

Synthesis and Characterisation of Helicate and Mesocate Forms of a Double-Stranded Diruthenium(ii) Complex of a Di(terpyridine) Ligand
Kate L. Flint A , J. Grant Collins B , Siobhan J. Bradley C , Trevor A. Smith C , Christopher J. Sumby A and F. Richard Keene A DA Department of Chemistry, School of Physical Sciences, The University of Adelaide, Adelaide, SA 5005, Australia.
B School of Physical, Environmental and Mathematical Sciences, UNSW Canberra, Australian Defence Force Academy, Canberra, ACT 2600, Australia.
C ARC Centre of Excellence in Exciton Science, School of Chemistry, The University of Melbourne, Vic. 3010, Australia.
D Corresponding author. Email: richard.keene@adelaide.edu.au
Australian Journal of Chemistry 72(10) 762-768 https://doi.org/10.1071/CH19220
Submitted: 13 May 2019 Accepted: 30 May 2019 Published: 1 July 2019
Abstract
A diruthenium(ii) complex involving the di(terpyridine) ligand 1,2-bis{5-(5″-methyl-2,2′:6′,2″-terpyridinyl)}ethane was synthesised by heating an equimolar ratio of RuCl3 and the ligand under reflux conditions in ethylene glycol for 3 days, realising double-stranded helicate and mesocate forms which were chromatographically separated. The two species were obtained in relatively low yield (each ~7–9 %) from the reaction mixture. X-Ray structural studies revealed differences in the cavity sizes of the two structures, with the helicate structure having a significantly smaller cavity. Furthermore, the helicate and mesocate forms pack with notably different arrangements of the structures with the helicate having large solvent and anion filled pores. 1D/2D NMR studies revealed rigidity in the mesocate structure relative to that of the helicate, such that the –CH2CH2– signal was split in the former and appeared as a singlet in the latter. In a manner analogous to the behaviour of the parent [Ru(tpy)2]2+ coordination moiety (tpy = 2,2′:6′,2″-terpyridine), photophysical studies indicated that both the helicate and mesocate forms were non-emissive at ~610 nm at room temperature, but at 77 K in n-butyronitrile, both isomers showed emission at ~610 nm (λex 472 nm). However, the temporal emission characteristics were very different: time-resolved studies showed the emission of the helicate species decayed with a dominant emission lifetime of ~10 μs (similar to the emissive properties of free [Ru(tpy)2]2+ under the same conditions), whereas for the mesocate the emission lifetime was at least three orders of magnitude lower (~4 ns).
Introduction
Helicity is widely prevalent in nature – for example, in nucleic acids and in polypeptide segments which are a major component of proteins and enzymes. It is also present in artificial supramolecular structures – including many examples based on metal–ligand interactions.[1] These metallosupramolecular assemblies are generally classified as ‘helicates’,[2] and have a wide range of structures – including double-, triple-, and circular-helices.[1]
The prevalence of the helical motif in biology raises the interesting possibility of therapeutic applications for the metallo-helicates. Certainly, the chemotherapeutic use of platinum-based drugs is well known, but there can be severe adverse reactions, and there has been considerable interest in the use of other transition metals for drug development, in particular ruthenium complexes.[3] To this point, studies of helical ruthenium complexes and their biological effects have been limited: the first reports of such complexes were made by Crane and Sauvage,[4] and by Ho and co-workers,[5] but in neither of these cases were biological interactions studied. Subsequently, Hannon et al. have investigated dinuclear single-stranded,[6] double-stranded,[7] and triple-stranded species,[8] which showed varying degrees of anti-cancer activity – the most significant being a double-stranded species which revealed significantly greater activity to some breast cancer cell lines than cisplatin. Glasson et al.[9] reported a triple-stranded helical species which showed affinity to DNA oligonucleotides – and revealed significantly different interactions by the M and P chiral forms of the helicate.[1] Kumar et al.[10] have also reported a triple-stranded helical species, but it showed ‘very modest’ in vitro activity against bacteria. More recently, Allison and co-workers[11] reported double-stranded mesocates and helicates of ruthenium which showed varying degrees of selectivity and cytotoxicity to cancer cells in vitro.
As part of a study of the biological effects of helicate ruthenium complexes, including the effect of the helical chirality on such interactions, we chose to investigate complexes of the ligand 1,2-bis{5-(5″-methyl-2,2′:6′,2″-terpyridinyl)}ethane (1) (Chart 1). The di-iron double-stranded helix of this ligand was first reported by Rapenne et al.[12] as part of a study to develop precursors for the formation of trefoil knots, and it was subsequently the target of the first chromatographically resolved helicate species.[13] The present paper reports the synthesis, characterisation, and properties of the diruthenium species of this di(terpyridine) ligand, for which there are both helicate and mesocate forms.
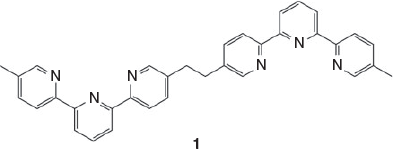
|
Results and Discussion
Synthesis
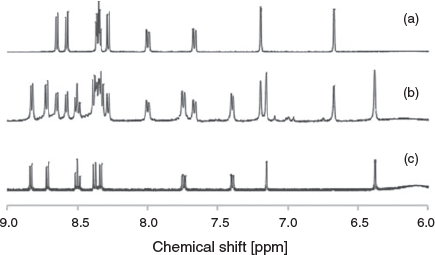
Initial attempts to synthesise the diruthenium(ii) double-stranded complex involving the di-terpyridine ligand 1,2-bis{5-(5″-methyl-2,2′:6′,2″-terpyridinyl)}ethane (1) were undertaken using a variety of ruthenium precursors {RuCl3·xH2O, [Ru(dmso)4Cl2][14] (dmso = dimethyl sulfoxide) and [Ru(H2O)6][15]}, heated with the ligand in a range of organic solvents (including isopropyl alcohol, 2-methoxyethanol, and ethylene glycol) for various reaction times and conditions (e.g. reflux, microwave, and in sealed tubes in the oven). All reactions yielded promising dark orange-red solutions, but the majority were found to contain predominantly polymeric material, from which the desired product could not be separated, and was generally not readily identified by NMR spectroscopy. However, the product from the reaction of RuCl3 and the ligand in ethylene glycol in a sealed tube in a 200°C oven for 4 days resulted in a more promising product, with broad peaks visible in the aromatic region of the 1H NMR spectrum corresponding closely to the coordinated ligand (Fig. S1, Supplementary Material). Separation of the double-stranded complex from polymeric material was achieved by preparative thin-layer chromatography (TLC) (silica) with a solvent mixture of 0.2 M NH4Cl in DMF/H2O (4 : 1). Upon isolation of the product, the 1H NMR spectrum revealed that two species were present (Fig. 1b) – one of which was the desired helicate and the other a secondary product. Separation of these complexes was achieved initially by crystallisation, with the helicate preferentially crystallising from the mixture using vapour diffusion of diethyl ether into an acetonitrile solution of the product and producing X-ray quality crystals. The secondary product, ultimately identified as the mesocate, was crystallised from the mixture using vapour diffusion of diisopropyl ether into an acetonitrile solution of the product, however X-ray quality crystals could not be obtained in this manner from the initial mixture. High-resolution ESI-MS revealed the two species to have ions of identical formula masses – m/z 311.0708 (corresponding to dinuclear {[Ru2(1)2]}4+) – indicating that the secondary product was likely the mesocate (Figs S19–S22, Supplementary Material).
|
Subsequent syntheses were performed under reflux conditions, heating for a 3-day time period, with separation of the complexes from the polymeric mixture achieved by vacuum liquid chromatography, utilising TLC grade silica.[16] The yields of the two complexes (~7–9 % of each) are relatively low, as expected because of the concomitant formation of the polymeric material, and consistent with other studies of similar helical complexes.[7,8,11] It is interesting to note that in these syntheses, while both the mesocate and helical forms were produced, the helicate/mesocate ratio increased with time. It would therefore appear that the helical form was the more thermodynamically favoured – although in the synthesis of inert complexes of ruthenium(ii) complexes involving polypyridyl ligands the observation of such thermodynamic equilibration is not common. It has, however, recently been reported in the isomerisation of the [Ru(Me4phen)(bb7)]2+ complex {where Me4phen is 3,4,7,8-tetramethyl-1,10-phenanthroline and bb7 is the tetradentate ligand bis[4(4′-methyl-2,2′-bipyridyl)]-1,7-heptane}.[17]
Recent work by Allison and co-workers,[11] involving ligands with two thiazole-bipyridine tridentate coordinating moieties, reported the first separation and characterisation of double-stranded diruthenium helicates and mesocates, and a modification of those methods allowed separation of the two species in this work using vacuum liquid chromatography on TLC grade silica[16] with the solvent mixture CH3CN/H2O/sat. aq. KNO3 (7 : 1 : 0.5). Crystallisation of the first fraction (containing the putative mesocate) by vapour diffusion of diisopropyl ether into an acetonitrile solution of the complex produced X-ray quality crystals from which the single crystal structure could be obtained, which confirmed the identity of the mesocate.
X-Ray Crystallography
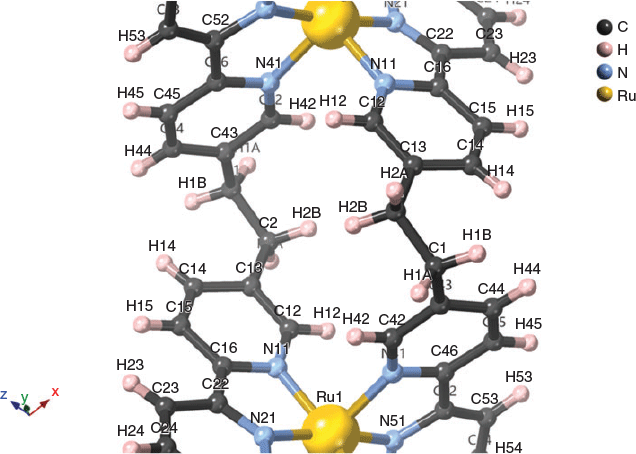
The structures of the ruthenium(ii) mesocate 2, and one of the two enantiomers of the ruthenium(ii) helicate 3 are shown in Fig. 2. For the mesocate 2, crystals suitable for X-ray diffraction were grown by vapour diffusion from diisopropyl ether/acetonitrile, and the resulting structure confirmed the formation of the mesocate complex of the type [Ru212]4+. The structure crystallises in the space group I2/a. Present in the asymmetric unit are half of a ruthenium(ii) mesocate cation and two PF6− anions. The two octahedral ruthenium(ii) centres are separated by 9.305 Å and bridged by two ligands such that the stereochemistry of the metal centres of each discrete unit is ΛΔ.[18] Inclusion of a van der Waals surface shows a central cavity in the structure due to the spatial orientation of the –CH2CH2– backbones (Fig. S13, Supplementary Material).

|
For the helicate 3, crystals suitable for X-ray diffraction were grown by vapour diffusion from diethyl ether/acetonitrile, and the resulting structure confirmed the formation of the helicate complex of the type [Ru212]4+. The structure crystallises in the space group P21/n. Present in the asymmetric unit are one ruthenium(ii) helicate cation and four PF6− anions. The two octahedral ruthenium(ii) centres are separated by 7.661 Å and bridged by two ligands such that the stereochemistry of the metal centres of each discrete unit is either ΔΔ or ΛΛ.[18] Inclusion of a van der Waals surface shows a significantly smaller central cavity than the mesocate, following the helical twist of the structure (Fig. S16, Supplementary Material). The closer interactions of the ligand backbone may contribute to the greater thermodynamic stability of 3 compared with 2.
Interestingly, the distance between ruthenium metal centres in the complexes described above varies significantly from the iron counterpart, as reported by Rapenne et al.[13] In the iron(ii) helicate the Fe–Fe distance is reported to be 8.31 Å for the ΛΛ double helix (and 7.93 Å for the racemic mixture). As reported above, for the racemate of the ruthenium(ii) helicate the Ru–Ru distance is 7.661 Å, and for the mesocate the Ru–Ru distance is 9.305 Å.
Comparison of the crystal packing of the helicate and mesocate structures revealed notably different densities – 1.319 and 1.708 g cm−3, respectively. The lower density of the helicate is in the main due to the use of the SQUEEZE routine of Platon[19] and exclusion of the solvent (which could not be identified) from the formula, artificially decreasing the density. This is supported by the packing when viewed down the a axis, which shows small voids, likely to contain disordered solvent and residual anions; while along the c axis the packing is arranged with alternating cation and anion layers (Fig. S17, Supplementary Material). In the ab plane the helicate molecules pack in a ‘loose’ herring-bone pattern, also incorporating anions, in order to maximise π-stacking and edge-to-face π-stacking interactions (Fig. S18, Supplementary Material). The packing of the mesocate shows layers comprised of integrated cation–anion entities (Fig. S14, Supplementary Material). Examination of the (101) plane shows the mesocate molecules form columns by interdigitation, allowing the formation of edge-to-face π-stacking interactions and methyl C–H⋯π contacts (Fig. S15, Supplementary Material).
NMR Studies
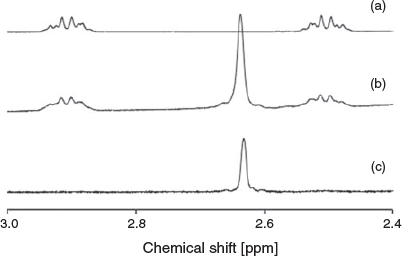
The observed 1H and 13C NMR resonances are consistent with the X-ray structures, with ROESY experiments assisting with assignment of all signals (Table S1, Supplementary Material). Interestingly, the signal corresponding to the bridge (–CH2CH2–) in the helicate 3 appears as a singlet, whereas in the mesocate 2 this signal is split (Fig. 3). This was supported by ROESY experiments, showing through-space correlations between the protons on the bridge and aromatic signals (Figs S5 and S12, Supplementary Material).
|
Analysis of the through-space correlations of the bridge (–CH2CH2–) proton signals to aromatic protons on the coordinated pyridine rings by ROESY NMR experiments, coupled with the spatial distances elucidated from the crystal structure, allowed assignment of the multiplet signals at 2.90 and 2.50 ppm for the mesocate. Using the annotation from the crystal structure, the multiplet at 2.90 ppm can be assigned as H1A/B and the multiplet at 2.50 ppm assigned as H2A/B (Fig. 4; Fig. S8, Supplementary Material). Based on the interatomic distances, H1A/B displays strong correlations with H44 and H14, while H2A/B has strong correlations with H42 and H12.
|
Significantly weaker correlations between H1A–H42 (2.90–7.19 ppm) and H2A–H14 (2.50–8.00 ppm) are also observed, which is consistent with the interatomic distances and spatial arrangement of the mesocate. The splitting of the signals, appearing as two multiplets rather than as a singlet (as in the case of the helicate), is due to the two CH2 groups becoming chemical-shift inequivalent, combined with each pair of geminal protons being magnetically inequivalent. As a consequence, the AA’XX’ system appears as a complicated non-first order pair of multiplets. An HSQC experiment was conducted to assist in assigning these multiplet signals to specific positions on the –CH2CH2– bridge, based on the 1H−13C coupling (Figs S6 and S7, Supplementary Material). However, these showed the bridge carbons to be equivalent, with both multiplets correlated with a signal carbon peak at ~30 ppm, preventing confirmation by this method.
The splitting of the 1H NMR signal corresponding to the ethylene bridge is not without precedent. Zhang and Dolphin reported similar observations for CoIII mesocate and helicate complexes, containing a methylene bridge.[20] In the case of the helicate the CH2 protons appeared as a singlet, while in the mesocate the authors describe the signal as two sets of doublets – although this could also be described as a non-first order multiplet.
Photophysics
Polypyridylruthenium(ii) complexes have an extensive and rich photophysical and photochemical literature, particularly in the case of tris(bidentate) species based on 2,2′-bipyridine (bpy) and 1,10-phenanthroline (phen) and their derivatives. Such mononuclear species are highly emissive, even at room temperature, with the triplet metal to ligand charge transfer (3MLCT) excited state having a high quantum yield and a significant lifetime (e.g. for [Ru(bpy)3]2+, emission occurs in aqueous solution at room temperature at ~625 nm with a lifetime of ~600 ns and a quantum yield of ~0.04).[21] Interestingly, the closely related bis(tridentate) complex [Ru(tpy)2]2+ (tpy = 2,2′:6′,2″-terpyridine) is not emissive at room temperature – attributed to non-radiative deactivation of the excited triplet state through coupling with a higher-energy metal-centred state – but shows significant emission (at ~610 nm) at 77 K in an n-butyronitrile or ethanol/methanol glass.[22]
We were interested to investigate the photophysical behaviour of the diruthenium helicate complexes, particularly in the case where the RuII coordination environment involved the ‘Ru(tpy)22+’ moiety – and to establish any variation between the helicate and mesocate forms. There are limited cases of photophysical studies of such complexes: in their studies of the double-stranded complex [Ru2(qpy)2(C2O4)]2+ (qpy = 2,2′:6′,2″:6″,2‴:6‴,2″″quinquepyridine) in which one metal centre is coordinatively bis(tridentate) and the other tris(bidentate), Ho et al.[5] reported an emission centred at 715 nm at 77 K in an EtOH/MeOH glass, attributed to the 3MLCT state. For two cases of triple-stranded diruthenium helicates involving tris(bidentate) coordination, Hannon et al.[8a] and Glasson et al.[9] reported emission at 705 and 604 nm, respectively, at room temperature. In the only analogous example to the present complex, Allison et al.[11] reported a diruthenium double-stranded complex involving a ligand with two thiazole-bipyridine domains separated by a triphenylene spacer unit which formed both helicate and mesocate forms – as in the present case. While the two forms were apparently non-emissive at room temperature, the authors noted that the helicate showed emission (~610 nm) at 77 K in an EtOH/MeOH glass: no comment was made regarding the mesocate form.
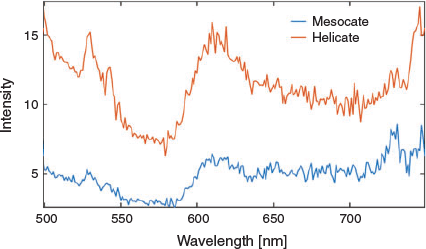
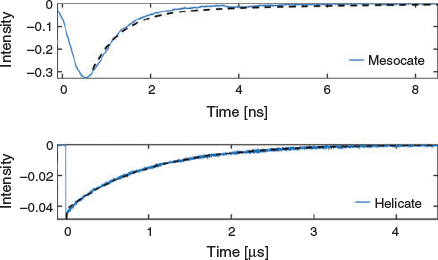
For the present complexes, photophysical studies revealed that both the helicate and mesocate forms showed no emission at ~610 nm at room temperature. However, at 77 K in n-butyronitrile, excitation at λex 472 nm revealed that both isomers showed emission at ~610 nm – as expected for the ‘Ru(tpy)22+’ moiety – but the temporal emission characteristics were very different. Time-resolved studies showed the emission of the helicate species at 610 nm decayed with a dominant emission lifetime of ~10 μs, which is analogous to the emissive properties of free [Ru(tpy)2]2+ under the same conditions,[22] while for the mesocate the emission lifetime was at least three orders of magnitude lower (~4 ns). The emission spectra and emission decay curves for the two isomers are shown in Figs 5 and 6, respectively. The reason for the large difference in emission timescales of the two compounds is not entirely understood at present, but we speculate that the short emission decay time of the mesocate relative to the helicate could be accounted for by a larger value of the radiative rate constant (kp) in the former compared with the latter. Since the emission intensities of the two compounds at 610 nm are reasonably similar (φp(mesocate) ~ φp(helicate)), and given that φp = kpτp, and τp = 1/(kp + kn) (where kn is the non-radiative rate constant and τp is the phosphorescence lifetime) an increase in the kp, along with little difference in kn, could lead to comparable emission intensities, but different emission decay times. The difference in the radiative rate constant is presumably linked to the electronic interactions between the metal and ligand that are in turn dependent on the structural differences, and the associated strain/metal–ligand distance and angle, of the respective compounds.
|
|
Further studies using excitation at λex 315 nm revealed significant additional emission at ~400 nm in both isomers (at both 77 K and room temperature), which by analogy with studies undertaken by Lakshmanan et al.[23] on mononuclear complexes of a series of 4′-aryl-substituted tpy ligands which showed similar emission, could be assigned to charge transfer between the ligand-centred (LC) transitions of the tpy ligands. Time-resolved studies with UV excitation (λex 315 nm) showed some of this emission decays on timescales of tens of nanoseconds at 77 K, but a detailed analysis of this behaviour will be published subsequently.
Conclusions
The synthesis of the diruthenium(ii) complex involving the di(terpyridine) ligand 1,2-bis{5-(5″-methyl-2,2′:6′,2″-terpyridinyl)}ethane resulted in the isolation of double-stranded helicate and mesocate forms which were chromatographically separated. X-Ray structural studies revealed differences in the cavity sizes of the two structures, with the helicate structure having a significantly smaller cavity, and 1D/2D NMR studies indicated rigidity in the mesocate structure relative to that of the helicate, such that the –CH2CH2– signal split in the former and appeared as a singlet in the latter. Preliminary photophysical studies of the two isomers showed both to be non-emissive at ~610 nm at room temperature, but at 77 K in n-butyronitrile, they exhibited emission at ~610 nm (λex 472 nm); however, the temporal emission characteristics were very different with the dominant emission lifetimes of the helicate and mesocate being ~10 μs and ~4 ns, respectively.
At this stage, the helical form has not been resolved into its chiral forms, but that aspect will be reported subsequently, along with the implications of their biological interactions.
Experimental
Materials and Instruments
Commercially available reagents and reagent grade solvents were used without further purification unless otherwise indicated. Lithium diisopropylamide (LDA) was freshly prepared by addition of n-butyllithium (2.5 M in hexanes) to a solution of distilled diisopropylamine in tetrahydrofuran (THF). All other solvents and reagents were purified according to the literature.[24] Where possible, reactions were monitored by TLC on MERCK aluminium-backed silica gel 60 F254 plates or MERCK aluminium-backed neutral aluminium oxide 150 F254 plates, and visualised under UV light at λ 254 nm. Flash column chromatography was performed using Fluka aluminium oxide (0.05–0.15 mm, pH 7). Vacuum liquid chromatography was performed using Sigma–Aldrich (2–25 micron) TLC silica gel (2–25 micron).[16] All 1H NMR (499.818 MHz) and 13C NMR (125.692 MHz) spectra were obtained using an Agilent 500 MHz NMR spectrometer at 26°C. Spectra of samples were recorded in solutions of CDCl3, using TMS as an internal standard, or CD3CN. The following abbreviations for hydrogen multiplicities were used: s, singlet; d, doublet; t, triplet; m, multiplet; and br, broad. 1H signals were assigned with assistance of ROESY 2D NMR experiments where required.
High resolution mass spectrometry (ESI, positive ion mode) experiments of samples dissolved in acetonitrile were run using a Bruker maXis 3G UHR-TOF mass spectrometer, at the University of Canterbury (Christchurch, New Zealand). UV/Vis absorption spectra were acquired at ambient temperature on a Cary 1E UV-Visible Spectrophotometer (scan range: 600–200 nm, scan rate: 600.00 nm min−1, SBW: 2.0 nm). Emission spectra and kinetics were acquired at 77 K using an Oxford Instruments cryostat (OptistatDN), at the University of Melbourne. Steady-state emission spectra were recorded using a fluorimeter (Agilent, Eclipse) with 5 nm bandwidth on both excitation and emission monochromators. Time-resolved emission measurements were performed using a tuneable nanosecond optical parametric oscillator (Eskpla NT340) as the excitation source (~5 ns pulses at 10 Hz), an emission monochromator (Acton SpectraPro 300i), long-pass filter (GG550), photomultiplier tube (Hamamatsu R928), and digital oscilloscope (Teledyne/LeCroy WaveSurfer 10). 5-Methyl-2-(tributylstannyl)pyridine and 5,5″-dimethyl-2,2′:6′,2″-terpyridine were prepared according to the literature procedures.[25]
Synthesis of 1,2-Bis{5-(5′-methyl-2,2′:6′,2′-terpyridinyl)}ethane (1)
Compound 1 was synthesised using a modified procedure of that described by Rapenne et al.[12] A solution of 5,5″-dimethyl-2,2′:6′,2″-terpyridine (0.50 g, 1.915 mmol; 1 equiv.) in anhydrous THF (62.5 mL) was cooled to −78°C under Ar. Freshly prepared LDA (1.925 mmol in 7.5 mL THF; 1 equiv.) was added dropwise and the reaction mixture stirred at −78°C for 2 h. The mixture was warmed to 0°C, and then cooled again to −78°C. 1,2-Dibromoethane (0.74 mL,1.61 g; 8.57 mmol; 4.5 equiv.) was added, and the reaction mixture stirred at −78°C for 4 h, after which it was warmed to room temperature. After stirring at room temperature for a further 4 h, deionised water (50 mL) was added, and the THF removed by rotary evaporation. The aqueous solution was extracted with dichloromethane (3 × 25 mL), washed with deionised water (2 × 25 mL), brine (1 × 25 mL), dried over MgSO4, filtered, and the product purified by column chromatography (neutral Al2O3: initial elution with dichloromethane removed unreacted starting material, while subsequent elution with dichloromethane/methanol (9 : 1 by volume) allowed isolation of crude product). The crude product was further purified by reprecipitation from dichloromethane using diethyl ether to give 1,2-bis{5-(5″-methyl-2,2′:6′,2″-terpyridinyl)}ethane (1) as a white crystalline solid (0.13 g, 26 %). δH (500 MHz, CDCl3) 8.57–8.46 (m, J 18.0, 7.8, 4H), 8.39 (d, J 7.9, 2H), 7.92 (t, J 7.8, 1H), 7.62 (dd, J 19.3, 7.7, 2H), 3.08 (s, 2H), 2.41 (s, 3H).
Synthesis of [Ru212]4+ Complex
A solution of RuCl3·3H2O (10 mg, 0.038 mmol; 1 equiv.) and 1,2-bis{5-(5″-methyl-2,2′:6′,2″-terpyridinyl)}ethane (20 mg, 0.038 mmol; 1 equiv.) in ethylene glycol (10 mL) was heated to reflux under N2 for 3 days. The reaction mixture was cooled to room temperature, deionised water added (20 mL), and filtered through Celite. NH4PF6 solution (4 %, 5 mL) was added, the resulting precipitate isolated using centrifugation (15652 g, 5 min), washed with water, and dried under vacuum to give the crude product.
Separation of Complexes from Polymeric Material
The crude product was purified from polymeric material by vacuum liquid chromatography,[16] on silica gel using 0.2 M NH4Cl in DMF/H2O (4 : 1 by volume) as the eluent. This yielded a mixture of the mesocate and helicate [Ru212]4+ complexes (Rf 0.57 for a TLC using the same adsorbent/eluent combination).
Separation of Mesocate and Helicate
The separation of the mesocate and helicate forms was achieved using a procedure modified from that outlined by Rice and co-workers for double-stranded diruthenium helicates and mesocates.[11] The mixed product obtained above was subjected to vacuum liquid chromatography,[16] using CH3CN/H2O/sat. aq. KNO3 (7 : 1 : 0.5 by volume) as the eluent. This yielded the mesocate 2 as the first-eluted species (Rf 0.36 for a TLC using the same adsorbent/eluent combination) as a red orange solid (2.4 mg, 7 %). Mp > 260°C. λmax (CH3CN)/nm (ϵ/M−1 cm−1) 472 (5.98 × 103), 315 (3.43 × 104), 273 (2.55 × 104), 252 (2.45 × 104). δH (500 MHz, CD3CN) 8.65 (d, J 8.1, 4H), 8.58 (d, J 8.1, 4H), 8.41–8.31 (m, J 8.1, 3.9, 8H), 8.28 (d, J 8.2, 4H), 8.00 (dd, J 8.4, 1.5, 4H), 7.67 (dd, J 8.2, 0.6, 4H), 7.19 (d, J 1.5, 4H), 6.67 (d, J 0.6, 4H), 2.97–2.84 (m, J 5.1, 4H), 2.56–2.44 (m, J 3.6, 4H,), 2.00 (s, 12H). δC (126 MHz, CD3CN) 158.28, 158.19, 157.86, 157.48, 156.78, 153.56, 141.72, 141.37, 141.32, 140.91, 138.33, 126.13, 125.57, 125.45, 125.40, 31.64, 20.09. m/z (ESI) 311.0708 ([Ru2(L)2]4+).
The second product was subsequently eluted, yielding the helicate 3 (Rf 0.28 for a TLC using the same adsorbent/eluent combination) as an orange solid (3.1 mg, 9 %). Mp > 260°C λmax (CH3CN)/nm (ϵ/M−1 cm−1) 476 (1.64 × 103), 315 (1.17 × 104), 276 (1.16 × 104), 251 (1.16 × 104). δH (500 MHz, CD3CN) 8.83 (d, J 8.1, 4H), 8.71 (d, J 8.1, 4H), 8.50 (t, J 8.1, 4H), 8.38 (d, J 8.3, 4H), 8.34 (d, J 8.2, 4H), 7.74 (d, J 7.3, 4H), 7.40 (d, J 10.4, 4H), 7.15 (s, 4H), 6.38 (s, 4H), 2.63 (s, 8H), 2.03 (s, 12H). δC (126 MHz, CD3CN) 158.47, 158.21, 157.93, 157.04, 155.22, 152.90, 141.80, 141.42, 141.16, 140.18, 138.65, 126.69, 126.21, 125.92, 125.41, 30.79, 19.88. m/z (ESI) 311.0709 ([Ru2(1)2]4+).
X-Ray Crystallography
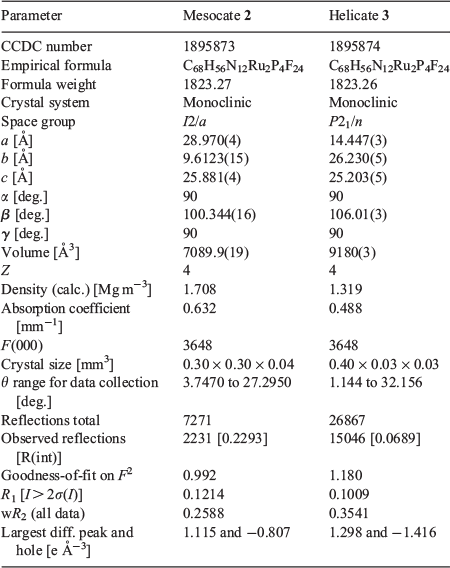
Crystals suitable for X-ray diffraction were grown by vapour diffusion from diisopropyl ether/acetonitrile (mesocate), and diethyl ether/acetonitrile (helicate). Single crystals were mounted in paratone-N oil on a plastic loop. X-Ray diffraction data were collected at 150(2) K on an Oxford X-Calibur single crystal diffractometer (λ 0.71073 Å) or on the MX2 beamline of the Australian Synchrotron at 100(2) K.[26] Datasets were corrected for absorption using a multi-scan method, and structures were solved by direct methods using SHELXS-2014,[27] or SHELXT,[28] and refined by full-matrix least-squares on F2 by SHELXL-2014/2018,[29] interfaced through the program X-Seed.[30] In general, all non-hydrogen atoms were refined anisotropically and hydrogen atoms were included as invariants at geometrically estimated positions. The crystallographic details for the structures of 2 and 3 are given in Table 1.
|
Full details of the structure determinations have been deposited with the Cambridge Crystallographic Data Centre as CCDC 1895873 (2) and 1895874 (3). Copies of this information may be obtained free of charge from The Director, CCDC, 12 Union Street, Cambridge CB2 1EZ, UK (fax: +441223-336-033, email: deposit@ccdc.cam.ac.uk).
Supplementary Material
NMR spectra and assignments, X-ray crystallography details, including packing diagrams, HR-ESMS spectra, and UV/vis spectra of the mesocate 2 and helicate 3 complexes are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors wish to thank Dr Marie Squire (University of Canterbury, New Zealand) for her assistance with ESI high-resolution mass spectrometry measurements. SJB and TAS acknowledge the support of the ARC Centre of Excellence in Exciton Science (CE170100026). Aspects of this research were undertaken on the MX1 beamline at the Australian Synchrotron, Victoria, Australia.
References
[1] M. Albrecht, Chem. Rev. 2001, 101, 3457.| Crossref | GoogleScholarGoogle Scholar | 11840991PubMed |
[2] J. M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier, D. Moras, Proc. Natl. Acad. Sci. USA 1987, 84, 2565.
| Crossref | GoogleScholarGoogle Scholar | 3472223PubMed |
[3] X. Li, A. K. Gorle, M. K. Sundaraneedi, F. R. Keene, J. G. Collins, Coord. Chem. Rev. 2018, 375, 134.
| Crossref | GoogleScholarGoogle Scholar |
[4] J. D. Crane, J. P. Sauvage, New J. Chem. 1992, 16, 649.
[5] P. K. K. Ho, K. K. Cheung, C. M. Che, Chem. Commun. 1996, 1197.
| Crossref | GoogleScholarGoogle Scholar |
[6] U. McDonnell, M. R. Hicks, M. J. Hannon, A. Rodger, J. Inorg. Biochem. 2008, 102, 2052.
| Crossref | GoogleScholarGoogle Scholar | 18664401PubMed |
[7] A. C. G. Hotze, B. M. Kariuki, M. J. Hannon, Angew. Chem. Int. Ed. 2006, 45, 4839.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) G. I. Pascu, A. C. G. Hotze, C. Sanchez-Cano, B. M. Kariuki, M. J. Hannon, Angew. Chem. Int. Ed. 2007, 46, 4374.
| Crossref | GoogleScholarGoogle Scholar |
(b) L. Cardo, I. Nawroth, P. J. Cail, J. A. McKeating, M. J. Hannon, Sci. Rep. 2018, 8, 13342.
| Crossref | GoogleScholarGoogle Scholar |
[9] C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, J. A. Smith, F. R. Keene, C. Motti, Chem. – Eur. J. 2008, 14, 10535.
| Crossref | GoogleScholarGoogle Scholar |
[10] S. V. Kumar, W. K. C. Lo, H. J. L. Brooks, J. D. Crowley, Inorg. Chim. Acta 2015, 425, 1.
| Crossref | GoogleScholarGoogle Scholar |
[11] S. J. Allison, D. Cooke, F. S. Davidson, P. I. P. Elliott, R. A. Faulkner, H. B. S. Griffiths, O. J. Harper, O. Hussain, P. J. Owen-Lynch, R. M. Phillips, C. R. Rice, S. L. Shepherd, R. T. Wheelhouse, Angew. Chem. Int. Ed. 2018, 57, 9799.
| Crossref | GoogleScholarGoogle Scholar |
[12] G. Rapenne, C. Dietrich-Buchecker, J. P. Sauvage, J. Am. Chem. Soc. 1999, 121, 994.
| Crossref | GoogleScholarGoogle Scholar |
[13] G. Rapenne, B. T. Patterson, J. P. Sauvage, F. R. Keene, Chem. Commun. 1999, 1853.
| Crossref | GoogleScholarGoogle Scholar |
[14] I. P. Evans, A. Spencer, G. Wilkinson, J. Chem. Soc., Dalton Trans. 1973, 204.
| Crossref | GoogleScholarGoogle Scholar |
[15] P. Bernhard, H. B. Buergi, J. Hauser, H. Lehmann, A. Ludi, Inorg. Chem. 1982, 21, 3936.
| Crossref | GoogleScholarGoogle Scholar |
[16] J. C. Coll, B. F. Bowden, J. Nat. Prod. 1986, 49, 934.
| Crossref | GoogleScholarGoogle Scholar |
[17] B. Sun, H. M. Southam, J. A. Butler, R. K. Poole, A. Burgun, A. Tarzia, F. R. Keene, J. G. Collins, Dalton Trans. 2018, 47, 2422.
| Crossref | GoogleScholarGoogle Scholar | 29379923PubMed |
[18] It is noted that for a bis(2,2′:6′,2″-terpyridine) metal centre, [M(tpy)2]n+, the molecule possesses C2v point group symmetry and is therefore achiral. While the title complexes in this study possess such a bis(tridentate) immediate coordination environment, it is noted that the ‘tpy’ groups are unsymmetrically substituted with a methyl group in the 5″-position of the ‘outer’ pyridine ring, and the ethyl bridge to the other tridentate unit in the 5-position of the ‘inner’ pyridine ring. This substitution pattern means the bis(tridentate) coordination moieties are asymmetric and therefore each metal centre will have a chirality. The chirality can be defined by the ‘bis(bipyridine)’ configuration using the two ‘outer’ pyridine rings of the tpy moieties – in which case the helicate will have ΔΔ or ΛΛ configurations (ΔΔ in Fig. 2), and the mesocate will have a ΛΔ configuration (Λ at top in Fig. 2).
[19] A. Spek, Acta Crystallogr. C 2015, 71, 9.
| Crossref | GoogleScholarGoogle Scholar |
[20] Z. Zhang, D. Dolphin, Chem. Commun. 2009, 6931.
| Crossref | GoogleScholarGoogle Scholar |
[21] A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser, A. von Zelewsky, Coord. Chem. Rev. 1988, 84, 85.
| Crossref | GoogleScholarGoogle Scholar |
[22] A. Amini, A. Harriman, A. Mayeux, Phys. Chem. Chem. Phys. 2004, 6, 1157.
| Crossref | GoogleScholarGoogle Scholar |
[23] R. Lakshmanan, N. C. Shivaprakash, S. Sindhu, J. Fluoresc. 2018, 28, 173.
| Crossref | GoogleScholarGoogle Scholar | 28956219PubMed |
[24] W. L. F. Armarego, C. Chai, in Purification of Laboratory Chemicals (7th edn) (Eds W. L. F. Armarego, C. Chai) 2013, pp. 103–554 (Butterworth-Heinemann: Boston, MA).
[25] (a) U. S. Schubert, C. Eschbaumer, M. Heller, Org. Lett. 2000, 2, 3373.
| Crossref | GoogleScholarGoogle Scholar | 11029214PubMed |
(b) T. Seckin, I. Özdemir, S. Köytepe, N. Gürbüz, J. Inorg. Organomet. Polym. 2009, 19, 143.
| Crossref | GoogleScholarGoogle Scholar |
[26] D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson, T. T. Caradoc-Davies, J. Synchrotron Radiat. 2018, 25, 885.
| Crossref | GoogleScholarGoogle Scholar | 29714201PubMed |
[27] G. Sheldrick, Acta Crystallogr. Sect. A 1990, 46, 467.
| Crossref | GoogleScholarGoogle Scholar |
[28] G. Sheldrick, Acta Crystallogr. Sect. A 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[29] G. Sheldrick, Acta Crystallogr. Sect. C 2015, 71, 3.
| Crossref | GoogleScholarGoogle Scholar |
[30] L. J. Barbour, J. Supramol. Chem. 2001, 1, 189.
| Crossref | GoogleScholarGoogle Scholar |