to new network topologies.

|
Conclusion
In conclusion, the synthesis of H3tat-benzyl was achieved as well as the syntheses of six other new MOFs. The structures of MUF-tat and MUF-tat-benzyl were determined by SCXRD analysis. These frameworks are isostructural and have chiral (10,3)-a networks. Gas adsorption measurements revealed that MUF-tat-benzyl retains its porosity after removal of guest molecules, although PXRD analysis showed that it exhibits poor stability under ambient conditions. The structure of MUF-tat collapsed on activation, possibly owing to its high porosity. MUF-tat-butyl-1 and MUF-tat-hexyl both are highly unstable when exposed to air. This made their structural characterization difficult. A phase transition was observed in the structure of MUF-tat-allyl after activation and the structures of both solvated and activated MUF-tat-allyl could not be solved owing to the loss of crystallinity during measurements.
Experimental
General
All starting materials and solvents were used as received from commercial sources without further purification unless otherwise noted. Column chromatography was carried out on silica gel (grade 60, mesh size 230–400, Scharlau). NMR spectra were recorded at room temperature (unless otherwise noted) on Bruker-400 and Bruker-500 Avance instruments, with the use of the solvent proton as an internal standard. Low-pressure gas adsorption isotherms were measured by a volumetric method using a Quantachrome Autosorb iQ2 instrument. TGA was performed on a TA Instruments Q50 instrument.
Br3tat-benzyl
Br3tat (438 mg, 0.75 mmol) and KOH (1.3 g) were combined in THF (50 mL). To this mixture, benzyl bromide (400 µL, 3.4 mmol) was added and the mixture was refluxed overnight. After cooling to room temperature, the suspension was filtered. The solvent was removed from the collected filtrate under vacuum, and the residue was dissolved in dichloromethane (50 mL) and washed with 10 % aqueous HCl (40 mL) and then with water (40 mL). The organic layer was separated, dried over MgSO4, and filtered through silica. The solvent was removed under reduced pressure and the crude material was purified on a silica column eluting with a gradient from hexane to 2 : 3 DCM/hexane. Yield: 468 mg (73 %). δH (500 MHz, CDCl3) 7.80 (d, J 7.8, 3H), 7.48–7.41 (m, 18H), 7.15 (d, J 7.7, 3H), 6.03 (s, 6H).
H3tat-benzyl
A mixture of Br3tat-benzyl (820 mg, 0.96 mmol) and dry THF (200 mL) was degassed and stirred in a liquid nitrogen–acetone bath (−70°C) for 15 min. To this mixture, n-BuLi (2.2 M in hexane, 10 mL, 22 mmol) was added dropwise. The mixture was slowly warmed to −10°C over a period of 4 h and an orange solution was observed. The mixture was cooled to −78°C, and dry CO2 was passed through the solution for 20 min. The reaction mixture was warmed to room temperature (r.t.) and quenched with water (20 mL). Then, 1 M HCl (30 mL) was added and the mixture stirred for 2 h. THF was removed and the yellow solid was filtered off, washed with water, and dried, then recrystallized from MeOH. Yield: 575 mg (80 %). δH (500 MHz, DMSO) 12.83 (br, 3H), 8.08 (d, J 8.5, 3H), 8.02 (s, 3H), 7.62 (d, J 8.4, 3H), 7.43–7.40 (m, 6H), 7.36–7.32 (m, 9H), 6.28 (s, 6H). δC (125 MHz, DMSO) 167.91, 141.48, 140.66, 138.08, 129.61, 128.11, 126.62, 126.02, 125.92, 121.84, 121.46, 112.82, 102.92, 50.92. m/z (electrospray ionization (ESI) negative mode, CH3OH) 746.2319 ([C48H32N3O6]−; calc. 746.2369.
General Method for MOF Synthesis
The ligand (10 µmol), benzoic acid (12 mg, 98 µmol), and Zn(NO3)2·4H2O (14 mg, 53 µmol) were dissolved in a mixed solvent of dry DMF (1 mL) and water (35 µL) in a 4-mL vial. The vial was placed in an 85°C isothermal oven for 20–30 h to obtain MOF crystals. The mother liquor was replaced with anhydrous DMF after cooling. For SCXRD measurements, DMF was replaced with DBF.
Powder X-Ray Diffraction Patterns
All PXRD measurements were carried out on a Rigaku Spider X-ray diffractometer with Cu Kα radiation (Rigaku MM007 microfocus rotating-anode generator), monochromated and focussed with high-flux Osmic multilayer mirror optics, and a curved image-plate detector. Samples were kept damp with solvent before and during measurements. The two-dimensional images of the Debye rings were integrated with 2DP to give 2θ versus I diffractograms.
Single Crystal X-Ray Diffraction
The SCXRD analyses for MUF-tat-benzyl and MUF-tat-allyl were performed on crystals in fresh DBF at room temperature. Individual crystals were selected under a microscope, then mounted on a polymer mount with a minimum amount of Fomblin® Y oil. A Rigaku Spider diffractometer equipped with a MicroMax MM007 rotating-anode generator (Cuα radiation, 1.54178 Å), high-flux Osmic multilayer mirror optics, and a curved image-plate detector was used to collect the data. The data were integrated, scaled and averaged with FS Process.[44] Using Olex2,[45] the structure was solved with the SHELXT[46] structure solution program using intrinsic phasing and refined with the SHELXL[47] refinement package using least-squares minimization.
All zinc, oxygen, and carbon atoms were found in the electron density difference maps and refined anisotropically except for the carbons of the benzyl ring on MUF-tat-benzyl. Unrestrained atomic displacement parameters sometimes produced high thermal ellipsoid (Ueq) values during the refinement, thus RIGU, SADI, ISOR, DELU, and SIMU commands were introduced as appropriate with careful adjustments of the standard deviations. In MUF-tat-benzyl, DFIX restraints were employed for the bond between bridging carbon and the ring carbon. All phenyl rings were modelled as ideal hexagons. Hydrogen atoms were calculated and refined as a riding model.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
We are grateful to the Royal Society of New Zealand Marsden Fund and the MacDiarmid Institute for Advanced Materials and Nanotechnology for supporting this study. A.A. would like to thank David Lun for technical expertise, Omid Q. Taheri for computational assistance, and David Perl and Seok June Lee for useful discussions on crystallographic data.
References
[1] H. Furukawa, K. E. Cordova, M. O’Keeffe, O. M. Yaghi, Science 2013, 341,| Crossref | GoogleScholarGoogle Scholar | 23990564PubMed |
[2] J. Zhang, S. Yao, S. Liu, B. Liu, X. Sun, B. Zheng, G. Li, Y. Li, Q. Huo, Y. Liu, Cryst. Growth Des. 2017, 17, 2131.
| Crossref | GoogleScholarGoogle Scholar |
[3] Y. Wu, H. Chen, D. Liu, J. Xiao, Y. Qian, H. Xi, ACS Appl. Mater. Interfaces 2015, 7, 5775.
| Crossref | GoogleScholarGoogle Scholar | 25700143PubMed |
[4] A. Herbst, A. Khutia, C. Janiak, Inorg. Chem. 2014, 53, 7319.
| Crossref | GoogleScholarGoogle Scholar | 25006999PubMed |
[5] R. J. Marshall, Y. Kalinovskyy, S. L. Griffin, C. Wilson, B. A. Blight, R. S. Forgan, J. Am. Chem. Soc. 2017, 139, 6253.
| Crossref | GoogleScholarGoogle Scholar | 28385019PubMed |
[6] J. Cornelio, T.-Y. Zhou, A. Alkaş, S. G. Telfer, J. Am. Chem. Soc. 2018, 140, 15470.
| Crossref | GoogleScholarGoogle Scholar | 30382705PubMed |
[7] W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle, M. Bosch, H.-C. Zhou, Chem. Soc. Rev. 2014, 43, 5561.
| Crossref | GoogleScholarGoogle Scholar | 24604071PubMed |
[8] S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen, I. D. Williams, Science 1999, 283, 1148.
| Crossref | GoogleScholarGoogle Scholar |
[9] H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O’Keeffe, O. M. Yaghi, D. Materials, G. Discovery, Nature 2004, 427, 523.
| Crossref | GoogleScholarGoogle Scholar | 14765190PubMed |
[10] Y.-B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim, O. M. Yaghi, J. Am. Chem. Soc. 2015, 137, 2641.
| Crossref | GoogleScholarGoogle Scholar | 25646798PubMed |
[11] T. J. Prior, M. J. Rosseinsky, Inorg. Chem. 2003, 42, 1564.
| Crossref | GoogleScholarGoogle Scholar | 12611524PubMed |
[12] C. J. Kepert, T. J. Prior, M. J. Rosseinsky, J. Am. Chem. Soc. 2000, 122, 5158.
| Crossref | GoogleScholarGoogle Scholar |
[13] B. F. Abrahams, P. A. Jackson, R. Robson, Angew. Chem. Int. Ed. 1998, 37, 2656.
| Crossref | GoogleScholarGoogle Scholar |
[14] Y. Peng, T. Gong, K. Zhang, X. Lin, Y. Liu, J. Jiang, Y. Cui, Nat. Commun. 2014, 5, 4406.
| Crossref | GoogleScholarGoogle Scholar | 25030529PubMed |
[15] J. Navarro-Sánchez, A. I. Argente-García, Y. Moliner-Martínez, D. Roca-Sanjuán, D. Antypov, P. Campíns-Falcó, M. J. Rosseinsky, C. Martí-Gastaldo, J. Am. Chem. Soc. 2017, 139, 4294.
| Crossref | GoogleScholarGoogle Scholar | 28274119PubMed |
[16] C. Zhu, Q. Xia, X. Chen, Y. Liu, X. Du, Y. Cui, ACS Catal. 2016, 6, 7590.
| Crossref | GoogleScholarGoogle Scholar |
[17] D. Dang, P. Wu, C. He, Z. Xie, C. Duan, J. Am. Chem. Soc. 2010, 132, 14321.
| Crossref | GoogleScholarGoogle Scholar | 20879732PubMed |
[18] F. Song, C. Wang, J. M. Falkowski, L. Ma, W. Lin, J. Am. Chem. Soc. 2010, 132, 15390.
| Crossref | GoogleScholarGoogle Scholar | 20936862PubMed |
[19] X. Huang, Q. Li, X. Xiao, S. Jia, Y. Li, Z. Duan, L. Bai, Z. Yuan, L. Li, Z. Lin, Y. Zhao, Inorg. Chem. 2018, 57, 6210.
| Crossref | GoogleScholarGoogle Scholar | 29774746PubMed |
[20] H. Furukawa, M. A. Miller, O. M. Yaghi, J. Mater. Chem. 2007, 17, 3197.
| Crossref | GoogleScholarGoogle Scholar |
[21] H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim, O. M. Yaghi, Science 2010, 329, 424.
| Crossref | GoogleScholarGoogle Scholar | 20595583PubMed |
[22] D. Zhao, D. J. Timmons, D. Yuan, H.-C. Zhou, Acc. Chem. Res. 2011, 44, 123.
| Crossref | GoogleScholarGoogle Scholar | 21126015PubMed |
[23] A. Alkaş, J. Cornelio, S. G. Telfer, Chem. Asian J. 2019, 14, 1167.
| Crossref | GoogleScholarGoogle Scholar | 30499184PubMed |
[24] R. A. Valentine, A. Whyte, K. Awaga, N. Robertson, Tetrahedron Lett. 2012, 53, 657.
| Crossref | GoogleScholarGoogle Scholar |
[25] T. Techajaroonjit, S. Namuangruk, N. Prachumrak, V. Promarak, M. Sukwattanasinitt, P. Rashatasakhon, RSC Adv. 2016, 6, 56392.
| Crossref | GoogleScholarGoogle Scholar |
[26] E. M. García-Frutos, B. Gómez-Lor, Á. Monge, E. Gutiérrez-Puebla, I. Alkorta, J. Elguero, Chem. – Eur. J. 2008, 14, 8555.
| Crossref | GoogleScholarGoogle Scholar | 18680132PubMed |
[27] J. Y. Lee, L. Pan, S. P. Kelly, J. Jagiello, T. J. Emge, J. Li, Adv. Mater. 2005, 17, 2703.
| Crossref | GoogleScholarGoogle Scholar |
[28] D. Sun, Y. Ke, D. J. Collins, G. A. Lorigan, H.-C. Zhou, Inorg. Chem. 2007, 46, 2725.
| Crossref | GoogleScholarGoogle Scholar | 17348646PubMed |
[29] W. Chen, J.-Y. Wang, C. Chen, Q. Yue, H.-M. Yuan, J.-S. Chen, S.-N. Wang, Inorg. Chem. 2003, 42, 944.
| Crossref | GoogleScholarGoogle Scholar | 12588123PubMed |
[30] Y. Ke, D. J. Collins, D. Sun, H.-C. Zhou, Inorg. Chem. 2006, 45, 1897.
| Crossref | GoogleScholarGoogle Scholar | 16499346PubMed |
[31] F.-X. Gao, Y.-J. Ye, L.-T. Zhao, D.-H. Liu, Y. Li, Inorg. Chem. Commun. 2018, 94, 39.
| Crossref | GoogleScholarGoogle Scholar |
[32] X.-M. Lin, T.-T. Li, Y.-W. Wang, L. Zhang, C.-Y. Su, Chem. Asian J. 2012, 7, 2796.
| Crossref | GoogleScholarGoogle Scholar | 23038041PubMed |
[33] T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza, M. Haranczyk, Microporous Mesoporous Mater. 2012, 149, 134.
| Crossref | GoogleScholarGoogle Scholar |
[34] C. Janiak, J. Chem. Soc., Dalton Trans. 2000, 3885.
| Crossref | GoogleScholarGoogle Scholar |
[35] S.-Y. Zhang, D. Li, D. Guo, H. Zhang, W. Shi, P. Cheng, L. Wojtas, M. J. Zaworotko, J. Am. Chem. Soc. 2015, 137, 15406.
| Crossref | GoogleScholarGoogle Scholar | 26606156PubMed |
[36] Q. Zhang, M. Lei, F. Kong, Y. Yang, Chem. Commun. 2018, 10901.
| Crossref | GoogleScholarGoogle Scholar |
[37] S. Parshamoni, S. Sanda, H. S. Jena, S. Konar, Dalton Trans. 2014, 7191.
| Crossref | GoogleScholarGoogle Scholar | 24676502PubMed |
[38] S. Parshamoni, S. Sanda, H. S. Jena, S. Konar, Chem. Asian J. 2015, 10, 653.
| Crossref | GoogleScholarGoogle Scholar | 25523149PubMed |
[39] T. K. Maji, R. Matsuda, S. Kitagawa, Nat. Mater. 2007, 6, 142.
| Crossref | GoogleScholarGoogle Scholar | 17259990PubMed |
[40] A. M. Silvestre-Albero, J. M. Juárez-Galán, J. Silvestre-Albero, F. Rodríguez-Reinoso, J. Phys. Chem. C 2012, 116, 16652.
| Crossref | GoogleScholarGoogle Scholar |
[41] J.-R. Li, R. J. Kuppler, H.-C. Zhou, Chem. Soc. Rev. 2009, 38, 1477.
| Crossref | GoogleScholarGoogle Scholar | 19384449PubMed |
[42] S. J. Geier, J. A. Mason, E. D. Bloch, W. L. Queen, M. R. Hudson, C. M. Brown, J. R. Long, Chem. Sci. 2013, 4, 2054.
| Crossref | GoogleScholarGoogle Scholar |
[43] D. Wang, B. Liu, S. Yao, T. Wang, G. Li, Q. Huo, Y. Liu, Chem. Commun. 2015, 15287.
| Crossref | GoogleScholarGoogle Scholar |
[44] FS Process 1996 (Rigaku Corporation: Tokyo).
[45] O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard, H. Puschmann, J. Appl. Cryst. 2009, 42, 339.
| Crossref | GoogleScholarGoogle Scholar |
[46] G. Sheldrick, Acta Crystallogr. Sect. A 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[47] G. Sheldrick, Acta Crystallogr. Sect. C 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |